
The NADPH Metabolic Network Regulates Human Cardiomyopathy and Reductive Stress in
Dominant mutations in the alpha-B crystallin (CryAB) gene are responsible for a number of inherited human disorders, including cardiomyopathy, skeletal muscle myopathy, and cataracts. The cellular mechanisms of disease pathology for these disorders are not well understood. Among recent advances is that the disease state can be linked to a disturbance in the oxidation/reduction environment of the cell. In a mouse model, cardiomyopathy caused by the dominant CryABR120G missense mutation was suppressed by mutation of the gene that encodes glucose 6-phosphate dehydrogenase (G6PD), one of the cell's primary sources of reducing equivalents in the form of NADPH. Here, we report the development of a Drosophila model for cellular dysfunction caused by this CryAB mutation. With this model, we confirmed the link between G6PD and mutant CryAB pathology by finding that reduction of G6PD expression suppressed the phenotype while overexpression enhanced it. Moreover, we find that expression of mutant CryAB in the Drosophila heart impaired cardiac function and increased heart tube dimensions, similar to the effects produced in mice and humans, and that reduction of G6PD ameliorated these effects. Finally, to determine whether CryAB pathology responds generally to NADPH levels we tested mutants or RNAi-mediated knockdowns of phosphogluconate dehydrogenase (PGD), isocitrate dehydrogenase (IDH), and malic enzyme (MEN), the other major enzymatic sources of NADPH, and we found that all are capable of suppressing CryABR120G pathology, confirming the link between NADP/H metabolism and CryAB.
Published in the journal:
The NADPH Metabolic Network Regulates Human Cardiomyopathy and Reductive Stress in. PLoS Genet 9(6): e32767. doi:10.1371/journal.pgen.1003544
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003544
Summary
Dominant mutations in the alpha-B crystallin (CryAB) gene are responsible for a number of inherited human disorders, including cardiomyopathy, skeletal muscle myopathy, and cataracts. The cellular mechanisms of disease pathology for these disorders are not well understood. Among recent advances is that the disease state can be linked to a disturbance in the oxidation/reduction environment of the cell. In a mouse model, cardiomyopathy caused by the dominant CryABR120G missense mutation was suppressed by mutation of the gene that encodes glucose 6-phosphate dehydrogenase (G6PD), one of the cell's primary sources of reducing equivalents in the form of NADPH. Here, we report the development of a Drosophila model for cellular dysfunction caused by this CryAB mutation. With this model, we confirmed the link between G6PD and mutant CryAB pathology by finding that reduction of G6PD expression suppressed the phenotype while overexpression enhanced it. Moreover, we find that expression of mutant CryAB in the Drosophila heart impaired cardiac function and increased heart tube dimensions, similar to the effects produced in mice and humans, and that reduction of G6PD ameliorated these effects. Finally, to determine whether CryAB pathology responds generally to NADPH levels we tested mutants or RNAi-mediated knockdowns of phosphogluconate dehydrogenase (PGD), isocitrate dehydrogenase (IDH), and malic enzyme (MEN), the other major enzymatic sources of NADPH, and we found that all are capable of suppressing CryABR120G pathology, confirming the link between NADP/H metabolism and CryAB.
Introduction
The maintenance and integrity of specialized functional structures such as sarcomeres, the basic unit of contractile force in striated muscles, are inextricably linked to the cellular machinery of molecular chaperones and protein quality control pathways. Evidence for this notion is provided by the identification of myopathic mutations in genes that encode proteins with chaperone function, such as CryAB and Bag3, and whose products have been localized to Z-discs. Moreover, an increasing number of genes encoding Z-disc associated proteins, such as desmin, ZASP, myotilin and filamin C are linked to myofibrillar diseases [1]. The Z-disc, which is formed by a complex network of diverse proteins, defines the structural boundaries of sarcomeres and integrates the actin filaments of neighboring contractile units. Major morphological and cellular hallmarks that define myofibrillar disorders include disintegration of the Z-disc lattice network, mitochondrial disruption, and ectopic protein aggregates.
The autosomal dominant R120G mutation in the αB-crystallin gene (CryABR120G) manifests adult-onset cataracts, skeletal muscle weakness and heart failure [2]. CryAB, a small molecular weight heat shock protein, is expressed constitutively in the lens and in non-lenticular tissues associated with high rates of oxidative metabolism, such as heart and type I and type II skeletal muscle fibers. A primary function of CryAB in these tissues is to prevent aggregation of intermediate filament proteins such as desmin, a characteristic subcellular phenotype of desmin-related myopathies [3]. Earlier studies by several laboratories supported a loss-of-function mechanism for the CryABR120G mutation, based on alterations of its secondary and quaternary structures, decreased interactions for client substrates with intermediate filaments and reduced stability on heat denaturation in vitro [4], [5]. Subsequently, expression of either the mouse or human CryABR120G allele in the mouse heart produced cardiomyopathy, heart failure and shortened lifespan, all of which phenocopy the disease condition in humans [6], [7]. Additional missense, truncation and autosomal recessive mutations have underscored the dominant inheritance patterns of disease-causing CryAB expression but their underlying molecular mechanism(s) have remained elusive.
An important concept in the pathogenesis of neurodegenerative and, perhaps, myofibrillar disease is that misfolding and aggregation of destabilized mutant proteins trigger ‘toxic’ gain of function etiologies [8]. It has been hypothesized that the accumulation of amyloidogenic aggregates is preceded by the appearance of soluble, oligomeric species whose toxicities arise from multiple, non-exclusive mechanism(s) mediated by (1) flexible hydrophobic surfaces that promote aberrant interactions and sequestration [9], (2) poor clearance mechanisms that disrupt central protein quality control and propagate folding defects [10], [11], and (3) perhaps by compromised lipid integrity as suggested by in vitro model membrane systems [12]. Although CryABR120G does not appear to result from a “classical” amyloid, recent studies have shown that aggregates of mutant CryAB are recognized by the anti-oligomer A11 [13], [14] providing a link with well-known amyloid protein aggregation diseases such as Alzheimer's and Huntington's diseases.
Our recent work on a mouse model of the inherited human CryABR120G cardiomyopathy has provided the first persuasive case for pathology resulting from “reductive”, as opposed to oxidative, stress in disease pathogenesis. The molecular events and damaging effects of reactive oxygen species (ROS) on biological molecules and systems are well known. When ROS are generated in excess of a cell's capacity to neutralize them, by enzymatic and non-enzymatic antioxidant pathways, the cell experiences oxidative stress. A large number of disease states have been attributed, in whole or in part, to damage from ROS or oxidative stress. However, there has been little appreciation for the possibility that an excess of reducing equivalents might also cause problems for the cell. Reductive stress can be induced by supplying strong reducing compounds, such as dithiothreitol, to cells in culture, but only recently has it been shown by Rajasekaran and coworkers that reductive stress may occur as a pathological state consequent to expression of a mutant protein. CryABR120G-induced cardiomyopathy was accompanied by a significant shift towards a more reduced intracellular environment, as measured by the glutathione redox couple [7]. The dominance of CryABR120G, and its unexpected link to an excessively reduced environment, suggested a ‘toxic’ gain of function mechanism.
