
Deficit lyzosomální kyselé lipázy v diferenciální diagnostice familiární hypercholesterolemie
Lysosomal acid lipase deficiency in differential diagnosis of familial hypercholesterolemia
Lysosomal acid lipase deficiency (LAL-D) is a rare autosomal recessive lysosomal storage disease caused by deleterious mutations in the LIPAgene. Occurrence is worldwide estimated to be 1 : 130 000 to 1 : 300 000 live births. Patients presenting in infancy have the most rapidly progressive disease, developing signs and symptoms in the first weeks of life and rarely surviving beyond 6 months of age (Wolman disease). Children and adults typically present with some combination of dyslipidemia, hepatomegaly, elevated transaminases, and microvesicular hepatosteatosis on biopsy – cholesterylester storage disease (CESD). Liver damage with progression to fibrosis, cirrhosis and liver failure occurs in a large proportion of patients. Elevated low-density lipoprotein cholesterol levels and decreased high-density lipoprotein cholesterol levels are common features, and cardiovascular disease may manifest as early as childhood. The lipid profile is very similar as we can see in familial hypercholesterolemia, so it is not surprising that LAL-D is under-recognized in clinical practice. This article provides practical guidance to lipidologists, on how to recognize individuals with this disease and, current management options are reviewed in light of the development of enzyme replacement therapy with sebelipase alfa, a recombinant human lysosomal acid lipase enzyme.
Keywords:
combination therapy – hepatomegaly – lysosomal acid lipase deficiency – Wolman disease
Authors:
Zuzana Urbanová 1; Věra Malinová 2
Authors‘ workplace:
Klinika dětského a dorostového lékařství, 1. LF UK a VFN v Praze
1; Centrum preventivní kardiologie III. interní kliniky 1. LF UK a VFN v Praze
2
Published in:
AtheroRev 2019; 4(1): 30-34
Category:
Reviews
Overview
Deficit kyselé lyzosomální lipázy (LAL-D) je vzácné autosomálně recesivní střádavé onemocnění, způsobené mutací v LIPA genu. Výskyt je celosvětově odhadován na 1 : 130 000 až 1 : 300 000 živě narozených dětí. Vede ke střádání cholesteryl esterů a triglyceridů zejména v játrech a k mikrovezikulární steatóze jater v bioptickém nálezu. Forma kojenecká (Wolmanova choroba) je nejprogresivnější, děti vzácně přežívají 6 měsíců a umírají na multiorgánové selhání. U starších dětí a dospělých se projevuje dyslipidemií, hepatomegalií, zvýšením hodnot jaterních testů (Cholesteryl Ester Storage Disease – CESD). V laboratorních nálezech je zvýšení celkového a LDL-cholesterolu (LDL-C) a snížení HDL-cholesterolu (HDL-C). Kardiovaskulární komplikace se mohou vyskytovat již v mladém věku a u mnoha pacientů dochází k progresi jaterní fibrózy, k cirhóze a jaternímu selhání. Lipidogram je velmi podobný jako u familární hypercholesterolemie a předpokládá se, že toto onemocnění je v klinické praxi velmi poddiagnostikované. Tento článek upozorňuje na tuto problematiku a na současné možnosti diagnostiky a léčby s ohledem na vývoj enzymové substituční terapie enzymem sebelipázou alfa – rekombinantní humánní lyzosomální kyselou lipázou.
