
Plicní arteriální hypertenze
Pulmonary arterial hypertension
Pulmonary arterial hypertension (PAH) is a potentially fatal disease of pulmonary arterioles. Genetic predisposition plays a role in the pathophysiology of the disease, as well as a number of exogenous factors which cause endothelial dysfunction and subsequent vasoconstriction, vascular remodelling and thrombosis primarily in the region of small pulmonary arteries. The symptomatology of PAH is non–specific, hence the high rate of late-diagnosed cases of the disease. Patients with higher risk of pulmonary hypertension (i.e. systemic sclerodermia and HIV patients, patients with portal hypertension preceding liver transplantation and first grade relatives of PAH patients) should have preventative echocardiography examinations on a regular basis. The treatment of pulmonary hypertension is extremely complex and expensive, and therefore it is confined to specialised centres. The choice of PAH pharmacotherapy depends on the outcome of the acute pulmonary vasodilator test. Only patients with a positive outcome (11 % of patients) are indicated for the treatment by high doses of calcium channel blockers. This is what we call „conventional“ treatment, together with anticoagulation treatment and the treatment of heart failure. In case of a negative outcome of the test, „specific“ pharmacotherapy (prostanoids, endothelin receptor antagonists, phosphodiesterase 5 inhibitors) with both vasodilator and antiproliferative and antiaggregation effect are indicated in addition to chronic anticoagulation treatment. In patients in NYHA functional class (II) and III, treatment is initiated with bosentan, alternatively with sildenafil. Prostacyclin is the basic therapy in NYHA functional class IV. In a number of cases, monotherapy does not provide optimum response. Combination therapy is a rational alternative in such case, using several specific drugs. Once the possibilities of pharmacotherapy have been exhausted, atrial septostomy and lung transplantation can be considered. In addition to research for better therapeutic procedures for PAH, a major challenge in PAH treatment today is timely and correct diagnosing based on a correctly formulated suspicion. Still toady, great part of PAH patients is referred to specialised centres in advanced stages of the disease.
Keywords:
pulmonary arterial hypertension – conventional treatment – specific treatment – calcium channel blockers – prostanoids – endothelin receptor antagonists – phosphodiesterase 5 inhibitors
:
P. Jansa; D. Ambrož; T. Paleček; P. Poláček; J. Marešová; L. Jelínková; M. Aschermann; A. Linhart
:
Kardiol Rev Int Med 2007, 9(3): 145-153
Plicní arteriální hypertenze (PAH) je potenciálně fatální onemocnění plicních arteriol. V patofyziologii se různou měrou uplatňuje genetická dispozice a dále řada faktorů zevních, které vedou k endoteliální dysfunkci s následnou vazokonstrikcí, cévní remodelací a trombózou, zejména v oblasti malých plicních tepen. Symptomatologie PAH je nespecifická, proto je také stanovení diagnózy často pozdní. Nemocné se zvýšeným rizikem vzniku plicní hypertenze (pacienti se systémovou sklerodermií, infekcí HIV, portální hypertenzí před transplantací jater a prvostupňoví příbuzní nemocných s PAH) je nutno preventivně echokardiograficky vyšetřovat. Léčba plicní hypertenze je značně komplikovaná a ekonomicky mimořádně náročná. Proto je soustředěna do specializovaných center. O způsobu farmakoterapie PAH rozhoduje test akutní plicní vazodilatace. Pouze nemocní s pozitivním testem (11 % pacientů) jsou indikováni k léčbě vysokými dávkami blokátorů kalciových kanálů. Tato terapie se spolu s antikoagulační léčbou a léčbou srdečního selhání označuje jako léčba konvenční. V případě negativního testu je vedle chronické antikoagulační léčby indikována tzv. specifická farmakoterapie (prostanoidy, antagonisté endotelinových receptorů, inhibitory fosfodiesterázy 5) s účinky nejen vazodilatačními, ale rovněž antiproliferačními a antiagregačními. U pacientů ve funkčním stadiu NYHA (II) a III se léčba zahajuje bosentanem, alternativně sildenafilem. Ve funkčním stadiu NYHA IV je základem terapie intravenózní prostacyklin. V řadě případů není odpověď na monoterapii optimální. Jednu z racionálních alternativ pak představuje kombinační léčba několika specifickými farmaky. V případě vyčerpání možností farmakoterapie padá v úvahu atriální septostomie a transplantace plic. Největší současnou výzvou u PAH je vedle hledání dokonalejších léčebných postupů zejména včasná a správná diagnóza na základě správně vysloveného podezření. I dnes je totiž velká část nemocných stále referována do specializovaných center až v pokročilých stádiích onemocnění.
Klíčová slova:
plicní arteriální hypertenze – konvenční léčba – specifická léčba – blokátory kalciových kanálů – prostanoidy – antagonisté endotelinových receptorů – inhibitory fosfodiesterázy 5
Úvod a klasifikace
Plicní arteriální hypertenze (PAH) je primární onemocnění plicních arteriol, které je hemodynamicky charakterizováno vzestupem středního tlaku v plicnici nad 25 mm Hg v klidu a 30 mm Hg při zátěži, normálním tlakem v zaklínění a plicní cévní rezistencí nad 3 WU (Woodovy jednotky). PAH bez léčby rychle progreduje a postupně vede k pravostrannému srdečnímu selhání a ke smrti.
První případ PAH (dříve označované jako primární plicní hypertenze) popsal v roce 1891 Romberg jako sklerózu plicní tepny. Termín primární plicní hypertenze zavedl v roce 1951 David Dresdale pro nález etiologicky nevysvětlitelné plicní hypertenze. Do 50. let minulého století se rovněž datují snahy o léčbu plicní hypertenze krátkodobým podáním vazodilatancií bez výraznějšího dlouhodobého úspěchu. Terapeutický nihilizmus byl důvodem relativně okrajového zájmu kliniků o toto onemocnění. Posléze se stala určitou nadějí pro nemocné s PAH transplantace plic nebo srdce a plic, bohužel se značně nepříznivými výsledky. Zásadní změnu přineslo až studium celulárních a subcelulárních mechanizmů vzniku onemocnění a zavedení specifické farmakoterapie v 90. letech 20. století.
Zvýšený zájem o plicní hypertenzi vedl rovněž ke vzniku nových klasifikačních schémat. Vedle klasické patofyziologické klasifikace rozdělující plicní hypertenzi na prekapilární (zvýšený tlak v plicnici, normální v zaklínění), postkapilární (zvýšený tlak v plicnici i v zaklínění) a hyperkinetickou (při vysokém minutovém výdeji) je dnes chronická plicní hypertenze dělena podle klinické (Benátské) klasifikace do 5 kategorií: na PAH, plicní hypertenzi při srdečních onemocněních, plicní hypertenzi při respiračních onemocněních, plicní hypertenzi při chronické trombotické a/nebo embolické nemoci a plicní hypertenzi z jiných příčin (tab. 1) [1,2]. Do skupiny PAH je řazena především idiopatická a familiární PAH a dále řada stavů, v jejichž patogenezi se pravděpodobně uplatňují podobné patofyziologické mechanizmy a které jsou podobným způsobem ovlivnitelné farmakoterapií (asociovaná PAH). Zvláštní skupinu PAH představuje plicní venookluzivní nemoc a plicní kapilární hemangiomatóza, které jsou charakterizované plicní okluzivní venopatií a mikrovaskulopatií. Tyto 2 klinické jednotky vykazují jak morfologické znaky typické pro PAH (hypertrofie médie, fibróza intimy, plexiformní léze), tak obraz typický pro postkapilární plicní hypertenzi (plicní edém, dilatace lymfatických cév). Klinicky nejsou odlišitelné od idiopatické PAH. Léčba prostacyklinem však v těchto případech na rozdíl od jiných případů PAH vede k rychlému rozvoji plicního edému.