To investigate the reductive stress hypothesis of CryABR120G pathology, we have developed the first Drosophila melanogaster model of human CryABR120G toxicity in multiple organs and tissues. Because there is a strong evolutionary conservation of key developmental and metabolic pathways between humans and Drosophila [15]–[18], studies of human conditions in Drosophila have been very productive. This has been especially true in the fields of heritable developmental defects, including congenital heart diseases, aging-related conditions and neurodegenerative diseases [19]–[22]. Recently, with advanced microscopic technology [23], Drosophila has been used to model human cardiac physiology and aging [24]–[27]. Interestingly, flies with a dilated heart have been reported in certain genetic backgrounds [28]. Strikingly, cardiac dilation is observed in patients with protein aggregation cardiomyopathy and in mice that over-express CryABR120G [7].
In this study, we demonstrate that CryABR120G pathologies in the fly heart and eye are regulated by key enzymes that reduce nicotinamide adenine dinucleotide phosphate (NADP) to NADPH. As was the case in the mouse heart, the deleterious effects of expressing CryABR120G were strongly ameliorated by knockdown or mutation of the gene encoding G6PD. We further exploited the fly model and report that reduced function of other major generators of NADPH also strongly suppressed the CryABR120G phenotype, implicating the entire cellular NADP/NADPH network in CryABR120G pathology.
Results
Drosophila heart dysfunction caused by human CryABR120G
To extend our tests of the reductive stress hypothesis of CyrABR120G pathology, we used the Gal4-UAS modular expression system [29], [30] to permit expression of the human CryABR120G allele in various cell types. We first generated transformants carrying either a wild-type UAS-CryAB+ or UAS-CryABR120G construct. Next, to determine whether CryABR120G expression would affect cardiac function in Drosophila, we drove its expression in the fly heart with a Hand-Gal4 driver. We confirmed cardiac-specific expression from Hand-Gal4 by examining GFP fluorescence using a UAS-CryABR120G-GFP fusion construct. Visual inspection of fluorescent micrographs confirmed cardiomyocyte restricted expression of the construct. Remarkably, the marked CryABR120G appeared targeted to a repetitive myofibrillar component of the cardiac fibers of flies, as found in higher organisms [31], likely the Z-discs (Figure S1).
To investigate the effects of human wild type or mutant CryAB expression on the simple, linear Drosophila cardiac tube (Figure 1A) we imaged surgically exposed beating hearts and tracked wall movements of semi-intact flies using direct immersion DIC optics in conjunction with a high speed digital video camera [32]. We characterized the effects of CryAB+ and CryABR120G on the contractile performance and general morphology of Drosophila hearts.
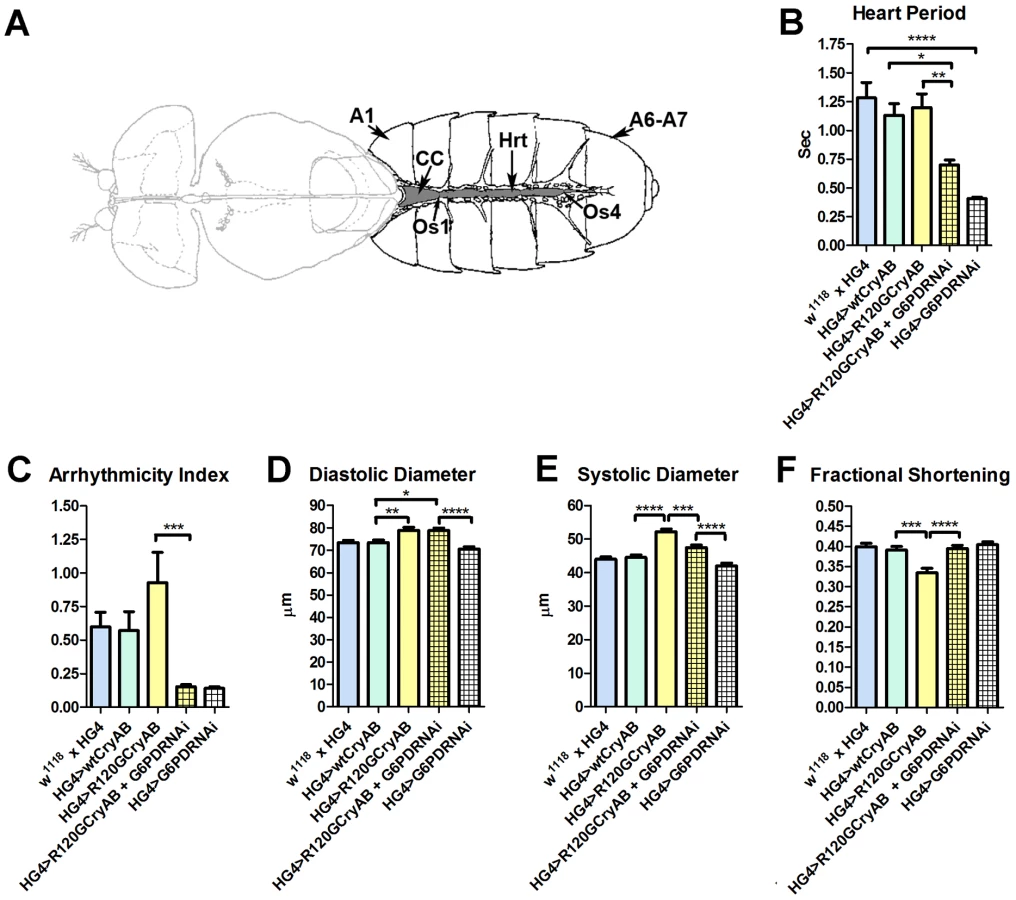
Heart period, which is defined as the length of time between the ends of two consecutive diastolic intervals, and arrhythmia indices, a quantitative measure that reflects cardiac rhythmicity and permits exploration of heart rhythm irregularities, were calculated for ∼45 three week old semi-intact Drosophila from each line. Cardiac diameters were measured directly from individual video frames at peak diastolic and systolic time points at multiple locations along the linear portion of abdominal segment three of each heart tube. These measurements revealed that expression human CryAB+ did not significantly perturb any analyzed index of cardiac function relative to control hearts (Figure 1, Figure S2). In contrast, expression of CryABR120G significantly affected several parameters of heart function. Arrhythmic beating patterns appeared to increase (Figure 1C), although these trends were not statistically significant. Diastolic and systolic diameters were significantly increased in response to CryABR120G expression, and fractional shortening of the fly heart was significantly reduced thus notably impairing Drosophila cardiac function (Figure 1D–F). In accord with the mouse observations [7], simultaneous RNAi-mediated knockdown of G6PD significantly improved several indices of cardiac function and overall heart performance. Hearts expressing both CryABR120G and RNAi targeted against Zw (the gene encoding G6PD) exhibited significantly shorter heart periods (increased heart rates; Figure 1B), significantly reduced arrhythmia indices and systolic diameters, and significantly greater fractional shortening (Figure 1C–F). Interestingly, cardiac restricted expression of G6PD RNAi alone significantly decreased heart periods relative to those of w1118 x HandGal4 control hearts (Figure 1B).