Klíčová slova:
dyslipidemie – hepatomegalie – cholesteryl ester storage disease – Wolmanova choroba
Úvod
Defekt lyzosomální kyselé lipázy (LAL-D) je vzácné autosomálně recesivní lyzosomální střádavé onemocnění charakterizované progresivní akumulací cholesteryl esterů a triglyceridů v játrech, slezině a ostatních orgánech. Typickým nálezem je dyslipidemie a dochází k časnému vývoji aterosklerózy. Odhadovaný výskyt v ČR 1 : 350 000, ale od roku 1975 bylo u nás diagnostikováno jen 19 pacientů s cholesterylester storage disease (CESD) a 1 pacient s Wolmanovou chorobou [1,2,3]. Protože laboratorní nálezy jsou podobné jako u jiných poruch metabolizmu lipidů, zejména jako u familiární hypercholesterolemie, předpokládáme, že je toto onemocnění poddiagnostikované a mnoho pacientů je sledováno pod diagnózou zejména familiární hypercholesterolemie (FH). Dalším klinickým projevem je hepatomegalie s progresivním postižením jater i postižením dalších orgánů, což není pro diagnózu FH typické. V minulosti byla k dispozici jen symptomatická léčba, která příliš nezlepšovala prognózu pacientů. Umírali v mladém věku na progredující hepatopatii a komplikace aterosklerózy. Protože máme v současné době dispozici specifickou enzymovou substituční léčbu – sebelipázu alfa (rekombinantní humánní lyzosomální kyselá lipáza) a je k dispozici jednoduchá dostupná diagnostika, chtěli jsme na tuto problematiku upozornit.
Patogeneze LAL-D
Molekulárně genetická podstata tohoto onemocnění spočívá v mutaci v LIPA genu, který kóduje lyzosomální kyselou lipázu (LAL), která hraje klíčovou roli v lipidovém metabolizmu. Zabezpečuje hydrolýzu cholesteryl esterů a triglyceridů v lyzosomech [4].
Cholesteryl estery a triglyceridy derivovoné z LDL-cholesterolu jsou degradovány pomocí LAL na neesterifikovaný cholesterol a volné mastné kyseliny, které jsou důležitými mediátory homeostázy nitrobuněčného cholesterolu.
Pokud je aktivita LAL snížena nebo zcela potlačena, cholesteryl estery a triglyceridy nejsou degradovány a akumulují se v lyzosomech buněk orgánů, nejvíce v játrech. Vlivem zvýšení koncentrace cholesterolu a up-regulace HMG-CoA reduktázy může LAL zpětně inhibovat aktivitu LDL-receptorů a snižovat clearance LDL-C z cirkulace. V laboratorních vyšetřeních nacházíme typicky zvýšení LDL-C, snížení HDL-C, triglyceridy mohou (ale nemusí být) také zvýšené [5].
Klinické projevy LAL-D
LAL-D je heterogenní onemocnění s různými klinickými projevy. Průměrný věk, v němž se nemoc projevuje, je 5 let, ale byl popsán i rozvoj prvních příznaků u muže ve věku 44 let a u ženy ve věku 68 let.
Nejvíce fulminantní a progresivní forma se projevuje v prvních týdnech po narození a poprvé byla popsána v roce 1956 jako Wolmanova choroba. O několik let později popsal Fredrickson případ 12letého chlapce se závažnou hypercholesterolemií, hepatomegalií a akumulací cholesterol esterů z nálezu jaterní biopsie. Tato pozdní forma byla popsána jako cholesteryl ester storage disease (CESD).
Různorodost a závažnost projevů je ovlivněna typem mutace a stupněm zbytkové aktivity enzymu.
Wolmanova choroba
Při této variantě nacházíme nulovou aktivitu LAL a klinicky se toto onemocnění projeví v prvních týdnech života neprospíváním, zvracením, distenzí bříška, průjmy. Typická je hepatosplenomegalie a hepatopatie vedoucí k selhání jater. Ke střádání cholesteryl esterů může docházet i ve slezině, lymfatických uzlinách a nadledvinách, v nichž v 50 % nacházíme kalcifikace, dítě postupně katechizuje a umírá zpravidla do 6. měsíce života na multiorgánové selhání. Onemocnění se může projevit dokonce i v těhotenství ascitem plodu a polyhydramnionem [6].
Cholesteryl ester storage disease
U dětí a dospělých se LAL-D projevuje méně fulminantně, někdy jen poruchami metabolizmu lipidů, dále bývá asymptomatická hepatosplenomegalie v první dekádě, nebo až v dospělosti, a progresivní hepatopatie s elevací jaterních testů, steatóza jater, periportální fibróza, cirhóza a tumory jater.
