
Syndróm konštitučného deficitu mismeč opravného systému (CMMR-D) – kazuistika rodiny s bialelickou MSH6 mutáciou
Constitutional Mismatch Repair-Deficiency Syndrome (CMMR-D) – a Case Report of a Family with Biallelic MSH6 Mutation
This work gives comprehensive information about new recessively inherited syndrome characterized by development of childhood malignancies. Behind this new described syndrome, called Constitutional mismatch repair-deficiency syndrome (CMMR-D), there are biallelic mutations in genes, which cause adult cancer syndrom termed Lynch syndrom (Hereditary non-polyposis cancer syndrom-HNPCC) if they are heterozygous mutations. Biallelic germline mutations of genes MLH1, MSH2, MSH6 and PMS2 in CMMR-D are characterized by increased risk of hematological malignancies, atypical brain tumors and early onset of colorectal cancers. An accompanying manifestation of the disease are skin spots with diffuse margins and irregular pigmentation reminiscent of Café au lait spots of NF1. This paper reports a case of a family with CMMR-D caused by novel homozygous MSH6 mutations leading to gliomatosis cerebri, T-ALL in an 11-year-old female and glioblastoma multiforme in her 10-year-old brother, both with rapid progression of the diseases. A literature review of brain tumors in CMMR-D families shows that they are treatment-resistant and lead to early death. Therefore, this work highlights the importance of early identification of patients with CMMR-D syndrome – in terms of initiation of a screening program for early detection of malignancies as well as early surgical intervention.
Key words:
constitutional mismatch repair-deficiency syndrome (CMMR-D) – MSH6 – high-grade brain tumors – childhood T-NHL
The author declares he has no potential conflicts of interest concerning drugs, pruducts, or services used in the study.
The Editorial board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Submitted:
24. 4. 2012
Accepted:
25. 6. 2012
Autori:
D. Ilenčíková
Pôsobisko autorov:
II. detská klinika, LF UK a DFNsP Bratislava, Slovenská republika
Vyšlo v časopise:
Klin Onkol 2012; 25(Supplementum): 34-38
Súhrn
Práca podáva prehľadné informácie o novom recesívne dedičnom syndróme, ktorý je charakteristický vývojom malignít v detskom veku. So vznikom tohto syndrómu, nazývaného Syndróm konštitučného deficitu mismeč opravného systému (CMMR-D), sa spájajú bialelické mutácie v génoch, ktoré sú v heterozygotnom stave zodpovedné za vznik Lynchovho syndrómu (Dedičný nádor hrubého čreva bez polypózy – HNPCC). Bialelické zárodočné mutácie génov MLH1, MSH2, MSH6 and PMS2 pri CMMR-D sú charakterizované zvýšeným rizikom hematologických malignít, atypických nádorov mozgu a včasným nástupom nádorov hrubého čreva. Sprievodným prejavom ochorenia je výskyt kožných makúl s difúznymi okrajmi a nerovnomernou pigmentáciou, podobných Café au lait škvrnám u neurofibromatózy 1. Práca predstavuje kazuistiku rodiny s CMMR-D, u ktorej bola objavená homozygotná mutácia MSH6 génu vedúca k vývoju gliomatózy mozgu a T-ALL u 11-ročného dievčatka a glioblastoma multiforme u jej 10-ročného brata. Oba mozgové tumory mali rýchlu progresiu ochorenia. Literárna rešerš mozgových tumorov u detí z CMMR-D rodín poukazuje na rezistenciu týchto mozgových tumorov voči liečbe a skorú smrť u detí. Preto táto práca upozorňuje na dôležitosť skorej identifikácie pacientov s CMMR-D syndrómom. Jednak z hľadiska včasného zahájenia screeningového programu, ako aj skorej chirurgickej intervencie.