Patofyziologie
Na rozvoji PAH se nepochybně podílí kombinace faktorů zevních a genetických, které vytvářejí podmínky pro vznik nemoci [3]. Familiární výskyt PAH byl v literatuře dokumentován velmi časně po prvním popisu onemocnění již v roce 1954 [4]. Během dalších desetiletí byly publikovány práce popisující další rodiny postižené tímto onemocněním. Vývoj v genetickém mapování genů pro dědičná onemocnění v 90. letech minulého století umožnil systematické hledání genů zodpovědných za výskyt PAH v již dříve identifikovaných rodinách. Za pomoci genetických map a genetických DNA markerů (mikrosatelity) bylo možno v těchto rodinách provést tzv. celogenomový scan, který reprodukovatelně prokazoval genetickou vazbu onemocnění s markery na chromozomu 2q31–32 [5,6]. Analýza 1 z kandidátních genů v tomto genomovém regionu prokázala u postižených probandů mutace v genu BMPR2 (Bone morphogenetic protein receptor 2), který patří do rodiny receptorů pro TGF–beta (Transforming growth factor – beta) [7]. Doposud dokumentované mutace v tomto genu svědčí pro tzv. privátní charakter mutací, tedy specifický výskyt mutací v každé rodině. Většina dokumentovaných mutací vede k předčasné terminaci translace proteinu a tím k poruše jeho funkce. U zdravých jedinců BMP inhibuje proliferaci a indukuje apoptózu buněk hladkého svalstva ve stěně plicních arteriol a má antiapoptický účinek na endoteliální buňky. U nemocných s PAH má BMP na buňky hladkého svalstva účinek proliferativní a proapoptotický účinek je zesílen. Antiapoptotický účinek na buňky endoteliální se mění rovněž v proapoptotický. V případě familiárního výskytu PAH se jedná o onemocnění přenášené autozomálně, dominantně s variabilní penetrancí (jen 10–20 % nositelů mutace má projevy onemocnění) a expresivitou (tzn. závažností projevů manifestního onemocnění). Ženy jsou familiárním i sporadickým onemocněním postiženy častěji. Zdá se, že je přítomna tzv. genetická anticipace, známá například u Huntingtonovy chorey, u níž se u každé následující zatížené generace projeví postižení dříve a/nebo je klinicky závažnější.
U idiopatické PAH může být přítomna mutace BMPR2 přibližně u 10–20 % pacientů a v tomto případě se pravděpodobně jedná o nové mutace, které mohou být dále předány potomkům. Zbylých 80–90 % těchto pacientů představuje důležitou skupinu pro další výzkum, neboť minimálně u části z nich lze předpokládat významnou genetickou komponentu. Také ve skupině pacientů s PAH, kteří anamnesticky užívali některá anorektika, byly nalezeny mutace v genu BMPR2, a to asi u 10 % nemocných.
U pacientů s hereditární hemoragickou teleangiektazií (HHT, M. Rendu–Osler–Weber typ 2) byla identifikována mutace v genu ALK1 (activin receptor–like kinase 1), který tyto pacienty kromě projevů HHT vystavuje riziku vzniku PAH [8].
U nemocných s idiopatickou PAH je také v 65 % případů přítomna v homozygotní konstituci varianta polymorfizmu v oblasti promotoru genu pro serotoninový transportér, jehož zvýšená aktivita souvisí s hyperplazií buněk hladkého svalstva ve stěně plicních arteriol.
Hlavní změny v oblasti plicní mikrocirkulace u pacientů s PAH zahrnují vazokonstrikci, cévní remodelaci a trombózu v důsledku relativně vyšší produkce faktorů s účinky vazokonstrikčními, růstovými a trombogenními než faktorů s účinky vazodilatačními, antiproliferačními a antitrombotickými při endoteliální dysfunkci.
Prostacyklin a tromboxan A2 jsou hlavní metabolity kyseliny arachidonové. Prostacyklin je silný vazodilatátor, inhibitor aktivace trombocytů a má rovněž významné účinky antiproliferační. Tromboxan A2 je naproti tomu účinný vazokonstriktor a destičkový agonista. U nemocných s PAH je metabolizmus posunut ve prospěch tromboxanu.
U PAH je dokumentována zvýšená plazmatická koncentrace vazokonstrikčního endotelinu–1 a serotoninu. Oba tyto faktory rovněž navozují buněčnou proliferaci. U pacientů s idiopatickou PAH nalézáme dále sníženou koncentraci serotoninu v krevních destičkách.
U pacientů s PAH je prokázána snížená aktivita endoteliální NO syntézy. Ta je však naopak zvýšena v plexiformních lézích, kde pravděpodobně facilituje proliferaci endoteliálních buněk. Také koncentrace dalšího vazodilatačního faktoru, vazoaktivního intestinálního peptidu, je u PAH snížena v séru i v plicní tkáni.
V patofyziologii PAH hrají důležitou roli draslíkové kanály [9]. Regulují koncentraci vápenatých a draslíkových iontů v krevních destičkách, buňkách hladkého svalstva a v endoteliálních buňkách. Koncentrace Ca2+ v cytozolu ovlivňuje cévní tonus, koncentrace K+ moduluje apoptózu a cévní remodelaci. Inhibice voltážově řízených draslíkových kanálů (Kv), např. hypoxií a deriváty fenfluraminu nebo jejich downregulace, vede k depolarizaci buněčné membrány a k influxu vápenatých iontů do cytozolu. V trombocytech dochází ke zvýšení efluxu serotoninu a k inhibici jeho zpětného vychytávání. Důsledkem je vazokonstrikce a proliferace.
U nemocných s PAH nalézáme v řadě případů pozitivitu autoprotilátek, infiltraci cévních lézí zánětlivými elementy a depozita komplementu ve stěně plicních cév. To může poukazovat na roli zánětu v patofyziologii PAH.
U PAH je známa řada abnormit koagulačního a fibrinolytického systému. Zvýšena bývá hladina von Willebrandova faktoru, fibrinopeptidu A, inhibitoru aktivátoru plazminogenu a tromboxanu. Naopak snížena bývá hladina tkáňového aktivátoru plazminogenu a trombomodulinu. Důsledkem je zvýšená pohotovost ke vzniku trombů.
Ze zevních faktorů podílejících se na rozvoji PAH je znám vliv hypoxie, anorektik, metamfetaminu kokainu a některých infekcí.
Akutní hypoxie vede k systémové vazodilataci a k vazokonstrikci v malém oběhu cestou zvýšení sekrece endotelinu, serotoninu a snížením aktivity voltážově řízených draslíkových kanálů v buňkách hladkého svalu v cévní stěně.
Vyšší riziko vzniku PAH u osob užívajících některá anorektika (aminorex fumarát, fenfluramin, dexfenfluramin) lze vysvětlit jejich působením na zvýšení efluxu serotoninu z trombocytů [10]. Elevace tlaků v plicnici může být patrna již po 3–4 týdnech užívání anorektik, k rozvoji PAH je však třeba zpravidla více než 6 měsíců.
V patofyziologii PAH asociované se systémovými onemocněními pojiva bude nepochybně zásadní role zánětu.
Přítomnost perivaskulární zánětlivé infiltrace ukazuje na roli zánětu také u PAH asociované s HIV infekcí. V patofyziologii se předpokládá působení cytokinů, růstových faktorů a endotelinu. V aktivaci zánětlivých mechanizmů hrají zřejmě zásadní roli některé proteiny viru HIV (proteiny Tat, Nef) [11].
U PAH asociované s vrozenými srdečními vadami je v patofyziologii rozhodující chronická expozice plicní cirkulace zvýšenému průtoku krve.
V rozvoji portopulmonální hypertenze může být jedním z klíčových mechanizmů přestup vazoaktivních mediátorů z portální do plicní cirkulace při otevřených porto–systémových zkratech. Takto může působit např. serotonin produkovaný enterochromafinními buňkami ve střevě.
Výskyt PAH je rovněž vyšší u některých hematologických onemocnění. U myelodysplastického syndromu je příčina vzniku plicní hypertenze multifaktoriální, může se na ní podílet splenektomie, portální hypertenze nebo infiltrace plicního parenchymu hematopoetickými buňkami. Byly rovněž popsány vzácné případy plicní hypertenze u esenciální trombocytemie, ß–talasemie a srpkovité anémie.
Epidemiologie a prognóza
Plicní arteriální hypertenzí trpí na celém světě nepochybně několik milionů obyvatel. Většina případů však, zejména v méně ekonomicky rozvinutých zemích, uniká diagnóze. V Evropě, USA, Kanadě a Japonsku se výskyt PAH odhaduje na několik set tisíc případů.
Podle údajů z francouzského národního registru je minimální prevalence PAH 15 nemocných na milion obyvatel, zhruba 43 % představují nemocní s idiopatickou a familiární PAH [12]. Nejvyšší výskyt PAH je u lidí ve věku 41–60 let, překvapivě častěji u mužů.