Many of these differences in contracting heart tubes were qualitatively visualized via M-mode traces, which display the dynamics of cardiac contractions of representative hearts from the various genotypes (Figure 2). These traces show the positions of the heart wall edges (Y direction) over time (X direction). M-modes from semi-intact heart preparations from control flies show fairly regular contractions. However, these traces reveal a subtly arrhythmic beating pattern in CryABR120G cardiac tubes relative to controls. Further, the CryABR120G hearts were dilated, and exhibited a lower extent of shortening. Co-expression of Zw RNAi increased heart rate, promoted rhythmic beating and rescued percent fractional shortening in the CryABR120G mutant hearts. Thus, overall, cardiac output of the mutant hearts is likely to be significantly enhanced by reducing the enzymatic activity of G6PD in flies as found in mouse models [7].

CryABR120G-induced defects in non-heart tissues
We also asked whether CryAB expression produced any deleterious effects if expressed in other tissues. When CryAB+ was expressed ubiquitously (using a Tub-Gal4 driver), or in the eye only (using either ey-Gal4 or GMR-Gal4 drivers) no abnormal phenotypes were observed in 32 of 33 lines, in spite of easily detectable expression (in eight lines examined by Western blotting; not shown). We did recover a single CryAB+ transformant that produced a rough eye phenotype. However, the insertion was located upstream of the escargot (esg) gene, a location where mis-expression elements are known to produce rough eyes by promoting esg expression [33]. We used qRT-PCR and verified that this particular CryAB+ insertion also drove overexpression of esg. We therefore attribute its phenotype to esg overexpression, and not to CryAB+.
In contrast, we identified four UAS-CryABR120G lines that produced complete or partial lethality with ubiquitous expression (using either A5C-Gal4 or Tub-Gal4 drivers). Western blotting of protein extracts from the single line with any survivors revealed abundant CryABR120G expression. No expression was detected in seven fully viable lines (Figure S3). We speculate that the high frequency of non-expressing transformant lines reflects a strong selection against lines that show even slightly leaky expression. Subsequent Western blots of flies with expression limited to the eye (which allows survival) revealed that the three lines that were lethal when constitutively expressed also had abundant Gal4-induced expression of CryABR120G (Figure 3).

When CryABR120G expression was driven in the wing, flies with deformed and mis-shapen wings were produced (Figure 4B), while expression in the eye throughout development (with ey-Gal4) resulted in variably rough and small eyes (Figure 4D). A stronger and more consistent eye phenotype was observed when expression was driven in differentiating eye discs (with GMR-Gal4), characterized by irregular patterning and loss of ommatidia and pigment (Figure 4E).

Cardiomyopathy caused by mutations in CryAB is correlated with the presence of cytoplasmic protein aggregates containing CryAB and a number of other proteins [7], [14]. To determine whether human CryABR120G in Drosophila was also found in aggregates we extracted Triton-X 100 soluble and insoluble protein fractions from eyes expressing CryAB+ and CryABR120G and used Western blotting to determine the distribution of CryAB between these fractions (Figure 3). We found a portion of the CryABR120G mutant protein in the insoluble pellet fraction (6.6%±2.9, 23%±13, 1.9%±1.0 and 4.9%±1.0 for lines 7B, 13A, 14A and 16A, respectively), while CryAB+ protein was found virtually exclusively in the soluble fraction (with only 0.3%±0.2% and 0.2%±0.1% in the pellet for lines 8B and 12A, respectively). Thus, CryABR120G in Drosophila shares the characteristic of being found, in part, in insoluble aggregates.
The eye is dispensable for normal development and viability, and is an established model for examining the cellular effects of human neurodegenerative disease genes [34], [35]. Therefore, we chose to use the phenotype produced by GMR-Gal4 UAS-CryABR120G in the eye as a model to test the reductive stress hypothesis. Eye phenotypes were quantitated by scoring into five groups, ranging from ∼wildtype (category 1), to having all or nearly all of the eye strongly affected (category 5; Figure 4E).
We then tested whether a reduction of G6PD could suppress the CryABR120G phenotype, as it does in mouse and fly hearts. Two RNAi lines that significantly reduced expression of Zw both suppressed the CryABR120G eye phenotype (Figure 5B,C; Table S1). To extend these observations we then asked whether overexpression of Zw would exacerbate the CryABR120G phenotype. Four overexpression lines were tested and, unlike the heart phenotype (Figure S2), all strongly enhanced the CryABR120G phenotype (Figure 5E–H; Table S1). The results shown in Figure 5 using the CryABR120G line 16A were confirmed with the independent CryABR120G insertion line 14A that also has a strong eye phenotype: responses to Zw knockdown and overexpression were extremely similar for the two lines (not shown). There was no phenotypic effect of altered G6PD levels on normal eyes.

We considered the possibility that the UAS-controlled RNAi constructs might titrate GAL4 and produce apparent suppression simply through reduced expression of CryABR120G. However, since the G6PD overexpression constructs are also driven by GAL4, but they enhance the CryABR120G phenotype, this concern appears unfounded.
The enzyme 6-phosphogluconate dehydrogenase (PGD) acts downstream of G6PD in the pentose phosphate pathway, and like G6PD, reduces NADP to NADPH. We tested whether mutations in the gene encoding PGD (Pgd) would also affect the CryABR120G phenotype. Two deletions that removed the gene (along with neighboring genes) suppressed the CryABR120G phenotype when tested as heterozygotes (Figure 6B,C; Table S1). In addition, flies carrying a chromosome with mutations in Zw and Pgd, in heterozygous condition, also showed strong suppression of the CryABR120G phenotype (Figure 6D). Finally, RNAi directed against Pgd produced very strong suppression of the CryABR120G phenotype (Figure 6E; Table S1). These mutants or RNAi knockdowns, by themselves, had no effect on the phenotype of normal eyes.