Kľúčové slová:
syndróm konštitučného deficitu mismeč opravného systému (CMMR-D) – MSH6 – high-grade mozgové tumory – T-bunkový lymfóm v detskom veku
Úvod
Opravný systém pre chybne spárované bázy (ang. MisMatch Repair – MMR) zabezpečuje integritu genómu a gény MLH1, MSH2, MSH6 a PMS2 zohrávajú v tomto procese kľúčovú úlohu. MMR systém koriguje chybne spárované bázy, ktoré vznikajú počas replikácie DNA [1,2]. Tento systém je okrem opravy chybne spárovaných báz zapojený aj do celulárnej odpovede voči mnohým agensom, ktoré poškodzujú DNA a tiež do prestavby imunoglobulínov [3]. Heterozygotné zárodočné mutácie v génoch MLH1, MSH2, MSH6 a PMS2 sú kauzálne asociované so vznikom Lynchovho syndrómu (LS), ktorý predstavuje autozómovo-dominantné dedičné nádorové ochorenie, známe aj pod názvom Dedičný nádor hrubého čreva a konečníka (HNPCC). Ochorenie sa prejavuje vznikom nádoru kolorekta, maternice, žalúdka a iných malignít v tretej až piatej dekáde života. Tumory asociované s LS sa prejavia po strate funkcie „wild type“ (alely divého typu) u horeuvedených MMR génov a prítomnosťou mikrosatelitovej instability [4].
U jedincov s bialelickou zárodočnou mutáciou v MMR génoch vzniká zriedkavé ochorenie, ktoré bolo prvýkrát rozpoznané v roku 1999 [5]. Z historického pohľadu výskyt skorého nástupu nádoru hrubého čreva, lymfómov, leukémií a znakov neurofibromatózy, podnietili Bandipalliam et al [6] v roku 2005 pomenovať ochorenie „CoLoN“ syndróm (Co – kolon, Lo – lymfómy a N – neurofibromatóza). V roku 2007 bolo Feltonom et al [7] nazvané Lynchov syndróm III, keďže sa u pacientov manifestovali nádory charakteristické pre Lynchov syndróm typu I a II. Skupina nemeckých autorov Krüger et al [8] mu priradila názov „Syndróm detských malignít“. Posledne bolo ochorenie pomenované podľa genetického mechanizmu svojho vzniku a dostalo na prvý pohľad značne krkolomný názov „Syndróm konštitučného deficitu Mismeč opravného systému – CMMR-D“ [9]. Doterajšie poznatky z 89 rešeršovaných publikácii o 92 pacientoch s CMMR-D za posledných 12 rokov charakterizujú ochorenie podľa hlavných rozpoznávacích znakov nasledovne [2]:
- viacpočetné „Café au lait“ makuly (CALM),
- malignity iné ako pri neurofibromatóze typ I (NF1),
- včasný výskyt malignity,
- špecifická genealógia,
- molekulovo-geneticky potvrdená bialelická zárodočná mutácie v MMR génoch.
- Viacpočetné „Café au lait“ makuly
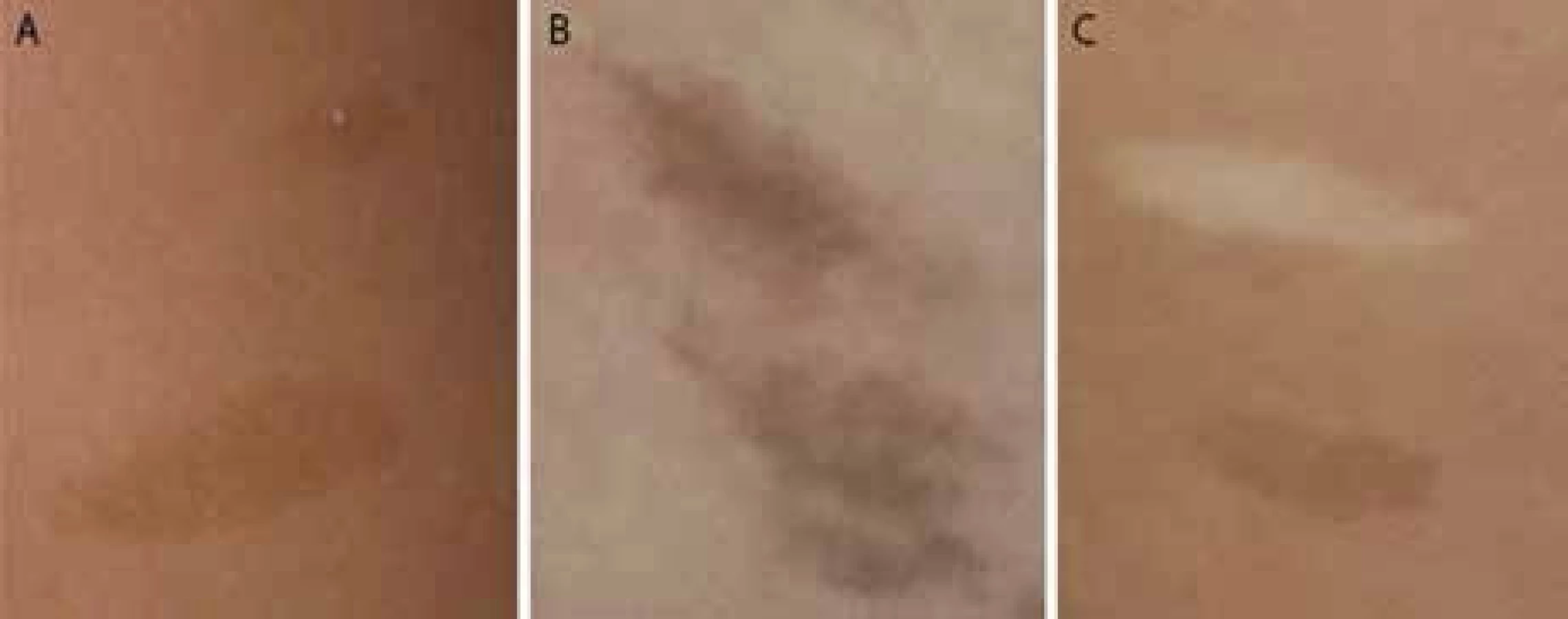
Kožné hyperpigmentácie sa podobajú „café au lait“ makulám u neurofibromatózy 1 (obr. 1A). Na rozdiel od NF1 makúl sa u detí s CMMR-D syndrómom nachádzajú na koži difúzne, neostro ohraničené okraje a nerovnomerná pigmentácia makúl, ako je to zobrazené na obr. 1B. U CMMR-D syndrómu sa spolu s hyperpigmentovanými ložiskami vyskytujú často aj hypopigmentované ložiská (obr. 1C). - Malignity iné ako pri NF1
CMMR-D vykazuje v porovnaní s NF1 rozdielne spektrum malignít. Z hematologických malignít u CMMR-D prevažujú lymfómy, z nádorov mozgu astrocytómy, glioblastómy a solidné tumory sú charakteristické pre LS. Pričom u NF1 z hematologických malignít sú najčastejšie zastúpené Juvenilná myelomonocytová leukémia, z nádorov mozgu gliómy a zo solidných nádorov neurofibrosarkómy a rhabdomyosarkómy [10]. - Včasný výskyt malignity
Výskyt prvých malignít (nádory mozgu, leukémie) je pre ochorenie charakteristický už od 2. roku života a rovnako vznik ďalších typov nádorov so zvyšujúcim sa vekom. Pre CMMR-D sú rozpoznávajúcim znakom viacpočetné malignity u pacienta. Tumory, ktoré sú asociované s Lynchovým syndrómom, sa vyskytujú často v súrodeneckej línii a v mladom veku. - Špecifická genealógia
Výskyt viacerých malignít u pacienta a jeho súrodencov, asociovaných s tumormi charakteristickými pre Lynchov syndróm s výskytom v rannom detskom veku poukazujú pri CMMR-D syndróme na autozómovo recesívny typ dedičnosti. Pri genealogickom vyšetrení je dôležité upriamiť pozornosť aj na vylúčenie prípadnej konsangvinity, ktorá zvyšuje pravdepodobnosť identifikácie CMMR-D syndrómu.