Idiopatická PAH se v populaci vyskytuje s roční incidencí 2–5 případů, podle NIH (National Institute of Health) registru jsou častěji postiženy ženy (1,7krát častěji). Familiární forma tvoří 6–10 % případů PAH.
Odhad prevalence PAH u nemocných se systémovými onemocněními pojiva je obtížný, neboť schází konzistentní epidemiologická studie. Publikované údaje se pohybují v širokém rozmezí vzhledem k nejednotně užívané definici PAH a rozdílnému metodickému přístupu při stanovení nebo odhadu tlaku v plicnici. Systémová sklerodermie, zejména její CREST varianta, je nejčastější příčinou PAH mezi systémovými onemocněními. V publikaci zpracovávající údaje z registru 722 nemocných se systémovou sklerodermií ve Velké Británii je prevalence PAH udávána zhruba 12 % [13]. V jiné skupině 930 pacientů se systémovou sklerodermií byla kumulativní incidence PAH 13 %. U systémového lupus erythematodes je plicní hypertenze přítomna u 5–10 % pacientů. Vzácně se PAH vyskytuje u revmatoidní artritidy, dermatomyozitidy, polymyozitidy a Sjögrenova syndromu. PAH u systémových onemocnění postihuje zejména ženy. Ve srovnání s idiopatickou PAH se onemocnění manifestuje ve vyšším věku, bývá charakterizováno horšími hemodynamickými parametry a závažnější prognózou.
K PAH vedou jak vrozené srdeční vady jednoduché (defekt síňového septa, defekt komorového septa, otevřená tepenná dučej), tak vady komplexní (truncus arteriosus, univentrikulární srdce). PAH vzniká prakticky vždy u truncus arteriosus, u 50 % nemocných s velkým defektem septa komor (průměr defektu nad 1 cm) a u 10 % pacientů s velkým defektem septa síní (průměr defektu nad 2 cm), častěji u defektu typu sinus venosus [14].
U nemocných infikovaných virem HIV je výskyt PAH 6–12krát častější než v běžné populaci. Na 200 infikovaných virem HIV připadá 1 nemocný s komplikující PAH. PAH se vyskytuje u 2–6 % nemocných s jaterní cirhózou. U kandidátů transplantace jater je výskyt PAH popisován u 4–15 % nemocných. PAH bývá často oligosymptomatická, významně však ovlivňuje prognózu a zvyšuje peritransplantační mortalitu, zvláště pokud je střední tlak v plicnici vyšší než 35 mm Hg. Riziko vzniku PAH roste s délkou trvání portální hypertenze.
Zásadní údaje o prognóze idiopatické PAH pocházejí z údajů amerického NIH registru [15]. V letech 1981–1985 bylo do registru zařazeno celkem 194 nemocných, jejich osud byl sledován do roku 1988. K dispozici nebyla specifická léčba, nemocní dostávali pouze diuretika a kyslík. 1 rok přežilo 68 %, 2 roky 48 % a 3 roky 34 % nemocných. Medián přežití byl 2,8 roku. Medián přežití u neléčené PAH při systémové sklerodermii se pohybuje kolem 12 měsíců. Podobně nepříznivou prognózu má PAH asociovaná s infekcí HIV. Naopak lepší prognózu než u idiopatické PAH pozorujeme u nemocných s PAH asociovanou s vrozenou srdeční vadou.
K závažným prediktorům prognózy patří tíže symptomů (hodnocená podle modifikované klasifikace NYHA), funkční zdatnost hodnocená testem 6minutovou chůzí a hemodynamické parametry. Podle registru NIH byl medián přežití nemocných ve funkčním stadiu NYHA I a II zhruba 6 let, zatímco u nemocných ve funkčním stadiu NYHA III 2,5 roku a ve funkčním stadiu NYHA IV 6 měsíců. Vzdálenost pod 380 m dosažená při testu 6minutovou chůzí při specifické léčbě je spojena s horší prognózou. Z hemodynamických parametrů je s prognózou nejtěsněji svázán tlak v pravé síni, střední tlak v plicnici a srdeční index. Horší prognózu mají nemocní s vyšší hodnotou tzv. biomarkerů: s vyšší hladinou kyseliny močové, s pozitivním troponinem, s vyšším BNP (nad 150 pg/ml) nebo NT–proBNP (nad 1400 pg/ml).
Významně odlišnou prognózu lze očekávat u nemocných s PAH, kteří jsou respondéry při testu akutní plicní vazodilatace. 5 let přežije více než 95 % nemocných, zejména pokud jsou současně antikoagulováni [16].
Klinický obraz a diagnostika
Většina příznaků u nemocných s plicní hypertenzí souvisí se zvýšením tlaku v plicnici. Nejsou specifické a často se vyskytují až při zvýšení tlaku v plicnici na dvojnásobek normálních hodnot [17]. Právě nespecifické projevy onemocnění jsou příčinou tak časté pozdní diagnózy.
Nejčastějším symptomem je postupně progredující námahová dušnost a únavnost. Závažnost dušnosti významně koreluje s prognózou. Anginózní bolesti na hrudi jsou důsledkem ischemie pravé komory, synkopy a presynkopy jsou projevem nízkého srdečního výdeje. Mezi vzácnější projevy onemocnění patří chrapot způsobený útlakem levého vratného nervu dilatovaným kmenem plicnice, kašel a hemoptýza.
Ve fyzikálním nálezu souvisí manifestace jednotlivých nálezů s tíží plicní hypertenze. Často bývá akcentace 2. srdeční ozvy nad plicnicí, přítomnost 4. ozvy a cvalového rytmu. 3. ozva bývá přítomna v pokročilých stadiích onemocnění. Může být slyšitelný šelest trikuspidální a pulmonální regurgitace. U většiny nemocných je zvýšená náplň krčních žil a hmatná systolická pulzace v prekordiu a v epigastriu při hypertrofii pravé komory. Známkou pokročilého onemocnění je přítomnost periferních otoků a cyanóza.
Cílem diagnostického snažení u PAH je průkaz nebo vyloučení plicní hypertenze a následně určení její etiologie a závažnosti. Přes veškerý zájem, který je problematice PAH v poslední době věnován, je správná diagnóza stále stanovována pozdě. Z údajů zmiňovaného francouzského národního registru plyne, že doba od manifestace prvních příznaků do stanovení správné diagnózy se pohybuje kolem 27 měsíců a většina nemocných je stále diagnostikována v klinickém stadiu NYHA III a IV [12]. Velká pozornost je proto věnována systematickému screeningu definovaných populací s vyšším rizikem vniku PAH [18].
Rutinní genetické poradenství není v současné době u PAH dostupné. V některých centrech je však možné v rámci výzkumných projektů nabídnout pacientům s familiárním výskytem PAH screening mutací v genu BMPR2, případně účast v programech cílených na výzkum molekulární patogeneze tohoto onemocnění.