Klinické projevy LAL-D
- hepatomegalie/hepatosplenomegalie
- průjem
- bolesti břicha
- zvracení
- anémie
- malabsorpce
- cholestáza
- steatorea
- neprospívání
- žlučníkové obtíže
- ICHS
- iktus
- adrenální kalcifikace
- jícnové varixy
Diferenciální diagnóza
Výše uvedené klinické a laboratorní nálezy mohou být přítomny i u jiných poruch metabolizmu lipidů – nejčastěji u FH, familiárního defektu apolipoproteinu B a u kombinované a polygenní hypercholesterolemie [7]. U dospělých je nutné uvažovat také o nealkoholové steatóze, steatohepatitidě a alkoholové hepatopatii. Hladiny LDL-cholesterolu bývají u pacientů s deficitem kyselé lipázy méně zvýšené než u pacientů s FH, nevídáme hepatopatii (zejména u dětí), na druhou stranu hepatopatie nemusí být přítomna u všech pacientů s LAL-D, někteří pacienti mohou mít také jen mírně zvýšené nebo normální hodnoty transamináz.
Vyšetření vedoucí k diagnóze LAL-D
Důležitá je podrobná osobní i rodinná anamnéza, která nás může vést k prvnímu podezření, že se o nejedná FH. Rodiče mají typicky normální nebo mírně patologický lipidogram a v rodině často není závažná kardiovaskulární zátěž. Ve fyzikálním vyšetření nacházíme hepatosplenomegalii, která je opět v rozporu s diagnózou FH, zejména u dětí.
Laboratorní vyšetření
V biochemickém vyšetření nacházíme elevaci jaterních testů (ALT, AST, GGT), hypercholesterolemii, zvýšení LDL-cholesterolu a někdy triglyceridů, snížení HDL-cholesterolu.
Zobrazovací techniky
Ultrasonografie (USG) a nukleární magnetická rezonance (NMR) prokazují hepatosplenomegalii, zvětšení nadledvin a jejich kacifikace. V poslední době je k dispozici 3T magnetická rezonance jako méně invazivní metoda než jaterní biopsie.
Měření LAL-aktivity
LAL-D může být diagnostikována biochemicky měřením enzymatické aktivity v různých buněčných typech: prakticky se však dnes využívá pouze hodnocení aktivity enzymu v leukocytech periferní krve. Reakce je specifická pro LAL, výsledky jsou tedy při použití této metody velmi spolehlivé.
Pro screening se využívá metoda suché kapky (dried blood spot – DBS), při které je aktivita LAL měřena fluorimetricky. Tato metoda je relativně jednoduchá, ale jde o vyšetření orientační, které může být falešně negativní (např. vlivem použité dezinfekce) a je třeba použít inhibitor LAL lalistat, který pomůže „odfiltrovat“ interferující aktivitu ostatních lipáz ve vzorku [9].
Jaterní biopsie
V současné době se biopsie provádí, jen pokud ostatní vyšetření nevedou k diagnóze. Nacházíme mikrovesikulární steatózu v jaterních lobulech, akumulaci lipidů v hepatocytech, pěnové buňky (makrofágy) s vysokým obsahem ceroidu lokalizovaným periferně, ztrátu lipopigmentu (lipofuscinu) v hepatocytech. Je pozitivní imunohistochemické barvení na lyzosomální markery katepsin D, LAMP1, LAMP2 (Lysosomal Associated Membrane Protein 1, Lysosomal Associated Membrane Protein 2).
Genetické vyšetření
DNA analýza – mutace genu LIPA, nejčastější je mutace E8SJM, 50–70 %.
Sledování a léčba pacientů s LAL
Pacienti by měli být sledováni ve specializovaných metabolických centrech, zaměřených na léčbu lyzosomálních chorob. Vzhledem k progresivnímu charakteru onemocnění by měli být pacienti 1krát ročně kontrolováni (jaterní funkce, krevní obraz, lipidový profil). Zobrazovacími metodami by se měla pravidelně měřit velikost jater a sleziny. Magnetická rezonanční spektroskopie umožňuje sledovat obsah tuku v játrech a progresi jaterní fibrózy. Co se týče rizikových faktorů aterosklerózy, k pacientům s LAL-D by se mělo přistupovat jako k vysoce rizikovým.