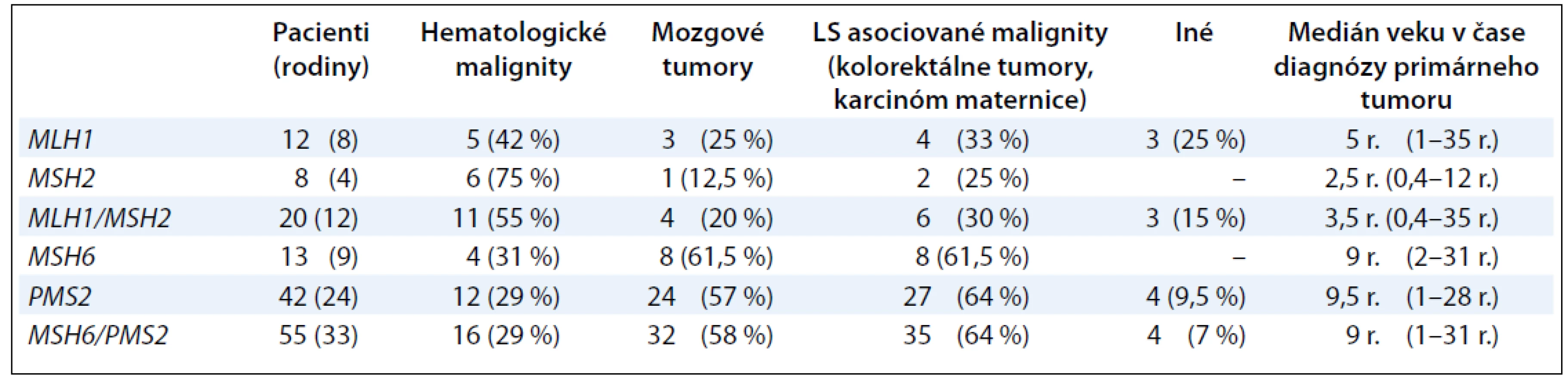
Súhrnná práca Wimmer a Kratz sumarizuje informácie o genotypovo--fenotypových koreláciách u pacientov s CMMR-D syndrómom (tab. 1) [10]. Väčšina pacientov s bialelickou mutáciou v géne MLH1 a MSH2 umiera na hematologické malignity alebo nádory mozgu v prvej dekáde života [10]. Naproti tomu pacienti s bialelickou mutáciou v géne PMS2 a MSH6 vyvíjajú najčastejšie mozgové tumory (55 %) a hematologické malignity (29 %) v prvej dekáde života a LS-asociované tumory (kolorektum a maternica) majú nástup vývoja v druhej až tretej dekáde života [10]. Analýzy in vitro ukázali, že niektoré liečivá, ako napríklad monofunkčné metylačné agensy, sú menej efektívne pri liečbe MMR deficientných bunkových línií [11,12].
Na príklade troch detí z jednej rodiny, z ktorých všetky boli nositelia bialelickej mutácie v géne MSH6, pričom dve vyvinuli infaustné malígne ochorenie, by sme chceli demonštrovať diagnostický postup a pragmatické preventívne odporúčania pre deti s CMMR-D.
Kazuistika
Predstavujeme rodinu s tromi postihnutými deťmi, ktoré sa zúčastnili genetickej konzultácie u toho istého klinického genetika a lekára. Dievčatko v 11. roku života (obr. 2, v rodokmeni IV-2), pochádzajúce z konsangvinného rómskeho manželstva, bolo odoslané na genetickú konzultáciu pri diagnostikovanom nádore mozgu s početnými kožnými hyperpigmentáciami typu „café au lait“, avšak bez ďalších príznakov neurofibromatózy typ I. CT a MR mozgu odhalili veľký tumorózny útvar v pravej hemisfére vo fronto-temporo-parietálnej oblasti. Stereotaxická biopsia bola vykonaná s diagnózou fibrilárny astrocytóm II. stupňa. Nasledovala chemoterapeutická liečba podľa protokolu POG 9233/34 pre low-grade nádory centrálneho nervového systému a ožiarenie neurokránia s celkovou dávkou 54 Gy.

O mesiac neskôr si sťažovala na dýchavičnosť, kašeľ a horúčku. RTG vyšetrenie zobrazilo fluidotorax na pravej strane s rozšíreným mediastínom. Histológia poukázala na lymfoblastický T-NHL lymfóm, trepanobiopsia kostnej drene bola infiltrovaná v rozsahu do 10 %. Pacientka podstúpila liečbu protokolom EURO-LB 2002.
Kontrolné CT mediastína a MR mozgu po prvej časti liečebného protokolu poukázali na redukciu tumorovej masy. Pôvodná histológia tumoru mozgu (fibrilárny astrocytóm II. stupňa) bola však najprv hodnotená podľa staršej klasifikácie a v čase publikácie bola prehodnotená podľa novej WHO klasifikácie. Histologická diagnóza bola definitívne stanovená ako gliomatosis cerebri III. stupňa. Približne v polovici plánovanej terapie pacientka začala trpieť bolesťami hlavy a zvracaním. MR ukázalo na nové nádorové ložisko v mozgu v ľavej hemisfére.