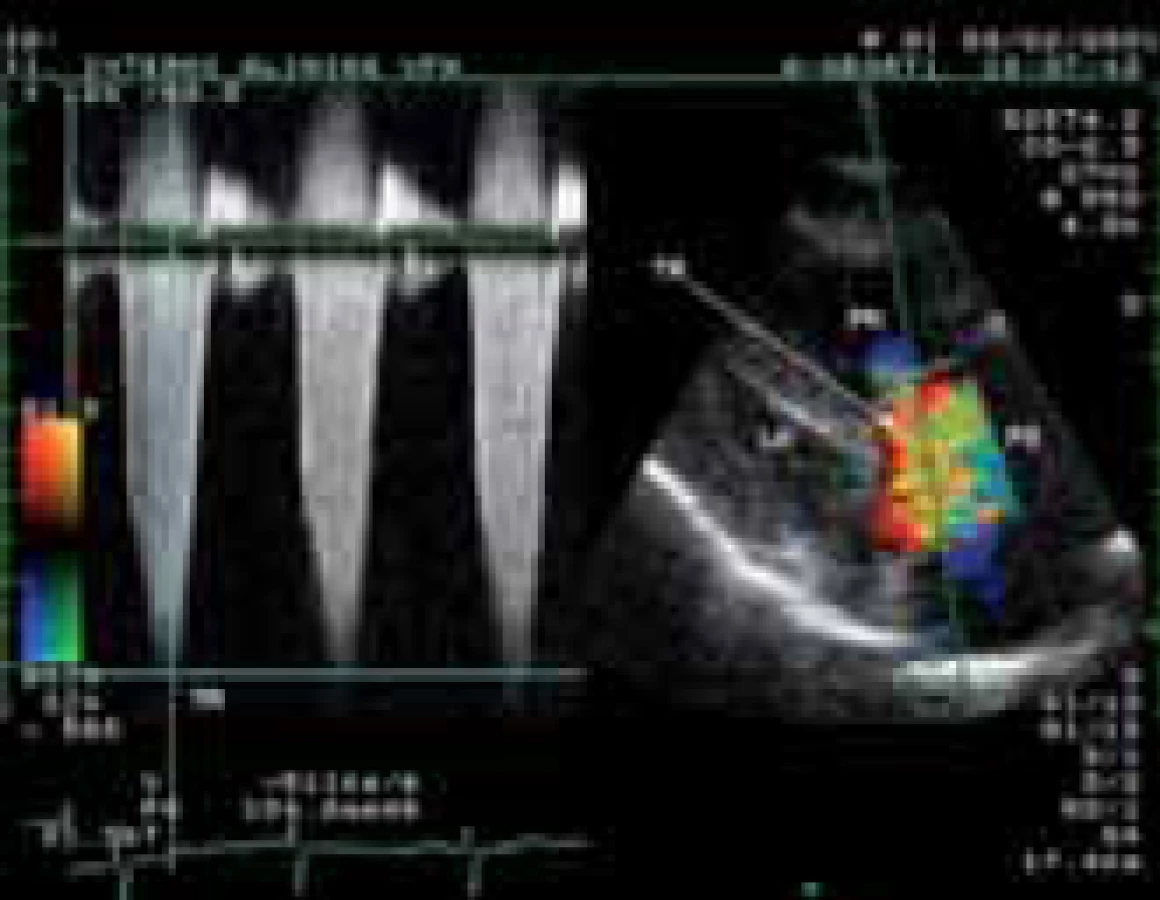
Klíčovým vyšetřením v detekci plicní hypertenze je echokardiografie [19]. Vedle základní diferenciální diagnostiky umožňuje zhodnotit velikost, tvar, a funkci pravé komory. Pro odhad stupně plicní hypertenze je nezbytné dopplerovské echokardiografické vyšetření. Systolický tlak v plicnici lze odhadnout z vrcholového gradientu trikuspidální regurgitace po přičtení tlaku v pravé síni (obr. 1). Za hranici normotenze v plicnici jsou u mladších osob považovány hodnoty 35 mm Hg, u starších osob 40 mm Hg. Přítomnost perikardiálního výpotku u PAH je považována za významný nepříznivý prognostický ukazatel. U symptomatických nemocných s hraničními tlaky v plicnici v klidu může být přítomna závažná plicní hypertenze při zátěži, kterou lze detegovat zátěžovým echokardiografickým vyšetřením. Specifickou skupinu tvoří osoby s jen lehkým stupněm plicní hypertenze nejasné etiologie. Jejich dlouhodobá prognóza je obtížně predikovatelná. Proto jsou doporučovány echokardiografické kontroly v intervalu 6–12 měsíců. Echokardiografický screeningplicní hypertenze je indikován u nemocných se systémovou sklerodermií každý rok, u ostatních systémových onemocnění pojiva a infekce HIV v případě manifestace symptomů signalizujících možnou plicní hypertenzi. U prvostupňových příbuzných nemocných s PAH má být ECHO vyšetření prováděno v intervalu 3–5 let, u nemocných s jaterním onemocněním vždy před plánovanou transplantací jater.
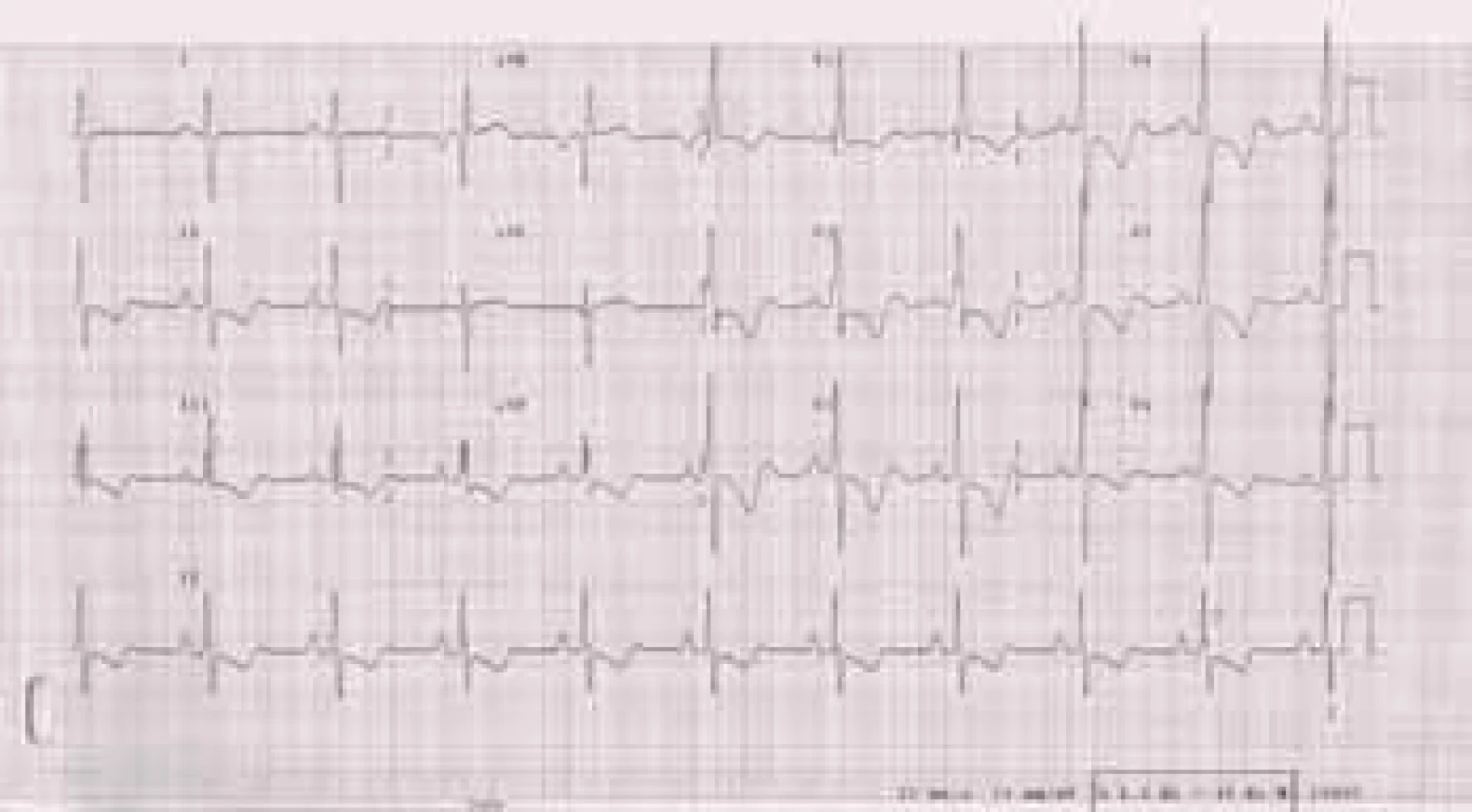
Přítomnost EKG–známek hypertrofie pravé komory je nález pro plicní hypertenzi specifický, ale málo senzitivní. Dále může být na EKG přítomna blokáda pravého raménka Tawarova, denivelace úseku ST, abnormity vlny T a P (obr. 2).