These results are consistent with the reductive stress hypothesis, but it is still conceivable that G6PD and PGD influence CryABR120G pathology via other products of the pentose phosphate pathway. To determine whether varying the enzymatic production of NADPH would affect the CryABR120G phenotype, regardless of the source of that variation, we tested whether alteration of isocitrate dehydrogenase (IDH) or malic enzyme (MEN) levels would affect the phenotype. We observed that RNAi-mediated knockdown of IDH (either the putative mitochondrial (CG6439) or cytoplasmic (CG7176) form) was capable of suppressing the CryABR120G phenotype (Figure 7B–F; Table S1). Knockdown of MEN also suppressed the CryABR120G phenotype (Figure 7G; Table S1). Knockdown of IDH or MEN had no effect on normal eyes. Although only one of three lines expressing RNAi against Idh (CG7176) achieved significant suppression of the CryABR120G phenotype, it is also the case that these lines only achieved an ∼20–30% reduction in Idh RNA levels. In the cases of Zw, Pgd and Men, where strong suppression was observed, ∼40–50% knockdowns of RNA levels were achieved (Table S1).

To assess the redox environment of cells in response to altered dosage of genes that mediate NADP/H metabolism we carried out two series of experiments to determine the ratio of reduced to oxidized glutathione (GSH∶GSSG). When RNAi-mediated knockdown of G6PD, PGD, MEN or IDH was driven in heads with GMR-Gal4 the GSH∶GSSG ratio was reduced in every case (Figure 8A), though the reduction as a result of PGD knockdown was not significant at P = 0.05 level. The results confirm our supposition that these knockdowns impair cells' ability to generate NADPH. We observed a slight, though not statistically significant, increase in the GSH∶GSSG ratio when G6PD was overexpressed in heads. However, when we assayed whole larvae, with expression driven ubiquitously, G6PD overexpression produced a very large and significant increase in the GSH∶GSSG ratio (Figure 8B). These results confirm the involvement of the NADP/H network in cellular dysfunction produced by CryABR120G expression and strongly implicate reductive stress as the causative agent for pathology.

One concern that arose when assessing the phenotypic effect of human CryABR120G expression was that two Drosophila lines (7B and 13A) showed only mild phenotypic effects in the eye (Table S2), despite exhibiting robust protein expression (Figure 3). However, these two lines do show rough eye phenotypes if subjected to a strong heat shock during late larval/early pupal development, a treatment that also strongly enhances the phenotype of lines 14A and 16A (Table S2). Flies carrying GMR-Gal4 alone showed no eye phenotype when subjected to this heat shock. Moreover, one of these lines (7B) also exhibits a wing phenotype when expression is driven in the wing (Table S2).
To test whether these particular insertions might be associated with suppression of the CryABR120G phenotype, we combined them with GMR-Gal4 and the CryABR120G line 16A that was used in the experiments of Figures 4, 5 and 7. The combination of line 7B or 13A with 16A resulted in an eye phenotype that was reduced from that of 16A alone, indicating that these two lines do suppress the CryABR120G eye phenotype to some degree (Figure S4). We have not determined why these two lines show little phenotypic consequence in the eye, but we speculate that it may have to do with subtle differences in timing of gene expression. Perhaps slightly earlier onset of expression in lines 7B or 13A results in a protective response, in the same way that mild heat shocks, by inducing synthesis of heat shock proteins, can protect against subsequent, more severe heat shocks [36], [37]. Alternatively, the expression of genes in the region of the 7B and 13A insertion loci might be altered to result in phenotypic suppression. In spite of these lines showing only mild effects, our experiments convincingly show that CryABR120G expression, but not CryAB+, can cause strong phenotypic effects in the fly heart, eyes and wings. Moreover, detailed examination of the heart and wing defects show that they are responsive to altered levels of the enzymes that reduce NADP to NADPH.
Discussion
Our results show that Drosophila provides a suitable model in which to study the pathology of the human CryABR120G mutation. Expression of the mutant allele, but not the wildtype, in fly hearts, causes heart dilation and dysfunction very reminiscent of the cardiomyopathy produced in humans carrying this dominant allele [2]. We also found that a reduced level of G6PD ameliorates many of the perturbed cardiac functional parameters in CryABR120G flies, just as it does in the CryABR120G mouse [7]. Despite a lack of reduction in CryABR120G diastolic diameters in response to Zw (G6PD) RNAi co-expression, systolic diameters in the double mutants were rescued and did not significantly differ from those found in fly hearts expressing wildtype human CryAB. Thus, fractional shortening was indistinguishable between wildtype CryAB-expressing and CryABR120G+Zw RNAi-expressing hearts. Furthermore, cardiac restricted expression of Zw RNAi, either with CryABR120G or alone, significantly increased heart rates relative to control hearts. This suggests G6PD deficiency can improve cardiac output in either mutant or non-mutant backgrounds and may be a potent modifier of cardiac function.
In Drosophila, overexpression of G6PD can extend lifespan and protect against oxidative stress [38]. In mammalian cells, overexpression of wildtype small heat shock proteins leads to increased G6PD expression and protection against oxidative stress [39]. Our finding, that reduction of G6PD can be beneficial in some circumstances, is also not without precedent. Some studies have suggested a link between G6PD deficiency and protection against cardiovascular disease in humans, although such findings have not been replicated in larger patient studies [40]–[42]. Previous work from one of our laboratories showed that G6PD reduction is highly beneficial in one specific case — when human CryABR120G is expressed in the mouse heart [7]. We do not see a conflict in these differing outcomes, but instead conclude that the effect of modifying G6PD levels may range from beneficial to deleterious, with the outcome determined by the constellation of genetic variation present in individuals' genomes and the environmental stressors that they experience.
To generate an experimental paradigm suited to rapid genetic exploration we expressed CryABR120G in the fly eye and found that it strongly disturbs normal development and pattern formation. The eye phenotype was also responsive to altered G6PD levels, validating it as a model for investigation of the underlying mechanism of CryABR120G pathology. Unlike a recent report [43], we saw no abnormal eye phenotype that could be attributed to expression of the wild-type human CryAB gene. In the one case where we did observe an eye phenotype it most likely resulted from induced expression of the esg gene that lay adjacent to that particular insertion of the CryAB transgene. Our findings indicate that CryABR120G induces cellular dysfunction in both the heart and the eye, or lethality if expressed ubiquitously, while wild-type CryAB is relatively benign.
The human disease produced by the CryABR120G allele is sometimes called Desmin-Related Myopathy (DRM), owing to the presence of desmin in the characteristic cytoplasmic protein aggregates, and to similarities with diseases caused by mutations in the gene encoding the intermediate filament desmin [2], [6], [44]. Although Drosophila do not have a desmin homolog, several lines of evidence argue that the cellular dysfunction in Drosophila resulting from CryABR120G expression is, nonetheless, a legitimate model for this disease. First, a number of other proteins have been identified within the aggregates, including (at least) another small heat shock protein and G6PD [7], [14], both of which have homologs in Drosophila. Second, CryABR120G causes the formation of cytoplasmic aggregates even when expressed in human cell types that do not express desmin [45], [46], and our results show that a portion of the CryABR120G is also found in aggregates in Drosophila. Third, the identical response of mouse and Drosophila CryABR120G pathologies to G6PD reduction strongly suggests an identical mechanism of action.