Biopsiou bol zistený anaplastický astrocytóm III. stupňa, ktorý bol chirurgicky odstránený. K udržiavacej liečbe pre T-NHL podľa HIT-AGG-2007 protokolu, bol pridaný temozolomid, používaný v liečbe „high-grade“ CNS nádorov. O tri mesiace neskôr kontrolné CT mozgu poukázalo na významnú progresiu nádorovej masy v mozgovom tkanive a pacientka umrela na progresiu ochorenia. Imunohistochemické vyšetrenie astrocytómu a T-NHL ukázalo normálne farbenie MLH1 a MSH2 a PMS2 proteínov, avšak absenciu MSH6 expresie. Molekulárna analýza mikrosatelitovej instability (MSI) v nádorovom tkanive bola vykonaná pomocou markerov pre hodnotenie MSI v kolorektálnom karcinóme (BAT26, BAT25, NR-21, NR-24, MONO-27). Výsledok bol pozitívny, MSI-L v BAT26 mikrosatelitnom markere.
Priame sekvenovanie identifikovalo mutáciu v géne MSH6 c.3261insC (p.F1088LfsX5) v homozygotnej forme. Sekvenačná analýza génu MSH6 zistila inzerciu C na pozíciu 3261, v kodóne 1088 v exóne 5. Táto mutácia bola v heterozygotnej forme popísaná Ollikainenom et al [13] v asociácii s neskorším nástupom karcinómu endometria a absenciou rakoviny hrubého čreva v rodine s Lynchovým syndrómom.
Brat tejto pacientky (obr. 2, v rodokmeni IV-4) bol odoslaný na genetickú konzultáciu po stanovení diagnózy nádoru u sestry a CT mu bol diagnostikovaný mozgový nádor – glióm. Molekulovo-genetickým vyšetrením sa potvrdila identická homozygotná mutácia v géne MSH6 ako u jeho sestry. Mal niekoľko Café au lait makúl na oboch nohách a na hrudníku. MRI mozgu odhalilo tumorovú masu v ľavej temporo--parietálnej oblasti a biopsia potvrdila diagnózu glioblastóm multiforme. Chlapec podstúpil chirurgickú resekciu a chemoterapiu temozolomidom podľa HIT-AGG-2007 protokolu. O 4 mesiace po diagnostikovaní ochorenia pacient umrel vo veku 11 rokov pre progresiu glioblastómu do pravej mozgovej komory.
1,5-ročný chlapček (obr. 2, v rodokmeni IV-5) je dieťa z druhého príbuzenského páru tej istej rodiny s Café au lait makulami na trupe, horných a dolných končatinách, charakteristickými pre CMMR-D. U tohto dieťaťa bola potvrdená rovnaká zárodočná mutácia v homozygotnom stave. Jeho rodičia sú prvostupňoví pokrvní príbuzní (bratanec a sesternica), rovnako ako u rodičov hore popísaných súrodencov. Rodičia prvého súrodeneckého páru, ako aj rodičia tohto chlapčeka, pochádzajú z veľkej rodiny bez výskytu nádoru hrubého čreva alebo maternice. Výnimkou je otcov strýko, ktorý mal viac kožných hyperpigmentácií a bol mu diagnostikovaný thymóm vo veku 15 rokov. V priebehu liečby mu bol zistený nádor pečene a vo veku 18 rokov ochoreniu podľahol. Následne bolo požadované genetické testovanie identifikovanej zárodočnej mutácie u všetkých pokrvne príbuzných. Rodičia z oboch manželských párov sú nositeľmi heterozygotnej mutácie, avšak doposiaľ všetci odmietli preventívny program pre Lynchov syndróm pozostávajúci predovšetkým z ročných kolonoskopií a vyšetrenia endometria maternice u žien.
Diskusia ku kazuistike
Konštitučné bialelické mutácie v géne MSH6 boli doposiaľ popísané u 19 osôb z 12 rodín. U 11 osôb boli prítomné len tumory CNS, pričom medián veku pri stanovení diagnózy bol 8 rokov [14–18]. Päť z týchto osôb vyvinulo druhú hematologickú malignitu alebo LS-asociované tumory. Mutácia v c.3261insC HMSH6 bola už objavená v heterozygotnej forme u jednej rodiny [13], v ktorej všetci nositelia mutácie vyvinuli nádor hrubého čreva alebo endometria vo veku 50–60 rokov.
V popísanej rodine nie je doposiaľ evidencia žiadneho nádorového ochorenia u rodičov ani starých rodičov, takže predpokladáme, že táto mutácia v heterozygotnom stave má nízku penetranciu. Táto skutočnosť u dospelých silne kontrastuje s vývojom mozgových nádorov a T-NHL u ich detí s homozygotnou mutáciou. Interval od diagnózy druhého CNS nádoru u dievčatka po obdobie jej smrti a od diagnózy prvého nádoru u jej brata k jeho smrti bol napriek intenzívnej liečbe veľmi krátky – 4 mesiace. Za normálnych okolností detí s high-grade CNS tumormi a mutáciou v géne HMSH6 prežívajú pri horeuvedenej liečbe 12–24 mesiacov [20]. Preto predpokladáme spojitosť veľmi agresívneho priebehu ochorenia a vývoj na liečbu rezistentných nádorov mozgu u detí s mutáciou MSH6.