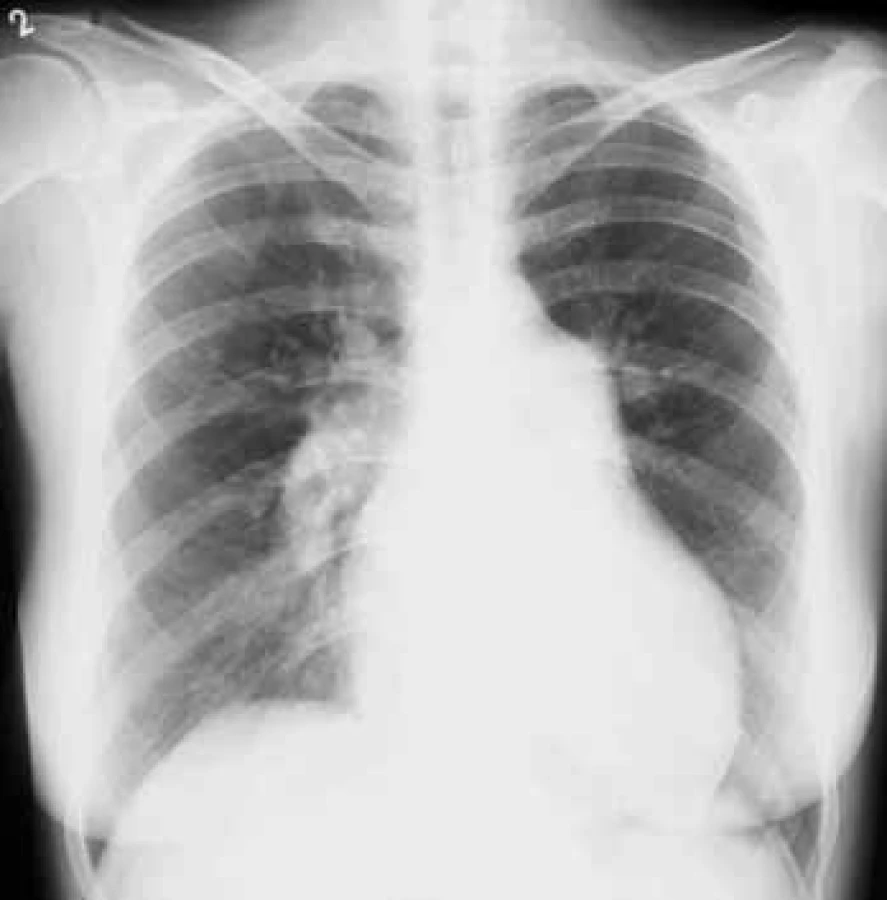
Na RTG snímku hrudníku bývá přítomna dilatace kmenů plicnice a náhlé zúžení cév na hranici lalokových a segmentárních tepen. Důsledkem spazmu periferních plicních cév je zvýšení transparence periferie plicních polí (obr. 3).
Ventilační a perfuzní scintigrafie plic a CT angiografie plic umožňuje odlišit chronickou tromboembolickou plicní hypertenzi.
Magnetická rezonance dovoluje posuzovat nejen morfologické, ale i funkční parametry plicního oběhu.
Funkční vyšetření plic je zásadní pro odlišení podílu onemocnění dýchacích cest nebo plicního intersticia na rozvoji plicní hypertenze. U nemocných s idiopatickou PAH může být redukována vitální kapacita zhruba na 80 % náležitých hodnot a difuzní kapacita pro CO asi na 60–80 % náležitých hodnot. Izolovaná redukce difuzní kapacity pro CO bez proporcionální redukce plicních objemů je nálezem značně charakteristickým pro PAH.
Polysomnografie by měla být provedena u pacientů s podezřením na syndrom obstrukční spánkové apnoe (obézní nemocní s excesivní denní spavostí a nočním chrápáním).
Základní laboratorní vyšetření při podezření na PAH zahrnují vyšetření autoprotilátek, sérologické vyšetření (HIV, hepatitidy) a vyšetření jaterních testů k vyloučení možných příčin PAH.
Nejjednodušším, levným a snadno opakovatelným zátěžovým testem je test 6minutovou chůzí, který je hlavním cílovým ukazatelem většiny dosud provedených randomizovaných klinických studií se specifickou léčbou PAH.
Plicní biopsie je indikována pouze při diagnostických rozpacích k vyloučení vaskulitidy nebo intersticiálního plicního procesu jako příčiny plicní hypertenze.
Definitivní diagnóza PAH nemůže být stanovena bez pravostranné srdeční katetrizace a invazivního hemodynamického vyšetření, které umožňuje přesně kvantifikovat tlaky v malém oběhu, určit plicní cévní odpor a vazodilatační odpověď při testu akutní plicní vazodilatace. K navození vazodilatace se používá intravenózní prostacyklin nebo adenozin a inhalační NO. Za kritérium pozitivity testu se považuje pokles středního tlaku v plicnici alespoň o 10 mm Hg vstupních hodnot, ale pod 40 mm Hg bez současného zhoršení srdečního výdeje.
Léčba
V 50. letech 20. století má svůj původ idea reaktivní plicní hypertenze, na základě níž byla u PAH zkoušena první vazodilatancia. Podání krátkodobě působících látek s vazodilatačním účinkem se nesetkalo s výraznějším úspěchem. Až v dalších desetiletích byly publikovány práce o účinku vazodilatačních blokátorů kalciových kanálů a antikoagulační léčby. Tyto postupy spolu s léčbou srdečního selhání se označují jako konvenční léčba. Podstatným pokrokem ve farmakoterapii PAH bylo však až zavedení tzv. specifické léčby (prostanoidy, antagonisté endotelinových receptorů a inhibitory fosfodiesterázy 5) s dokumentovanou účinností i v pokročilejších stadiích onemocnění (po ztrátě vazoreaktivity).
Režimová opatření
Fyzická zátěž je u nemocných s PAH vhodná podle individuální tolerance. Optimální je lehké aerobní cvičení (chůze). Není vhodný pobyt ve vyšších nadmořských výškách. Při cestě letadlem je nezbytná inhalace kyslíku. Doporučuje se očkování proti chřipce.
Těhotenství je u PAH kontraindikováno, neboť mateřská mortalita během gravidity a porodu dosahuje 30–50 %. Nutná je účinná antikoncepce. Z hlediska rizika tromboembolizmu je akceptovatelná hormonální antikoncepce při současné antikoagulační léčbě.
U nemocných s PAH je rovněž nutné pečlivě vážit indikaci chirurgických výkonů. Zejména operace u více symptomatických nemocných a anestezie trvající déle než 3 hodiny představují významné rizikové faktory.
Konvenční léčba
Diuretika zlepšují symptomy související s městnáním při srdečním selhání. Vzhledem ke zhoršenému vstřebávání perorálních léků při kongesci trávicího traktu je při dekompenzaci většinou nutná léčba intravenózní.
Cílem oxygenoterapie je dosáhnout saturace tepenné krve kyslíkem nad 90 %. Indikace léčby kyslíkem u nemocných s Eisenmengerovým syndromem je sporná a obecně se nedoporučuje.
Digitalis není v současné době běžnou součástí farmakoterapie PAH.
Chronická antikoagulační léčba warfarinem je u PAH indikována, zejména pokud je střední tlak v plicní tepně vyšší než 30–35 mm Hg [20]. Cílové INR se má pohybovat kolem 2, při pozitivitě antifosfolipidových protilátek kolem 3.
Léčba vysokými dávkami blokátorů kalciových kanálů (nifedipin, diltiazem, amlodipin) je indikována pouze v případě zachovalé vazoreaktivity. Pozitivní vazodilatační test pozorujeme u necelých 11 % nemocných s idiopatickou PAH a podstatně méně často u PAH asociované s ostatními stavy. Dlouhodobé odpovědi na vazodilatační léčbu blokátory kalciových kanálů, která je charakteristická mj. zlepšením symptomů do stadia NYHA I a II, dosáhneme pouze asi u poloviny akutních respondérů. Při selhání léčby blokátory kalciových kanálů je nezbytná specifická farmakoterapie.
Prostanoidy
Prostaglandin I2 (prostacyklin) je hlavní produkt metabolizmu kyseliny arachidonové v cévním endotelu. Je to účinný vazodilatátor v plicní i systémové cirkulaci, dále se vyznačuje vlastnostmi protidestičkovými, antiproliferativními a pozitivně inotropními. U nemocných s PAH je syntéza prostacyklinu v plicních cévách významně snížena [21].