The pyridine nucleotides NADH and NADPH are essential co-factors of oxidative and reductive enzymatic processes involved in energetics, oxidative metabolism, redox homeostasis, calcium homeostasis, macromolecular biosynthesis, mitochondrial functions, gene expression, aging and cell death [47]. In this study, we examined the effect of altered levels of the four enzymes that are primarily responsible for reducing NADP to NADPH (Figure 9): two enzymes of the pentose phosphate pathway, G6PD and PGD which together account for ∼40% of NADPH levels in the adult; MEN which generates pyruvate for import into mitochondria and accounts for another ∼30%; and, IDH, which accounts for ∼20% of NADPH [48]–[50]. These enzymes constitute a metabolic network linked by a common substrate (NADP) and interacting regulation [50]. A major finding of our study is that, even though G6PD, PGD, MEN and IDH carry out varied metabolic reactions, alterations to any of their activities have significant consequences for the phenotypes produced by CryABR120G expression, implying a common mechanism of action through NADP/H.

In our experiments, reduction of IDH was less effective at CryABR120G suppression than reductions of G6PD, PGD or MEN, a result that is not entirely surprising. Alteration of either G6PD or PGD activity is likely to affect both activities coordinately since they constitute sequential steps in the tightly regulated pentose phosphate pathway, and MEN by itself produces more NADPH than any of the other three enzymes of this network. IDH produces less NADPH than either MEN or the G6PD/PGD couple. Additionally, our RNAi-mediated knockdowns of IDH were relatively ineffective. What was surprising was that knockdown of the mitochondrial NAD-dependent IDH resulted in significant suppression of the CryABR120G phenotype. We surmise that mitochondrial metabolism affects the cytoplasmic NADP/H network.
The NADP∶NADPH redox couple, and the linked glutathione redox couple (GSSG∶GSH), participate in a diverse array of biological processes. Therefore, we envision a number of possible mechanisms through which CryABR120G could alter the cellular redox potential and thereby contribute to toxicity. Most obviously, redox-sensitive sequestration of both existing and newly synthesized proteins could seriously disturb cellular regulation. The function of many proteins depends on the reduced or oxidized state of thiol-containing cysteine residues. It is conceivable that structurally flexible hydrophobic protein surfaces, which are normally buried within a folded protein, could engage in non-productive protein-protein interactions, in part, due to alterations of intra-chain disulfide links. Partially folded proteins might, in this way, become soluble toxic intermediates [9]. Alternatively, misfolding might occur as a result of alterations in other redox-sensitive post-translational modifications, for example glutathionylation, nitrosylation, and (de)acetylation. Co-aggregation of several distinct polypeptides might cripple multiple disparate functions within the cell. As mentioned, our results also suggest that alterations in mitochondrial homeostasis and energy metabolism could affect the levels of oxidized or reduced NADP/H. The reciprocal is most certainly true as well, with normal mitochondrial function dependent upon the function of the NADP-reducing enzyme MEN. Additionally there are scores of enzymes that use NADP/H as a cofactor, and the activity of one or more of these enzymes could be affected to generate the phenotypes we observed. It will require significant further work to identify the critical determinants of NADP/H involvement in CryABR120G pathology. Drosophila provides powerful tools for genetic screening to identify such factors and the model for CryABR120G pathology that we describe here provides a context for carrying out such screens. We anticipate that such efforts may ultimately lead to the identification of potential targets for therapy and the promise of useful treatments for the human disorders.
Materials and Methods
Construction of transgenic flies
P{UASP-CryAB+} and P{UASP-CryABR120G}: A fragment DNA containing human CryAB cDNA was released from donor plasmids (pCMVHA-wtCryAB [51] or pCDH1-MCS1-R120GhCryAB (unpublished); by NcoI and DraI digestion, followed by Klenow filling in to make blunt ends. The vector pUASP [52] was digested with EcoRI or NotI respectively and filled in to make blunt ends. Ligation of respective inserts and vectors produced P{UASP-CryAB+} and P{UASP-CryABR120G}. Each was verified by restriction enzyme mapping and sequencing, then was used in P-element mediated transformation by standard methods [53], [54].
P{UASTattB-GFP-CryABR120G}: The CryABR120G cDNA was released from pCDH1-MCS1-R120GhCryAB by EcoRI digestion and was ligated in frame to the same site at the C terminus of GFP coding region an intermediate vector. The fusion protein was released using flanking XbaI sites and sub-cloned into pUASTattB vector provided by J. Bischof [55]. The plasmid was injected into y w P{ry+t7.2, hsFLP}1; M{3xP3-RFP.attP}ZH-86Fb; M{vas-int.B}ZH-102D (from the Bloomington Drosophila stock center) to integrate the fusion protein construct onto chromosome 3R cytological location 86F. The X chromosome carrying FLP was removed by crossing after transformation.
Mapping of transformed P elements
Insertion sites of transformed P elements were mapped either by inverse PCR or Splinkerette PCR method [56].
Other Drosophila strains and culture conditions
P{tub-GAL4}, P{ey-GAL4} and P{Act5C(FRT.y+)Gal4} flies were obtained from the Bloomington, IN, USA, Drosophila stock center (lines #5138, #5535 and #25374, respectively). After being exposed to FLP, P{Act5C(FRT.y+)Gal4} lost the y+ marker and became recessive lethal. It was then kept as the balanced stock y w; P{Act5C-GAL4}/S2 CyO. P{GMR-GAL4} flies are discussed in [57]. Fly lines carrying P{UAST-G6PD} along with control y w2 were provided by W. C. Orr [38]. Mutations for Zw and Pgd were obtained from Bloomington stock center. RNAi fly stocks were from Vienna Drosophila RNAi Center [58]. Table S1 lists the stock numbers of all UAS-RNAi lines (Vienna Drosophila RNAi Center), Zw overexpression lines (W. C. Orr), and mutants (Bloomington Drosophila Stock Center). Flies were raised at 25°C, unless otherwise specified, on standard cornmeal-agar medium in standard 25×90 mm vials.
Western blotting
Assay for CryABR120G protein expression: The UASP-CryABR120G lines were crossed to flies carrying the tub-GAL4 driver. One male and one female were taken from seven crosses with surviving progeny. Each fly was homogenized in 100 µl of 1X sample buffer. Eight µl of lysate was loaded in each lane of a 12% SDS-PAGE gel, separated by electrophoresis and then examined by Western blotting to detect the CryAB protein. A lysate of mammalian cells expressing CryABR120G was included as a positive control. Rabbit antiserum (1∶5000 dilution) against human CryAB protein was the primary antibody [7].