V literatúre bolo doposiaľ opísaných len málo prípadov detí s bialelickou mutáciou v niektorom z MMR génov zameraných na detailný popis priebehu ochorenia a liečbu pacienta, preto sa po takýchto prácach zvyšuje dopyt. Prezentovaná kazuistika má prispieť k lepšiemu pochopeniu a zlepšeniu štandardnej terapie u high-grade CNS nádorov u detí s CMMR-D syndrómom.
Odporúčania pre klinický manažment
Preventívny program pre rodičov (heterozygotov) je zostavený podľa medzinárodných odporúčaní pre Lynchov syndróm [21]. Avšak u detí (homozygotov) je ťažké zostaviť súbor preventívnych vyšetrení. Doposiaľ u nich neexistujú senzitívne a pragmatické odporúčania, nakoľko sa u nich môže objaviť široké spektrum nádorových ochorení, a to už od 2. roku života spolu s rizikom vzniku sekundárnych malignít.
Z literárnych rešerší a klinických skúseností sa pre nositeľov bialelickej mutácie v niektorom z MMR génov už od dvoch rokov odporúčajú pravidelné klinické vyšetrenia pozostávajúce z vyšetrenia krvného obrazu, sledovania hladiny imunoglobulínov, ročnej magnetickej rezonancie mozgu a kolonoskopie [2]. Cieľom tohto odporúčania je včasné odhalenie malígneho ochorenia a zahájenie chirurgickej či chemoterapeutickej liečby. Toto opatrenie by malo zabezpečiť zníženie úmrtnosti detí s CMMR-D.
Diferenciálna diagnostika
Čo sa týka diferenciálnej diagnostiky CMMR-D syndrómu, fenotypovo podobné genetické ochorenie so zvýšeným rizikom malignity (5–10 %) a prítomnosťou „café au lait“ je neurofibromatóza 1 (NF 1). Vzhľadom na incidenciu 1 : 3 000 živorodených patrí medzi najčastejšie monogénovo podmienené ochorenie s predispozíciou k malignite, kde sú CAL makuly hlavným príznakom klinického spektra ochorenia. Podľa diagnostických kritérií je podmienkou pre stanovenia diagnózy prítomnosť minimálne 2 zo 7 príznakov [23].
Je spôsobené mutáciou, ktorá inaktivuje gén NF1, ktorý je mapovaný v oblasti 17q11.2 a skladá sa z 60 exónov. Proteínový produkt tumorsupresorového NF1 génu je neurofibromín, ktorý funguje ako negatívny regulátor Ras protoonkogénu. Na Slovensku sa molekulovo-genetické vyšetrenie realizuje v spolupráci Laboratória klinickej a molekulovej genetiky 2. detskej kliniky LF UK a DFNsP a Oddelenia molekulárnej biológie, Prírodovedeckej fakulty Univerzity Komenského v Bratislave použitím štandardného protokolu založeného na vyšetrení mRNA [23]. Výhodou tejto diagnostickej metódy je vysoký záchyt patognomických mutácií.
Diferenciálna diagnostika NF1 patrí bezpochyby do rúk odborníka. Najčastejšie je potrebné uvažovať o neurofibromatóze typu 2. Vzhľadom na frekvenciu 1 : 40 000 je menej častá. Hlavnou diagnostickou známkou je bilaterálny tumor VIII. hlavového nervu (vestibulárny schwanóm, predtým neurinóm akustiku), manifestujúci sa v 95 % prípadov do 30. roku života. CAL makúl pribúdajúcich od detstva býva podstatne menej. Dokázala sa kauzálna súvislosť s mutáciou tumorsupresorového génu lokalizovaného na chromozóme 22q, ktorého produktom je merlín.
U Watsonovho syndrómu (WS) je prítomnosť CAL makúl asociovaná s pulmonálnou valvulárnou stenózou, nízkym vzrastom a makrocefaliou. Keďže sa u niektorých pacientov zistila mutácia NFl génu, vznikol predpoklad, že Watsonov syndróm a neurofibromatóza 1 sú alelické ochorenia. V oblasti 17q však existuje séria susediacich génov pre pulmonálnu stenózu, neurokutánne anomálie, nízky vzrast a mentálnu retardáciu, ktoré podporujú hypotézu jeho vzniku ako syndrómu naliehajúcich génov [24].