Epoprostenol je syntetický analog prostacyklinu. Vzhledem ke krátkému biologickému poločasu je nutno jej podávat formou dlouhodobé kontinuální infuze do centrálního žilního katétru. Roztok epoprostenolu je termolabilní, proto je nezbytné jej během aplikace chladit. Epoprostenol byl poprvé použit pro léčbu PAH v 80. letech 20. století. V několika randomizovaných studiích u nemocných s idiopatickou PAH a PAH asociovanou se systémovými onemocněními pojiva prokazatelně zlepšil symptomy, hemodynamiku, funkční zdatnost a prognózu nemocných [22,23]. Obdobný účinek byl rovněž doložen v řadě observačních studií u nemocných s PAH asociovanou s vrozenými srdečními vadami, infekcí HIV a portální hypertenzí. Léčba se zahajuje dávkou 2 ng/kg/min. V důsledku tachyfylaxe je nezbytné postupné zvyšování dávky léčiva. Za optimální dávku při dlouhodobé léčbě se považuje 25–40 ng/kg/min, při které se u řady nemocných stabilizuje klinický stav a kontinuální zvyšování dávek pak není nutné. Vedle vlastních nežádoucích účinků epoprostenolu (bolesti čelistí, flush, bolesti hlavy, nauzea, hypotenze, tachykardie, bolesti na hrudi, trombocytopenie) jsou hlavním rizikem léčby lokální a systémové infekční komplikace v důsledku permanentního centrálního žilního katétru; dalším rizikem jsou poruchy infuzní pumpy. Intravenózní epoprostenol je lékem volby u nemocných v klinickém stadiu NYHA IV.
Treprostinil je analog prostacyklinu, stabilní za pokojové teploty. To umožňuje jeho podání v podobě subkutánní infuze. Treprostinil u nemocných s PAH zlepšuje hemodynamiku, funkční zdatnost a prognózu [24, 25]. Účinek je však významně závislý na dávce, za minimální účinnou dávku se považuje 10 ng/kg/min. Nejčastějším nežádoucím účinkem je lokální bolestivá reakce v místě podkožní infuze. Vyskytuje se až u 85 % léčených. V případě intolerance lokálního podání lze treprostinil rovněž podávat alternativně intravenózně. Ve srovnání s epoprostenolem odpadá nutnost chlazení infuzní soustavy a zejména riziko plynoucí z náhlého přerušení infuze. Treprostinil je vzhledem ke své stabilitě vhodný také pro inhalační podání, které se v současnosti studuje. K navození účinku postačují 4 inhalační aplikace denně. Další možností aplikace treprostinilu je podání per os.
Iloprost je stabilní analog prostacyklinu s poločasem 20–30 minut schválený pro inhalační léčbu idiopatické PAH. Pro navození dostatečného efektu na plicní cirkulaci je zapotřebí minimálně 6–12 inhalací denně. Několikaměsíční léčba iloprostem vede ke zlepšení funkční zdatnosti, hemodynamiky a funkční třídy podle NYHA [26]. Dlouhodobý efekt monoterapie zůstává sporný [27]. Iloprost se spíše jeví jako vhodný lék do kombinačních schémat.
Beraprost je perorální stabilní analog prostacyklinu s poločasem 35–40 minut. Účinek spočívající ve zlepšení vzdálenosti při testu šestiminutovou chůzí je doložen po 3 a 6 měsících léčby, při déletrvající monoterapii však již není přesvědčivý [28]. Beraprost je pro léčbu PAH zaregistrován v Japonsku a Korey.
Antagonisté endotelinových receptorů
Aktivovaný endotelinový systém u PAH lze ovlivnit duální nebo selektivní blokádou endotelinových receptorů [29]. Otázka superiority jedné ze strategií je nadále předmětem diskusí.
Bosentan je duální antagonista receptorů pro endotelin s výraznější afinitou k receptoru ETA. Bosentan je první schválený perorální lék u PAH. Působí antiproliferačně a vazodilatačně. Jeho efekt na signifikantní zlepšení funkční zdatnosti hemodynamických parametrů a přežívání nemocných byl prokázán v řadě klinických studií, zejména u idiopatické PAH, ale také u PAH asociované se systémovými onemocněními pojiva, vrozenými srdečními vadami a infekcí HIV [30–32]. V poslední době byly prezentovány výsledky první randomizované klinické studie u nemocných s PAH ve funkčním stadiu NYHA II [33]. Dosud totiž scházela jednoznačná evidence o efektu specifické léčby u méně pokročilého onemocnění. V léčené skupině došlo k signifikantnímu poklesu plicní cévní rezistence, byl patrný významný trend ke zlepšení vzdálenosti při testu 6minutovou chůzí a především ve srovnání s placebovou skupinou byla zásadní redukce klinických příhod. Toto zjištění vede k závěru, že PAH již v klinickém stadiu NYHA II bez léčby zřetelně progreduje v horizontu několika měsíců a vyžaduje léčbu. Doporučené dávkování bosentanu je 62,5 mg 2krát denně po dobu prvních 4 týdnů, dále 125 mg 2krát denně. K hlavním nežádoucím účinkům bosentanu patří reverzibilní a na dávce závislá hepatopatie. Vyskytne se asi u 11 % léčených. Kontrola jaterních testů je nutná po 2 týdnech od zvýšení dávky a dále v měsíčních intervalech během léčby. K normalizaci testů vede redukce dávky nebo přerušení léčby. Bosentan může zejména v prvních týdnech léčby vést ke zvýšení retence tekutin, což vyžaduje posílení diuretické léčby. Dále může navozovat anémii, je teratogenní a interaguje s metabolizmem warfarinu. Bosentan je indikován jako lék první volby u pacientů s PAH ve stadiu NYHA III, kteří nemají zachovalou vazoreaktivitu nebo u nichž došlo k selhání léčby blokátory kalciových kanálů.
Sitaxsentan je prakticky selektivní antagonista endotelinového receptoru ETA. Jeho účinek u PAH byl studován v dávkách 50, 100 a 300 mg 1krát denně per os [34,35]. Dávka 300 mg je z hlediska hepatotoxicity neakceptovatelná. Dávka 100 mg 1krát denně vede ke zlepšení hemodynamických parametrů a k signifikantnímu zlepšení vzdálenosti při testu 6minutovou chůzí. Spektrum nežádoucích účinků je obdobné jako u bosentanu. K elevaci transamináz nad 3násobek normy dochází u 3–5 % pacientů léčených 100 mg sitaxsentanu. Sitaxsentan je v současné době zaregistrován v EU pro léčbu PAH ve funkční třídě NYHA III.
Ambrisentan je vysoce selektivní antagonista endotelinového receptoru ETA. V dávkách 1 mg, 2,5 mg, 5 mg a 10 mg 1krát denně zlepšuje vzdálenost při testu 6minutovou chůzí, hemodynamiku a prodlužuje dobu do klinického zhoršení [36]. Elevace transamináz nad 3násobek normy se vyskytuje zhruba u 3 % léčených nemocných. Ambrisentan neovlivňuje významněji metabolizmus warfarinu. Ambrisentan byl v USA nedávno zaregistrován pro léčbu PAH.
Inhibitory fosfodiesterázy 5
Sildenafil je účinný a prakticky selektivní inhibitor PDE–5 (fosfodiesterázy 5) specifické k cGMP (cyklický guanozinmonofosfát). Inhibice degradace cGMP jako druhého posla v regulační kaskádě NO zesiluje relaxaci hladkých svalových vláken a vazodilataci navozenou cGMP [37].
Sildenafil byl s úspěchem zkoušen v několika malých nerandomizovaných studiích u různých typů plicní hypertenze [38,39]. U pacientů s PAH v klinickém stadiu NYHA II a III byl testován v rozsáhlé multicentrické randomizované a placebem kontrolované studii [40]. V léčené skupině při dávkování 3krát denně 20, 40 nebo 80 mg zlepšil po 12 týdnech funkční zdatnost a hemodynamické parametry. Zlepšení funkční zdatnosti přetrvává i po 12 měsících. Ve většině případů však je nutné zvýšit dávkování na 3krát denně 80 mg. Z dlouhodobého sledování je také zřetelný příznivý vliv sildenafilu na přežívání nemocných. K hlavním nežádoucím účinkům patří bolesti hlavy, flush, dyspepsie a epistaxe. Sildenafil je zaregistrován pro léčbu PAH v dávce 20 mg 3krát denně.
V léčbě PAH se rovněž zkouší tadalafil v monoterapii nebo v kombinaci s bosentanem.
Kombinační farmakoterapie
Je obecně známou skutečností, že monoterapie PAH nevede u řady nemocných k takové dlouhodobé kontrole onemocnění, za kterou považujeme dosažení funkční třídy NYHA I–II, vzdálenosti při testu 6minutovou chůzí nad 380 m a kompenzace pravostranného srdečního selhání. Kombinační léčba umožňuje postihnout více patogenetických mechanizmů, které se podílejí na rozvoji onemocnění. Je proto logickou strategií v případě nedostatečného účinku monoterapie. Otevřená však zůstává otázka volby kombinačních schémat a jejich načasování. Dnes máme k dispozici údaje z řady nekontrolovaných studií a ze 3 randomizovaných studií s kombinační farmakoterapií u PAH [41].