Assay for CryAB protein solubility: To determine whether CryAB expressed in eyes existed in a soluble or insoluble form we modified the procedure described by Carbone et al. [59]. Ten heads from females expressing CryAB (wildtype or mutant) were collected and homogenized for 10 minutes on ice in 100 µl lysis buffer (10 mM Tris pH 7.5, 5 mM EDTA, 1% NP40, 0.5% deoxycholate, 150 mM NaCl and 1% Triton X-100). After incubation in lysis buffer for 30 minutes on ice, samples were frozen at −20°C overnight, then thawed out. Cuticle and debris in the lysate were separated by brief centrifugation at 1,000 rpm for 1 minute. Supernatant was collected. Soluble and insoluble fractions were further separated by centrifugation at 14,000 rpm for 15 minutes. After collecting the soluble fraction, the insoluble fraction was washed three times with 200 µl lysis buffer each, and then solubilized in 40 µl 1X sample buffer for Western. The soluble fraction was TCA precipitated and washed before being resuspended in 40 µl 1X sample buffer. 2.5 head's worth of soluble or insoluble fraction (10 µl) was loaded into each lane.
Western blotting was carried out following standard procedures using the Odyssey Western Blot Kit (Li-cor). Rabbit antiserum against Human CryAB and mouse anti-β-tubulin (Developmental Studies Hybridoma Bank clone E7) were used as primary antibodies. After incubation with fluorescent anti-rabbit and anti-mouse secondary antibodies, the membrane was scanned on an infrared Odyssey scanner by Li-cor. The Western signal was quantified on the Li-cor scanner and the results from four independent experiments were averaged. The insoluble CryAB is reported as mean percent of total CryAB ± standard error.
Scoring eye phenotypes
Stocks that carried the GMR-Gal4 driver and UAS-CryAB elements in homozygous condition were generated. Males from this stock were crossed to females from control lines (y w2 or w1118), or from lines carrying modifying elements or mutations. The eyes of daughters from these crosses were scored for the severity of the eye phenotype by assigning the eye to one of five categories (Figure 1E). All transgenes were hemizygous in the scored females. Mann-Whitney tests for significance were performed using GraphPad Prism software.
Quantitative RT-PCR
Total RNA from 15–25 female fly heads was harvested using Tri reagent and protocol (Sigma-Aldrich). cDNA was synthesized from total RNA using RevertAid First Strand cDNA Synthesis Kit (Fermentas). Quantitative PCR of the cDNA was carried out on an iQ-PCR machine (Bio-Rad) using Maxima SYBR green/Fluorescein qPCR Master Mix (Fermentas). Relative copy number was calculated against a set of common standard templates for each PCR reaction. For each cDNA sample, the relative copy numbers of gene of interest (X) and ribosomal protein L32 (RPL) were both obtained. Abundance of X in the sample was calculated by dividing the copy number of X by that of RPL. The average of three independent experiments was used to represent the abundance of X in a given genotype.
Drosophila cardiac performance analysis
Two independent wildtype UAS-CryAB+ controls and two UAS-CryABR120G mutant fly lines, as well as UAS-CryABR120G combined with UAS-Zw or UAS-Zw RNAi were crossed to Hand-Gal4 (II) driver flies (Hand is a direct target of Tinman and GATA factors during Drosophila cardiogenesis and hematopoiesis, [60]). As an additional control, Hand-Gal4 (II) driver-flies were crossed to w1118 flies. The progeny were raised at 25°C on standard cornmeal-agar medium. All flies were transferred to fresh food every 2–3 days. At three weeks of age, 45–50 female offspring from each cross were anaesthetized and dissected. All procedures were done at room temperature (18–22°C) as previously described [17], [24], [32], [61], [62]. Briefly, each head, ventral thorax and ventral abdominal cuticle was removed exposing the abdomen [62]. All internal organs and abdominal fat were removed leaving only the heart and associated muscles for each fly. Dissections were performed in oxygenated adult hemolymph. The semi-intact preparations were allowed to equilibrate with oxygenation for 20–30 min prior to filming. Analysis of heart morphology and physiology was performed using high speed movies of the semi-intact Drosophila preparations. 30 sec. movies were taken at rates of 100–200 frames/sec. using a Hamamatsu EM-CCD digital camera on a Leica DM LFSA microscope with a 10× immersion lens. All images were acquired and contrast enhanced using Simple PCI imaging software (Compix, Inc.). M-modes were generated and determination of cardiac parameters, including heart periods, diastolic and systolic diameters, fractional shortening and arrhythmicity indices for each group was performed using a MatLab-based image analysis program [63]. The “arrhythmicity index”, which is defined as the standard deviation of the heart period normalized to the median of each fly allowed us to quantify the average severity of arrhythmic beating patterns for each line. One-way ANOVAs of genotype as a function of each measured cardiac parameter, with Bonferroni multiple comparison tests, were employed to determine if significant differences among all Drosophila lines were present. P values<0.05 were considered significant.
Fluorescent Imaging of Drosophila cardiac tubes
Fluorescent imaging of Drosophila heart tubes were performed according to [64]. The UAS-GFP-CryABR120G fly line was crossed to Hand-Gal4 (II) driver flies. The progeny were aged to 1 and 3 weeks. Beating hearts of semi-intact Drosophila were placed in artificial Drosophila hemolymph containing 10 mM EGTA. Cardiac tubes were examined to ensure contractions were inhibited. Hearts were fixed in 1×PBS containing 4% formaldehyde at room temperature for 20 minutes with gentle shaking. Washing of hearts was performed three times for ten minutes with PBSTx (PBS containing 0.1% Triton-X-100) at room temperature with continual shaking. After washing, the hearts were incubated with Alexa584-phalloidin in PBSTx (1∶1000) for 20 minutes with continual agitation. Washing of the hearts was again carried out three times for ten minutes with PBSTx at room temperature. The hearts were rinsed in 100 µl of PBS for 10 minutes. The specimens were mounted on microscope slides and viewed at 10–25× magnification using a Zeiss Imager Z1 fluorescent microscope equipped with an Apotome sliding module.
Glutathione assay of adult fly heads
Female flies from transgenic or control lines were crossed to w1118; GMR-Gal4 males. 30–60 heads were collected from flies of the appropriate genotype for each cross and then immediately put in 60–120 µl (2 µl per head) 5% SSA (5-sulfosalicylic acid; Sigma #S-7408) and homogenized with a small eppendorf dounce pestle. Samples were then frozen at −80° and maintained frozen until assayed as described [65].
Larval glutathione assay
Female flies from UAS-RNAi, UAS-G6PD or control stocks were collected and crossed to y w; tub-Gal4/T(2;3)SM6, Cy; TM6,Tb males. Resulting third instar Tb+ larvae were collected and washed with 0.7% NaCl solution. Each collection was weighed and frozen on dry ice. The collected larvae were then stored at −80°C until ready for testing. At least 30 mg of larvae from the same genotype were pooled for each sample. Larvae were homogenized in 1X GSH MES buffer from the Glutathione Assay Kit by Cayman Chemical Company (10 µl/mg sample). The protocol provided with the kit was followed. Total GSH and GSSG measurements were calculated based on the Kinetic Method with minimal time course of 30 minutes.