V ostatnom čase Brems et al [25] identifikovali ako prví u pacientov s fenotypom podobným NF1 mutáciu v inom géne (SPRED1) nachádzajúcom sa v lokuse 15q25 a nazvali ho NF1 like syndróm (NF1 like). Niektorí z nich mali dysmorfiu tváre podobnú Noonanovej syndrómu, kongenitálne VVCH srdca a problémy s učením. Doterajšie publikované prípady vykazovali autozómovo dominantný typ dedičnosti [25].
Záver
Pod klinickým obrazom Café au lait makúl na koži dieťaťa sa môže skrývať závažná forma dedičného ochorenia s malignitami – CMMR-D syndróm. Precízna klinická diagnostika a cielená detekcia asociovaných príznakov s využitím tímu odborníkov, napr. v oblasti dermatológie, pediatrie, klinickej genetiky, onkológie, neurológii, rádiológii umožňuje včasné rozpoznanie dedičného ochorenia s rizikom malignity a zabezpečiť čo najoptimálnejší manažment zdravotnej starostlivosti [2,20,22,26]. Na príklade hore uvedenej kazuistiky je možné skonštatovať, že klinická diagnostika syndrómov s CAL makulami, veľmi podobných NF1, napr. Watsonovho, NF1-like syndrómu a Syndrómu konštitučného deficitu mismeč opravného systému si vyžaduje klinické skúsenosti [22,26]. K exaktnej diagnóze významne prispieva klinický genetik, ktorý indikuje použitie adekvátnych genetických testov na úrovni DNA. Práve včasná identifikácia heterozygotov/homozygotov ochorení s vysokým rizikom malígnych nádorov pomáha optimalizovať ich prevenciu, liečbu a ďalšiu starostlivosť [27].
Autor deklaruje, že v souvislosti s předmětem studie nemá žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
doc. MUDr. Denisa Ilenčíková, PhD.
II. detská klinika
LF UK a DFNsP Bratislava
Limbová 1
833 40 Bratislava
Slovenská republika
e-mail: denisa.ilencikova@dfnsp.sk
Obdrženo: 24. 4. 2012
Přijato: 25. 6. 2012
Zdroje
1. Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol 2006; 7(5): 335–346.
2. Wimmer K, Kratz CP. Constitutional mismatch repair-deficiency syndrome. Haematologica 2010; 95(5): 699–701.
3. Gardès P, Forveille M, Alyanakian MA et al. Human MSH6 deficiency is associated with impaired antibody maturation. J Immunol 2012; 188(4): 2023–2029.
4. Ilenčíková D, Bartošová Z, Babál P. Lynchov syndróm – novinky v diagnostike a liečbe. Onkologia 2010; 5(2): 70–76.
5. Ricciardone MD, Ozcelik T, Cevher B et al. Human MLH1 deficiency predisposes to hematological malignancy and neurofibromatosis type 1. Cancer Res 1999; 59(2): 290–293.
6. Bandipalliam P. Syndrome of early onset colon cancers, hematologic malignancies & features of neurofibromatosis in HNPCC families with homozygous mismatch repair gene mutations. Fam Cancer 2005; 4(4): 323–333.
7. Felton KE, Gilchrist DM, Andrew SE. Constitutive deficiency in DNA mismatch repair: is it time for Lynch III? Clin Genet 2007; 71(6): 499–500.
8. Krüger S, Kinzel M, Walldorf D et al. Homozygous PMS2 germline mutations in two families with early-onset haematological malignancy, brain tumours, HNPCC-associated tumours, and signs of neurofibromatosis type 1. Eur J Hum Genet 2008; 16(1): 62–72.
9. Scott RH, Mansour S, Pritchard-Jones K et al. Medulloblastoma, acute myelocytic leukemia and colonic carcinomas in a child with biallelic MSH6 mutations. Nat Clin Pract Oncol 2007; 4(2): 130–139.
10. Wimmer K, Etzler J. Constitutional mismatch repair-deficiency syndrom: have we so far seen only the tip of an iceberg? Hum Genet 2008; 124(2): 105–122.