První randomizovaná studie hodnotila efekt kombinace bosentanu a epoprostenolu. U nemocných, kteří dostávali kombinaci, byl trend k výraznějšímu zlepšení hemodynamiky patrnější než v případě monoterapie epoprostenolem. Výsledek však nebyl statisticky signifikantní.
Další randomizovaná studie prokázala zlepšení funkční zdatnosti při kombinační léčbě bosentanem a inhalačním iloprostem ve srovnání se samotným bosentanem.
Nedávno byly prezentovány výsledky rozsáhlé multicentrické studie se sildenafilem u pacientů léčených nejméně 3 měsíce intravenózním epoprostenolem. U nemocných léčených oběma preparáty se ve srovnání s monoterapií bosentanem významně zlepšila funkční zdatnost, hemodynamika, doba do klinického zhoršení a rovněž prognóza.
Nefarmakologické metody léčby PAH
K nefarmakologickým postupům v léčbě PAH patří balónková atriální septostomie a transplantace plic [42].
Atriální septostomie je intervenční metoda spočívající ve vytvoření umělé komunikace na úrovni síní se vznikem pravolevého zkratu. Cílem intervence je zvýšení srdečního výdeje za cenu systémové desaturace. V zemích s dostupnou farmakoterapií je atriální septostomie indikována jako paliativní metoda, případně jako most k transplantaci u nemocných s refrakterním pravostranným srdečním selháním a synkopami. V zemích, kde specifická léčba PAH není k dispozici, je atriální septostomie často jedinou možnou terapeutickou intervencí.
Transplantace plic představuje účinnou léčbu u nemocných v terminálním stadiu PAH po vyčerpání všech ostatních dostupných léčebných možností. Většina center indikuje transplantaci obou plic. Transplantace srdce a plic je indikována téměř výlučně u komplexních vrozených srdečních vad. Jednoroční přežití po transplantaci plic pro PAH se pohybuje mezi 66–75 %.
Strategie léčby
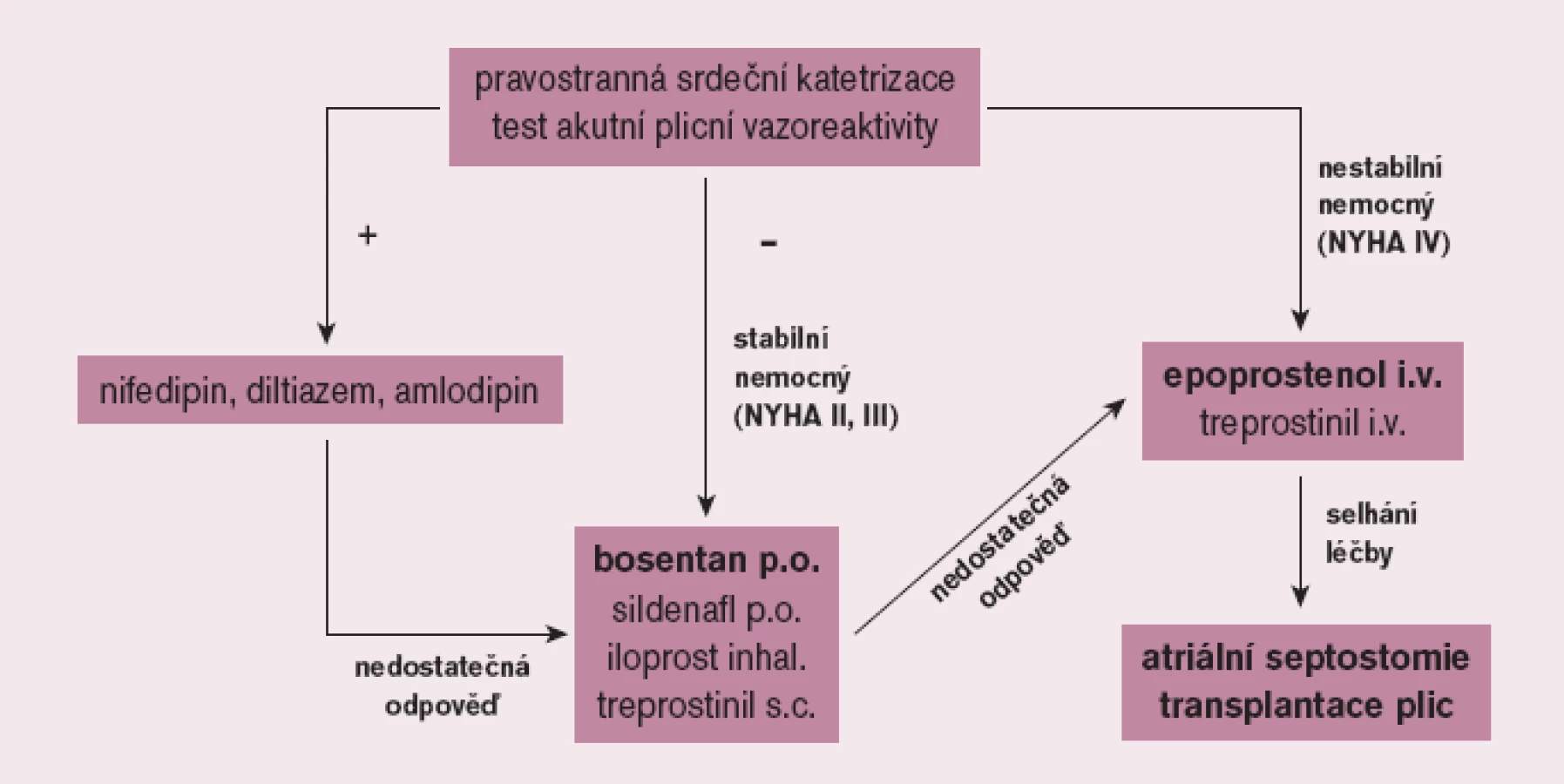
Cílem léčby PAH je ovlivnit nejen symptomy, ale především kvalitu života a prognózu nemocných. Před zahájením léčby PAH je nezbytné pečlivé vyšetření nemocného ve specializovaném centru včetně invazivního hemodynamického vyšetření s testováním akutní vazorekativity se záměrem identifikovat nepočetnou skupinu nemocných, kteří mohou profitovat z dlouhodobé léčby vysokými dávkami blokátorů kalciových kanálů. Většina nemocných však nesplňuje kritéria vazoreaktivity; tito pacienti jsou pak vedle antikoagulační léčby kandidáty léčby specifické. Její indikace vyžaduje striktně individuální přístup. U symptomatických nemocných má být zahájena bezprostředně po stanovení diagnózy. U pacientů ve funkčním stadiu NYHA (II) a III je metodou volby perorální léčba, kterou ve většině případů zahajujeme bosentanem, alternativně sildenafilem. Ve funkčním stadiu NYHA IV je základním lékem intravenózní prostacyklin a při selhání farmakoterapie pak atriální septostomie a transplantace plic (obr. 4).
Závěr
Zkoumání problematiky PAH v posledních letech značně pokročilo. Studium epidemiologie učinilo z PAH onemocnění hodné zřetele. Výrazný posun v poznání patofyziologie ovlivnil rozvoj terapie. Tu tvoří v současné době zejména prostanoidy, antagonisté endotelinových receptorů a inhibitory fosfodiesterázy 5. Nejedná se sice o léčbu kauzální, přesto však významně ovlivňující nejen symptomy, ale také prognózu nemocných.
Stejná pozornost jako terapii by však měla být věnována správné a časné diagnostice. Vzhledem ke komplikovanosti definitivní diagnostiky PAH je zapotřebí ji soustředit do specializovaných center s multidisciplinárním zázemím a bohatými zkušenostmi. Alarmující skutečností je však zjištění, že většina nemocných je do těchto center referována ve značně pokročilých stadiích onemocnění. Zásadní dluh vůči nemocným s PAH tak v dnešní době nespočívá ve stále nedostatečném poznání patofyziologie onemocnění nebo nedostupnosti kauzální terapie, ale především v jeho nedokonalé a pozdní detekci.
Ve snaze zlepšit detekci PAH v České republice vznikla z iniciativy Centra pro plicní hypertenzi ve Všeobecné fakultní nemocnice v Praze síť více než 30 spolupracujících renomovaných echokardiografických laboratoří, které se zaměřují na detekci plicní hypertenze a umožní včas a kvalifikovaně rozhodnout, zda je u nemocného s podezřením na plicní hypertenzi nezbytné podrobnější vyšetření ve specializovaném centru.