Dříve byla k dispozici jen symptomatická léčba. Dieta a hypolipidemika snížily hladiny lipidů, ale statiny, stejně jako ezetimib, nezastavily progresi jaterního postižení. Transplantace kmenových buněk a transplantace jater byly zatíženy velkým rizikem a dlouhodobá prognóza nebyla dobrá.
V současné době máme k dispozici specifickou substituční enzymovou léčbu (Enzyme Replacement Therapy – ERT). Cílem ERT je zvýšit aktivitu kyselé jaterní lipázy, zabránit hromadění cholesteryl esterů a triglyceridů v buňkách, a tím docílit zlepšení funkce postižených orgánů. Sebelipáza alfa, vede k poklesu hodnot jaterních transamináz, celkového, LDL-cholesterolu i triglyceridů a zvýšení hodnot HDL-cholesterolu. Sebelipázu podáváme formou nitrožilních infuzí v intervalu 2 týdnů [10].
Kazuistika
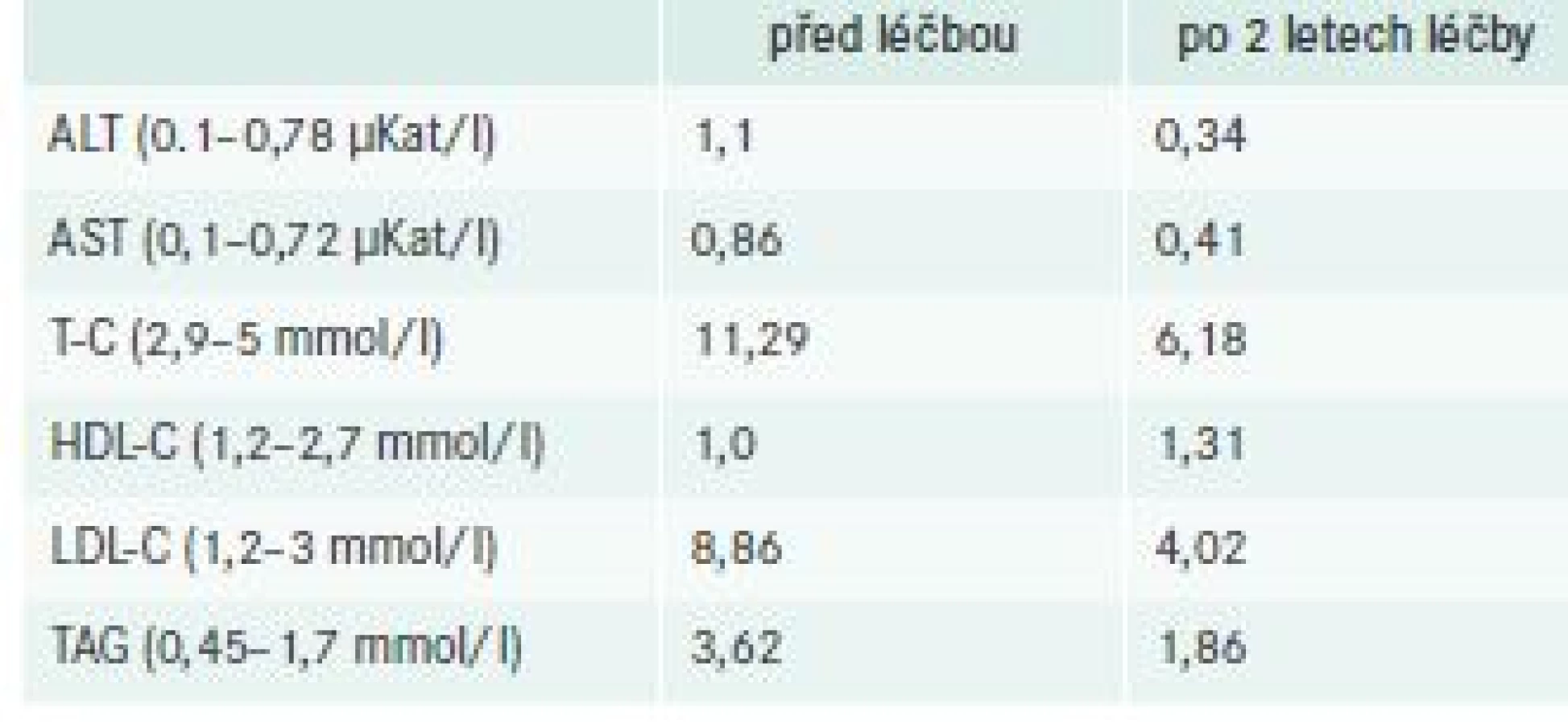
10 letá dívka přichází do naší lipidologické ambulance z gastroenterologického oddělení jiného pracoviště, na kterém byla sledována pro hepatopatii, hepatomegalii, kombinovanou hyperlipidemii s dg. suspektní familiární hypercholesterolemie. V osobní anamnéze nebylo nic pozoruhodného, fyziologický porod a těhotenství, porodní hmotnost byla 3 600 g/52 cm, vývoj v normě a nemocnost běžná. V roce 2008 (v 8 letech věku) prodělala mononukleózu, v krajské nemocnici byla zjištěna hypercholesterolemie 10 mmol/l a hepatopatie. V březnu 2010 pro zhoršení nálezů u ní byla zahájena léčba atorvastatinem, která vedla k výraznému zhoršení jaterních funkcí. Na základě jaterní biopsie byl popsán nespecifický nález – těžká portální fibróza, inkompletní cirhóza s lobulární zánětlivou aktivitou a dominujícími metabolickými změnami parenchymu, zduření hepatocytů, difuzní malokapénková steatóza, glykogenovaná jádra hepatocytů. V diferenciální diagnostice se uvažovalo o glykogenóze. Antropometricky byla na 50. percentilu, ve fyzikálním vyšetření dominovala hepatosplenomegalie, potvrzená magnetickou rezonancí. V laboratorním