11. Yip S, Miao J, Cahill DP et al. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res 2009; 15(14): 4622–4629.
12. Ripperger T, Beger C, Rahner N et al. Constitutional mismatch repair deficiency and childhood leukemia/lymphoma – report on a novel biallelic MSH6 mutation. Haematologica 2010; 95(5): 841–844.
13. Ollikainen M, Abdel-Rahman WM, Moisio AL et al. Molecular analysis of familial endometrial carcinoma: a manifestation of hereditary nonpolyposis colorectal cancer or a separate syndrome? J Clin Oncol 2005; 23(21): 4609–4616.
14. Peters A, Born H, Ettinger R et al. Compound heterozygosity for MSH6 mutations in a pediatric lymphoma patient. J Pediatr Hematol Oncol 2009; 31(2): 113–115.
15. Ostergaard JR, Sunde L, Okkels H. Neurofibromatosis von Recklinghausen type I phenotype and early onset of cancers in siblings compound heterozygous for mutations in MSH6. Am J Med Genet 2005; 139A(2): 96–105.
16. Etzler J, Peyrl A, Zatkova A et al. RNA-based mutation analysis identifies an unusual MSH6 splicing defect and circumvents PMS2 pseudogene interference. Hum Mutat 2008; 29(2): 299–305.
17. Rahner N, Höefler G, Högenauer C et al. Compound heterozygosity for two MSH6 mutations in a patient with early onset colorectal cancer, vitiligo and systemic lupus erythematosus. Am J Med Genet A 2008; 146A(10): 1314–1319.
18. Scott RH, Mansour S, Pritchard-Jones K et al. Medulloblastoma, acute myelocytic leukemia and colonic carcinomas in a child with biallelic MSH6 mutations. Nat Clin Pract Oncol 2007; 4(2): 130–134.
19. Auclair J, Leroux D, Desseigne F et al. Novel biallelic mutations in MSH6 and PMS2 genes: gene conversion as a likely cause of PMS2 gene inactivation. Hum Mutat 2007; 28(11): 1084–1090.
20. Ilencikova D, Sejnova D, Jindova J et al. High-grade brain tumors in siblings with biallelic MSH6 mutations. Pediatric Blood Cancer 2011; 57(6): 1067–1070.
21. Vasen HF, Möeslein G, Alonso O et al. Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer). J Med Genet 2007; 44(6): 353–262.
22. Ďurovčíková D. Neurofibromatosis typ 1 ochorenie multidisciplinárneho charakteru. Revue Med Prax 2008; 6(4): 10–12.
23. Messiaen L, Wimmer K. NF1 Mutational Spectrum. In: Kaufmann D (ed). Neurofibromatoses. Monogr Hum Genet Basel Karger 2008; 16: 63–77.
24. Allanson JE, Upadhyava M, Watson GH et al. Watson syndrome: is it a subtype of type 1 neurofibromatosis? J Med Genet 1991; 28(11): 752–756.
25. Brems H, Chmara M, Sahbatou M et al. Germline loss-of-function mutations in SPRED1 cause neurofibromatosis 1-like phenotype. Nat Genet 2007; 39(9): 1120–1126.
26. Hlavatá A, Rybárová A, Košťálová Ľ et al. Dlhodobé komplexné sledovanie detí s neurofibromatózou v detskom veku. Pediatr Prax 2009; 10(2): 75–80.
27. Ilenčíková D, Ďurovčíková D, Rybárová A et al. Nový syndróm malignity v diferenciálnej diagnostike s neurofibromatózou 1 a jej podobným chorobám. Lek Obzory 2009; 57(5): 221–227.
Štítky
Detská onkológia Chirurgia všeobecná OnkológiaČlánok vyšiel v časopise
Klinická onkologie

2012 Číslo Supplementum
- Metamizol jako analgetikum první volby: kdy, pro koho, jak a proč?
- Nejasný stín na plicích – kazuistika
- Fixní kombinace paracetamol/kodein nabízí synergické analgetické účinky
- Antidepresivní efekt kombinovaného analgetika tramadolu s paracetamolem
- Kombinace metamizol/paracetamol v léčbě pooperační bolesti u zákroků v rámci jednodenní chirurgie
Najčítanejšie v tomto čísle
- Syndrom Birt-Hogg-Dubé
- Klinický význam analýz genů středního rizika pro hodnocení rizika vzniku karcinomu prsu a dalších nádorů v České republice
- Hereditární difuzní karcinom žaludku
- Klinické dysmorfické syndrómy s tumorigenézou