Adresa pro korespondenci:
MUDr. Pavel Jansa
MUDr. David Ambrož
MUDr. Tomáš Paleček
MUDr. Pavel Poláček
MUDr. Jana Marešová,
MUDr. Ludmila Jelínková,
prof. MUDr. Michael Aschermann, DrSc., FESC
prof. MUDr. Aleš Linhart, DrSc.
Centrum pro plicní arteriální hypertenzi, II. interní klinika 1. LF UK a VFN, Praha
Email: jansapavel@yahoo.com
Sources
1.Riedel M. Klasifikace a nomenklatura plicní hypertenze. Kapitoly z kardiologie 2002; 4 : 46–49.
2.Simonneau G, Galie N, Rubin LJ et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 2004; 43 : 5–12.
3.Farber HW, Loscalzo J. Pulmonary arterial hypertension – mechanism of disease. N Engl J Med 2004; 351 : 1655–1665.
4.Dresdale DT, Michtom RJ, Schultz M. Recent studies in primary pulmonary hypertension, including pharmacodynamic observations on pulmonary vascular resistance. Bull N Y Acad Med 1954; 30 : 195–207.
5.Nichols WC, Koller DL, Slovis B et al. Localization of the gene for familial primary pulmonary hypertension to chromosome 2q31–32. Nat Genet 1997; 15 : 277–280.
6.Morse JH, Jones AC, Barst RJ et al. Mapping of familial primary pulmonary hypertension locus (PPH1) to chromosome 2q31–q32. Circulation 1997; 95 : 2603–2606.
7.Deng Z, Morse JH, Slager SL et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor–II gene. Am J Hum Genet 2000; 67 : 737–744.
8.Trembath RC, Thomson JR, Machado RD et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2001; 345 : 325–334.
9.Yuan JJ, Aldinger AM, Juhaszova M et al. Dysfunctional voltage–gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation 1998; 98 : 1400–1406.
10.Simonneau G, Fartoukh M, Sitbon O et al. Primary pulmonary hypertension associated with the use of fenfluramines derivatives. Chest 1998; 114(Suppl 3): 195–199.
11.HIV–1 Nef is associated with complex pulmonary vascular lesions in SHIV–nef–infected macaques. Am J Respir Crit Care Med 2006; 174 : 437–445.
12.Humbert M, Sitbon O, Chaouat A et al. Pulmonary arterial hypertension in France. Results from a national registry. Am J Respir Crit Care Med 2006; 173 : 1023–1030.
13.Mukerjee D, St George D, Coleiro B et al. Prevalence and outcome in systemic sclerosis associated pulmonary arterial hypertension: application of a registry approach. Ann Rheum Dis 2003; 62 : 1088–1093.
14.Vongpatanasin W, Brickner ME, Hillis LD et al. The Eisenmenger syndrome in adults. Ann Intern Med 1998; 128 : 745–755.
15.D'Alonzo GE, Barst RJ, Ayres SM et al. Survival in patients with primary pulmonary hypertension: results from a national prospective registry. Ann Intern Med 1991; 115 : 343–349.
16.Sitbon O, Humbert M, Jais X et al. Long–term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 2005; 111 : 3105–3111.
17.Rubin LJ. Primary pulmonary hypertension. Chest 1993; 104 : 236–250.
18.McGoon M, Gutterman D, Steen V et al. Screening, early detection, and diagnosis of pulmonary arterial hypertension. Chest 2004; 126(Suppl): 14–34.
19.Daniels LB, Krummen DE, Blanchard DG. Echocardiography in pulmonary vascular disease. Cardiol Clin 2004; 22 : 383–399.
20.Fuster V, Steele PM, Edwards WD et al. Primary pulmonary hypertension and the importance of thrombosis. Circulation 1984; 70 : 580–587.
21.Tuder RM, Cool CD, Geraci MW et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med 1999; 159 : 1925–1932.
22.Badesch DB, McLaughlin VV, Delcroix M et al. Prostanoid therapy for pulmonary arterial hypertension. J Am Coll Cardiol 2004 : 43(Suppl): 56–61.
23.Barst RJ, Rubin LJ, Long WA et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N Engl J Med 1996; 334 : 296–301.
24.Simonneau G, Barst RJ, Galie N et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double–blind, randomized, placebo – controlled trial. Am J Respir Crit Care Med 2002; 165 : 800–804.
25.Barst RJ, Galie N, Naeije R et al. Long–term outcome in pulmonary arterial hypertension patients treated with subcutaneous treprostinil. Eur Respir J 2006; 28 : 1195–1203.
26.Olschewski H, Simonneau G, Galie N et al. Inhaled iloprost in severe pulmonary hypertension. N Engl J Med 2002; 347 : 322–329.
27.Opitz CF, Wensel R, Winkler J et al. Clinical efficacy and survival with first–line inhaled iloprost therapy in patients with idiopathic pulmonary arterial hypertension. Eur Heart J 2005; 26 : 1895–1902.
28.Barst RJ, McGoon MD, McLaughlin VV et al. Beraprost therapy for pulmonary arterial hypertension. J Am Coll Cardiol 2003; 41 : 2119–2125.
29.Nishida M, Eshiro K, Okada Y et al. Roles of endothelin ETA and ETB receptors in the pathogenesis of monocrotaline–induced pulmonary hypertension. J Cardiovasc Pharmacol 2004; 44 : 187–191.
30.Channick R, Sunomeau G, Sitbon O et al. Effects of the dual endothelin–receptor antagonist bosentan in patients with pulmonary hypertension: a randomized placebo controlled study. Lancet 2001; 358 : 1119–1123.
31.Rubin LJ, Badesch DB, Barst RJ et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002; 346 : 896–903.
32.McLaughlin VV, Sitbon O, Badesch DB et al. Survival with first–line bosentan in patients with primary pulmonary hypertension. Eur Respir J 2005; 24 : 244–249.
33.Successful study with Tracleerr in patients with mildly symptomatic Pulmonary Arterial Hypertension. Actelion, Media release, 18. Dec. 2006. !!PROSÍM O KONTROLU, ZDA BYLO SPRÁVNĚ DOPLNĚNO!!
34.Barst RJ, Langleben D, Frost A et al for the STRIDE–1 study group. Sitaxsentan therapy for pulmonary arterial hypertension. Am J Respir Crit Care Med 2004; 169 : 441–447.
35.Barst RJ, Langleben D, Badesch D et al. Treatment of pulmonary arterial hypertension with the selective endothelin–A receptor antagonist sitaxsentan. J Am Coll Cardiol 2006; 47 : 2049–2056.
36.Galie N, Badesch D, Oudiz R et al. Ambrisentan therapy for pulmonary arterial hypertension. J Am Coll Cardiol 2005; 46 : 529–535.
37.Cohen AH, Hanson K, Morris K et al. Inhibition of cyclic 3'–5'–guanosin monophosphate–specific phosphodiesterase selectivity vasodilates the pulmonary circulation in chronically hypoxic rats. J Clin Invest 1996; 97 : 172–179.
38.Ghofrani HA, Schermuly RT, Rose F et al. Sildenafil for long–term treatment of nonoperable chronic thromboembolic pulmonary hypertension. Am J Respir Crit Care Med 2003; 167 : 1139–1141.
39.Perimenis P. Sildenafil for the treatment of altitude–induced hypoxaemia. Expert Opin Pharmacother 2005; 6 : 835–837.
40.Galie N, Ghofrani HA, Torbicki A et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 2005; 353 : 2148–2157.
41.Benza RL, Park MH, Keogh A et al. Management of pulmonary arterial hypertension with a focus on combination therapies. J Heart Lung Transplant 2007; 26 : 437–446.
42.Doyle RL, McCrory D, Channick RN et al. Surgical treatments/interventions for pulmonary arterial hypertension. Chest 2004; 126(Suppl): 63–71.
Labels
Paediatric cardiology Internal medicine Cardiac surgery CardiologyArticle was published in
Cardiology Review

2007 Issue 3
Most read in this issue
- Monitoring peroral anticoagulation therapy in outpatient practice
- Importance of implantable loop recorder in patients with unexplaided syncope
- CURRENT MEDICAL LITERATURE LTD, LONDON 1998, 438S.
- Pulmonary arterial hypertension