vyšetření byl celkový cholesterol 11,29, HDL-C 1,04, LDL-C 5,18, TAG 4,15, ALT 1,32, AST 1,30, GGT 0,22, ALP 2,99, glu 3,5. Protože tyto nálezy neodpovídaly diagnóze familiární hypercholesterolemie a budily podezření na jinou poruchu metabolizmu lipidů, byla doporučena do ambulance pro vrozené vady metabolizmu naší kliniky. Bylo vyžádáno druhé čtení bioptického materiálu. Prof. MUDr. Milan Eledeler DrSc. popsal mikrovezikulární steatózu jater s intralyzosomálním střádáním a vyslovil podezření na CESD. Tuto diagnózu pak potvrdilo enzymatické vyšetření aktivity kyselé lipázy – kyselá lipáza v leukocytech byla 10,3 nmol/mg/h (norma 190–650 nmol/mg/h). Byla zahájena dieta s omezením tuků a cholesterolu a v roce 2013 (ve 13 letech) byla dívka zařazena do observační studie pro léčbu rekombinantní formou lidské kyselé lipázy – sebelipase alfa (SBC-102), infuze 1krát za 2 týdny. V současné době je dívka 18letá, bez obtíží, s normální antropometrií, došlo k výrazné významné redukci jaterních testů, celkového a LDL-cholesterolu, triglyceridů a k vzestupu HDL-cholesterolu (tab).
Závěr
Prognóza pacientů s LAL-D je velmi individuální, od těžkého jaterního postižení v kojeneckém věku, vyžadujícího transplantaci jater, až po těžké kardiovaskulární komplikace v dospělém věku, průběh může být i mírný [11]. V každém případě je důležitá časná diagnostika, aby mohla být včas zahájena specifická léčba, která může zabránit vývoji orgánových změn. Dyslipidemie je nejčastějším prvním nálezem u dětí a dospělých s LAL-D, a mnoho pacientů, stejně jako výše referovaná dívka, může být odesláno do lipidologických ambulancí s diagnózou FH.
Proto je důležité o diagnóze LALD uvažovat, zvláště je-li přítomna hepatomegalie a hepatopatie, protože děti s FH tyto příznaky nemají. Včasnou diagnostiku LAL-D umožňuje vyšetření metodou suché kapky, která je dobře dostupná pro všechny lipidology a lékaře, kteří se touto problematikou zabývají.