Data processing
At least three independent samples were assayed for each genotype. The GSH/GSSG ratio for each sample was calculated independently. Statistical analysis of GSH/GSSG ratios was conducted using Graphpad Prism.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. SelcenD, EngelAG (2011) Myofibrillar myopathies. Handbook of clinical neurology/edited by PJ Vinken and GW Bruyn 101 : 143–154.
2. VicartP, CaronA, GuicheneyP, LiZ, PrévostMC, et al. (1998) A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet 20 : 92–95.
3. GoldfarbLG, DalakasMC (2009) Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. The Journal of Clinical Investigation 119 : 1806–1813.
4. BovaMP, YaronO, HuangQ, DingL, HaleyDA, et al. (1999) Mutation R120G in alphaB-crystallin, which is linked to a desmin-related myopathy, results in an irregular structure and defective chaperone-like function. Proc Natl Acad Sci U S A 96 : 6137–6142.
5. PerngMD, MuchowskiPJ, van DenIP, WuGJ, HutchesonAM, et al. (1999) The cardiomyopathy and lens cataract mutation in alphaB-crystallin alters its protein structure, chaperone activity, and interaction with intermediate filaments in vitro. The Journal of Biological Chemistry 274 : 33235–33243.
6. WangX, OsinskaH, KlevitskyR, GerdesAM, NiemanM, et al. (2001) Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ Res 89 : 84–91.
7. RajasekaranNS, ConnellP, ChristiansES, YanLJ, TaylorRP, et al. (2007) Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell 130 : 427–439.
8. WinklhoferKF, TatzeltJ, HaassC (2008) The two faces of protein misfolding: gain - and loss-of-function in neurodegenerative diseases. The EMBO Journal 27 : 336–349.
9. CampioniS, ManniniB, ZampagniM, PensalfiniA, ParriniC, et al. (2010) A causative link between the structure of aberrant protein oligomers and their toxicity. Nature Chemical Biology 6 : 140–147.
10. BalchWE, MorimotoRI, DillinA, KellyJW (2008) Adapting proteostasis for disease intervention. Science 319 : 916–919.
11. BolognesiB, KumitaJR, BarrosTP, EsbjornerEK, LuheshiLM, et al. (2010) ANS binding reveals common features of cytotoxic amyloid species. ACS Chemical Biology 5 : 735–740.
12. LashuelHA, LansburyPTJr (2006) Are amyloid diseases caused by protein aggregates that mimic bacterial pore-forming toxins? Quarterly Reviews of Biophysics 39 : 167–201.
13. KayedR, HeadE, ThompsonJL, McIntireTM, MiltonSC, et al. (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300 : 486–489.
14. SanbeA, OsinskaH, SaffitzJE, GlabeCG, KayedR, et al. (2004) Desmin-related cardiomyopathy in transgenic mice: a cardiac amyloidosis. Proc Natl Acad Sci U S A 101 : 10132–10136.
15. RubinGM, YandellMD, WortmanJR, Gabor MiklosGL, NelsonCR, et al. (2000) Comparative genomics of the eukaryotes. Science 287 : 2204–2215.
16. OlsonEN (2006) Gene regulatory networks in the evolution and development of the heart. Science 313 : 1922–1927.
17. CammaratoA, AhrensCH, AlayariNN, QeliE, RuckerJ, et al. (2011) A Mighty Small Heart: The Cardiac Proteome of Adult Drosophila melanogaster. PLoS ONE 6: e18497.
18. NeelyGG, KubaK, CammaratoA, IsobeK, AmannS, et al. (2010) A global in vivo Drosophila RNAi screen identifies NOT3 as a conserved regulator of heart function. Cell 141 : 142–153.
19. PaternostroG, VignolaC, BartschDU, OmensJH, McCullochAD, et al. (2001) Age-associated cardiac dysfunction in Drosophila melanogaster. Circ Res 88 : 1053–1058.
20. TickooS, RussellS (2002) Drosophila melanogaster as a model system for drug discovery and pathway screening. Curr Opin Pharmacol 2 : 555–560.
21. BierE, BodmerR (2004) Drosophila, an emerging model for cardiac disease. Gene 342 : 1–11.
22. LessingD, BoniniNM (2009) Maintaining the brain: insight into human neurodegeneration from Drosophila melanogaster mutants. Nat Rev Genet 10 : 359–370.
23. ChomaMA, IzattSD, WessellsRJ, BodmerR, IzattJA (2006) Images in cardiovascular medicine: in vivo imaging of the adult Drosophila melanogaster heart with real-time optical coherence tomography. Circulation 114: e35–36.
24. OcorrK, ReevesNL, WessellsRJ, FinkM, ChenHS, et al. (2007) KCNQ potassium channel mutations cause cardiac arrhythmias in Drosophila that mimic the effects of aging. Proc Natl Acad Sci U S A 104 : 3943–3948.
25. OcorrK, AkasakaT, BodmerR (2007) Age-related cardiac disease model of Drosophila. Mech Ageing Dev 128 : 112–116.
26. AkasakaT, KlinedinstS, OcorrK, BustamanteEL, KimSK, et al. (2006) The ATP-sensitive potassium (KATP) channel-encoded dSUR gene is required for Drosophila heart function and is regulated by tinman. Proc Natl Acad Sci U S A 103 : 11999–12004.
27. WolfMJ, AmreinH, IzattJA, ChomaMA, ReedyMC, et al. (2006) Drosophila as a model for the identification of genes causing adult human heart disease. Proc Natl Acad Sci U S A 103 : 1394–1399.
28. Taghli-LamallemO, AkasakaT, HoggG, NudelU, YaffeD, et al. (2008) Dystrophin deficiency in Drosophila reduces lifespan and causes a dilated cardiomyopathy phenotype. Aging Cell 7 : 237–249.
29. FischerJA, GinigerE, ManiatisT, PtashneM (1988) GAL4 activates transcription in Drosophila. Nature 332 : 853–856.
30. BrandAH, PerrimonN (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118 : 401–415.
31. McLendonPM, RobbinsJ (2011) Desmin-Related Cardiomyopathy: An Unfolding Story. American Journal of Physiology Heart and Circulatory Physiology 301: H1220–1228.
32. OcorrK, FinkM, CammaratoA, BernsteinSI, BodmerR (2009) Semi-automated Optical Heartbeat Analysis of Small Hearts. Journal of Visualized Experiments 31 : 1435.
33. RørthP, SzaboK, BaileyA, LavertyT, RehmJ, et al. (1998) Systematic gain-of-function genetics in Drosophila. Development 125 : 1049–1057.
34. BilenJ, BoniniNM (2005) Drosophila as a model for human neurodegenerative disease. Annu Rev Genet 39 : 153–171.
35. LuB, VogelH (2009) Drosophila models of neurodegenerative diseases. Annu Rev Pathol 4 : 315–342.
36. McAlisterL, FinkelsteinDB (1980) Heat shock proteins and thermal resistance in yeast. Biochem Biophys Res Commun 93 : 819–824.