Kontakt pro konzultaci pacientů:
MUDr. Věra Malinová
Klinika dětského a dorostového lékařství a ÚMDP, 1 LF UK a VFN v Praze
e-mail: vera.malinova@vfn.cz
doc. MUDr. Zuzana Urbanová, CSc.
Doručeno do redakce/Doručené do redakcie/Received 6. 1. 2019
Přijato po recenzi/Prijaté po recenzii/Accepted 31. 1. 2019
Sources
- Elleder M, Poupětová H, Ledvinová J et al. Deficit kyselé (lyzozomální) lipázy: přehled českých pacientů. Čas Lék česk 1999; 138(23): 719–724.
- Elleder M, Chlumska A, Hyanek J et al. Subclinical course of cholesteryl ester storage disease in an adult with hypercholesterolemia, accelerated atherosclerosis, and liver cancer. J Hepatol 2000; 32(3): 528–534.
- Hulkova H, Elleder M. Distinctive histopathological features that support a diagnosis of cholesterol ester storage disease in liver biopsy specimens. Histopathology 2012; 60(7): 1107–1113. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1365–2559.2011.04164.x>.
- Reiner Ž, Guardamagna O, Nair D et al. Lysosomal acid lipase deficiency – An under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis 2014; 235(1): 21–30. Dostupné z DOI: <http://dx.doi.org/10.1016/j.atherosclerosis.2014.04.003>.
- Fouchier SW, Defesche JC. Lysosomal acid lipase A and the hypercholesterolaemic phenotype. Curr Opin Lipidol 2013; 24 (4):332–338. Dostupné z DOI: <http://dx.doi.org/10.1097/MOL.0b013e328361f6c6>.
- Grabowski GA, Du H, Charnas L. Lysosomal acid lipas e deficiencies: the Wolman disease/cholesteryl ester storage disease spectrum. In: Valle D (ed), Beaudet AL, Vogelstein B et al. The Online Metabolic and Molecular Bases of Inherited Disease. McGraw Hill 2006. Dostupné z DOI: <http://dx.doi.org/10.1036/ommbid.172>.
- Valle D, Beaudet L,Vogelstein B (eds) et al. The Online Metabolic and Molecular Bases of Inherited Disease. McGraw Hill 2006. Dostupné z DOI: <https://ommbid.mhmedical.com>.
- Chatrath H, Keilin S, Attar BM. Cholesterol ester storage disease (CESD) diagnosed in an asymptomatic adult. Dig Dis Sci 2009; 54(1): 168–173. Dostupné z DOI: <http://dx.doi.org/10.1007/s10620–008–0310–2>.
- Malinová V, Honzík T. Lysosomální onemocnění – současné možnosti diagnostiky a terapie Pediatr Praxi 2013; 14(2): 99–103.
- Burton BK, Balwani M, Feillet F et al. A Phase 3 Trial of Sebelipase Alfa in Lysosomal Acid Lipase Deficiency, N Engl J Med 2015; 373(11): 1010–1020. Dostupné z DOI: <http://dx.doi.org/10.1056/NEJMoa1501365>.
- Bernstein DL, Hulkova H, Bialer MG et al. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol 2013; 58(6): 1230–1243. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jhep.2013.02.014>.
Labels
Angiology Diabetology Internal medicine Cardiology General practitioner for adultsArticle was published in
Athero Review

2019 Issue 1
- Memantine Eases Daily Life for Patients and Caregivers
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Hope Awakens with Early Diagnosis of Parkinson's Disease Based on Skin Odor
- Spasmolytic Effect of Metamizole
Most read in this issue
- Nutraceutika s hypolipidemickým účinkem v klinické praxi: shrnutí stanoviska Mezinárodního expertního panelu lipidologů vypracované výborem České společnosti pro aterosklerózu
- Deficit lyzosomální kyselé lipázy v diferenciální diagnostice familiární hypercholesterolemie
- Terapie dyslipidemie u pacienta se svalovou dystrofií: kazuistika
- Psoriáza ako rizikový faktor kardiovaskulárnych príhod: kazuistika