37. Plesofsky-VigN, BramblR (1985) Heat shock response of Neurospora crassa: protein synthesis and induced thermotolerance. J Bacteriol 162 : 1083–1091.
38. LeganSK, RebrinI, MockettRJ, RadyukSN, KlichkoVI, et al. (2008) Overexpression of Glucose-6-phosphate Dehydrogenase Extends the Life Span of Drosophila melanogaster. J Biol Chem 283 : 32492–32499.
39. PrevilleX, SalveminiF, GiraudS, ChaufourS, PaulC, et al. (1999) Mammalian small stress proteins protect against oxidative stress through their ability to increase glucose-6-phosphate dehydrogenase activity and by maintaining optimal cellular detoxifying machinery. Exp Cell Res 247 : 61–78.
40. MeloniL, MancaMR, LoddoI, CiogliaG, CoccoP, et al. (2008) Glucose-6-phosphate dehydrogenase deficiency protects against coronary heart disease. J Inherit Metab Dis 31 : 412–417.
41. CoccoP, ToddeP, ForneraS, MancaMB, MancaP, et al. (1998) Mortality in a cohort of men expressing the glucose-6-phosphate dehydrogenase deficiency. Blood 91 : 706–709.
42. LongWK, WilsonSW, FrenkelEP (1967) Associations between red cell glucose-6-phosphate dehydrogenase variants and vascular diseases. Am J Hum Genet 19 : 35–53.
43. TueNT, ShimajiK, TanakaN, YamaguchiM (2012) Effect of alphaB-crystallin on protein aggregation in Drosophila. J Biomed Biotechnol 2012 : 252049.
44. GoebelHH (1997) Desmin-related myopathies. Curr Opin Neurol 10 : 426–429.
45. SongS, HansonMJ, LiuBF, ChylackLT, LiangJJ (2008) Protein-protein interactions between lens vimentin and alphaB-crystallin using FRET acceptor photobleaching. Mol Vis 14 : 1282–1287.
46. MollR, FrankeWW, SchillerDL, GeigerB, KreplerR (1982) The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. Cell 31 : 11–24.
47. YingW (2008) NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxidants & redox signaling 10 : 179–206.
48. GeerBW, LindelDL, LindelDM (1979) Relationship of the oxidative pentose shunt pathway to lipid synthesis in Drosophila melanogaster. Biochem Genet 17 : 881–895.
49. GeerBW, KrochkoD, WilliamsonJH (1979) Ontogeny, cell distribution, and the physiological role of NADP-malic enzyme in Drosophila melanogaster. Biochem Genet 17 : 867–879.
50. MerrittTJ, KuczynskiC, SezginE, ZhuCT, KumagaiS, et al. (2009) Quantifying interactions within the NADP(H) enzyme network in Drosophila melanogaster. Genetics 182 : 565–574.
51. ZhangH, RajasekaranNS, OroszA, XiaoX, RechsteinerM, et al. (2010) Selective degradation of aggregate-prone CryAB mutants by HSPB1 is mediated by ubiquitin-proteasome pathways. J Mol Cell Cardiol 49 : 918–930.
52. RorthP (1998) Gal4 in the Drosophila female germline. Mech Dev 78 : 113–118.
53. RubinGM, SpradlingAC (1982) Genetic transformation of Drosophila with transposable element vectors. Science 218 : 348–353.
54. SpradlingAC, RubinGM (1982) Transposition of cloned P elements into Drosophila germ line chromosomes. Science 218 : 341–347.
55. BischofJ, MaedaRK, HedigerM, KarchF, BaslerK (2006) An optimized transgenesis system for Drosophila using germ-line-specific φC31 integrases. PNAS 104 : 3312–3317.
56. PotterCJ, LuoL (2010) Splinkerette PCR for Mapping Transposable Elements in Drosophila. PLoS ONE 5: e10168.
57. FreemanM (1996) Reiterative use of the EGF receptor triggers differentiation of all cell types in the Drosophila eye. Cell 87 : 651–660.
58. DietzlG, ChenD, SchnorrerF, SuK-C, BarinovaY, et al. (2007) A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448 : 151–156.
59. CarboneMA, AyrolesJF, YamamotoA, MorozovaTV, WestSA, et al. (2009) Overexpression of Myocilin in the Drosophila Eye Activates the Unfolded Protein Response: Implications for Glaucoma. PLoS ONE 4: e4216.
60. HanZ, OlsonEN (2005) Hand is a direct target of Tinman and GATA factors during Drosophila cardiogenesis and hematopoiesis. Development 132 : 3525–3536.
61. OcorrKA, CrawleyT, GibsonG, BodmerR (2007) Genetic Variation for Cardiac Dysfunction in Drosophila. PLoS ONE 2: e601.
62. VoglerG, OcorrK (2009) Visualizing the Beating Heart in Drosophila. J Vis Exp 31: pii: 1425.
63. FinkM, Callol-MassotC, ChuA, Ruiz-LozanoP, BelmonteJCI, et al. (2009) A new method for detection and quantification of heartbeat parameters in Drosophila, zebrafish, and embryonic mouse hearts. BioTechniques 46 : 101–113.
64. AlayariNN, VoglerG, Taghli-LamallemO, OcorrK, BodmerR, et al. (2009) Fluorescent Labeling of Drosophila Heart Structures. J Vis Exp e1423.
65. ScarbroughPM, MapuskarKA, MattsonDM, GiusD, WatsonWH, et al. (2012) Simultaneous inhibition of glutathione - and thioredoxin-dependent metabolism is necessary to potentiate 17AAG-induced cancer cell killing via oxidative stress. Free Radical Biol Med 52 : 436–43.
66. Miller A (1950) The Internal Anatomy and Histology of the Imago of Drosophila melanogaster. In: Demerec M, editor. Biology of Drosophila. New York: John Wiley and Sons, Inc. pp. 420–534.
Štítky
Genetika Reprodukčná medicínaČlánok vyšiel v časopise
PLOS Genetics
2013 Číslo 6
- Gynekologové a odborníci na reprodukční medicínu se sejdou na prvním virtuálním summitu
- Je „freeze-all“ pro všechny? Odborníci na fertilitu diskutovali na virtuálním summitu
Najčítanejšie v tomto čísle
- BMS1 Is Mutated in Aplasia Cutis Congenita
- Sex-stratified Genome-wide Association Studies Including 270,000 Individuals Show Sexual Dimorphism in Genetic Loci for Anthropometric Traits
- Distinctive Expansion of Potential Virulence Genes in the Genome of the Oomycete Fish Pathogen
- Distinct Neuroblastoma-associated Alterations of Impair Sympathetic Neuronal Differentiation in Zebrafish Models