
Noví hráči na šachovnici protidestičkové léčby – co můžeme očekávat?
New players on the antiplatelet therapy chequerboard – what can we expect?
Antithrombotic therapy plays an important role in the prophylaxis of cardiovascular diseases. Over the recent years, we have been witnessing an arrival of new antiplatelet therapies. A number of new antibody‑based molecules inhibiting thrombocyte adhesion are at the stage of clinical evaluation. Similarly, we see the new platelet P2Y12 receptor antagonists (prasugrel, ticagrelor, cangrelor, elinogrel), thromboxane receptor antagonists (terutroban) or PAR 1‑type thrombin receptor antagonists (SCH 530348). Some from within this array of drugs, e. g. prasugrel, have now been released for clinical use, while others have just entered the clinical testing stage.
Key words:
antiplatelet agents – P2Y12 receptor antagonists – PAR 1‑type receptor antagonists – TXA2 receptor antagonists – prasugrel – ticagrelor – cangrelor – SCH 530348 – terutroban
:
J. Bultas; D. Karetová
:
Ústav farmakologie, 3. LF UK Praha, II. interní klinika kardiologie a angiologie, 1. LF UK Praha
:
Kardiol Rev Int Med 2010, 12(1): 38-45
Antitrombotická léčba hraje v profylaxi pandemie kardiovaskulárních chorob významnou úlohu. V posledních letech jsme svědky nástupu nových protidestičkových léků. Řada nových molekul na bázi protilátek inhibujících adhezi trombocytů je ve fázi klinického hodnocení. Podobně můžeme pozorovat nové inhibitory destičkových receptorů P2Y12 (prasugrel, ticagrelor, cangrelor, elinogrel), receptorů tromboxanových (terutroban) či receptorů trombinových typu PAR-1 (SCH 530348). Některé z této plejády léků, např. prasugrel, jsou již uvolněny ke klinickému užití, u jiných teprve klinické hodnocení probíhá.
Klíčová slova:
protidestičkové léky – inhibitory receptorů P2Y12 – inhibitory receptorů PAR-1 – inhibitory receptorů TXA2 – prasugrel – ticagrelor – cangrelor – SCH 530348 – terutroban
Úvod
V posledním desetiletí se v etiopatogenezi aterotrombotických příhod prosadila koncepce „tří nestabilních faktorů“. Prvním faktorem je nestabilní plát s obnaženými vysoce trombogenními subendoteliálními strukturami, zejména kolagenem, který iniciuje adhezi a aktivaci trombocytů. Druhým je nestabilní céva charakterizovaná dysfunkčním endotelem s potlačenou schopností kontrolovat aktivovanou hemostázu se zvýšenou vazospastickou pohotovostí a s potlačenou obranou vůči aterogenním podnětům. Třetím je pak nestabilní trombocyt aktivovaný např. vykouřenou cigaretou, interakcí s aktivovanými endoteliemi či cytokiny uvolněnými ze zánětlivého ložiska. Zásah na kterémkoli místě snižuje riziko kardiovaskulární příhody. První dva faktory optimálně ovlivníme zásahem do aterogeneze – úpravou dyslipidemie a hypertenze, abstinencí kouření, kontrolou diabetu apod., v prevenci aterotrombotických příhod hraje prim protidestičková léčba.
Zatímco stávající protidestičkové léky, jako jsou acetylsalicylová kyselina (ASA), tiklopidin či klopidogrel nebo inhibitory rec. IIb/ IIIa, přicházely s mnohaletými pauzami, v posledních letech jsme svědky nástupu celé řady nových protidestičkových léků. Důvodů je mnoho. Prvním je jistě vysoký výskyt aterotrombotických komplikací, jehož ovlivnění je velkou příležitostí pro farmaceutické firmy. Za druhé je to skutečnost, že vlastní aktivace trombocytu je pod kontrolou řady receptorů, z nichž dosud umíme ovlivnit jen některé. Vývoj nových inhibitorů by mohl potencovat protidestičkový účinek a zlepšit tak prognózu nemocných. Posledním důležitým faktorem je, že stávající protidestičkové léky mají významné omezení – část populace je k jejich působení zcela či částečně rezistentní. Nové léky tak mají šanci dosavadní léky buď nahradit, či v kombinaci s nimi zvýšit protidestičkový účinek.
Jaký je současný stav a proč nejsme spokojeni?
Naše současné spektrum protidestičkových léků (nepřesně označovaných jako antiagregancia) je reprezentováno několika skupinami léků lišícími se mechanizmem účinku. Nejdéle užíváme inhibitory aktivace trombocytů – a to buď aktivace indukované tromboxanem A2 (do této skupiny patří acetylsalicylová kyselina (ASA) a prakticky neužívané inhibitory tromboxanových receptorů), či aktivace zprostředkované adenozin‑difosfátem (ADP), tedy skupina blokátorů destičkových receptorů P2Y12 (thienopyridiny – tiklopidin a klopidogrel). Inhibitory vlastní agregace jsou reprezentovány blokátory receptorů IIb/ IIIa (abciximab, integrilin či tirofiban). Menší význam mají léky stabilizující efekt trombocytu zvýšením nabídky cyklického adenozin monofosfátu (cAMP), jako je dipyridamol, nebo cyklického guanozin monofosfátu (cGMP), jakými jsou donátory NO (molsidomin).
Protidestičkové léky určené k profylaxi aterotrombotických příhod, tj. thienopyridiny a ASA, významně – asi o 20 – 25 % – sníží výskyt trombotických příhod a mortality, mají nicméně řadu nedostatků. Nejvýznamnějším a často diskutovaným problémem dvou nejčastěji užívaných protidestičkových léků – kyseliny acetylsalicylové (ASA) či klopidogrelu – je rezistence čili nedostatečná odpověď na léčbu, s níž se setkáváme asi u čtvrtiny až třetiny nemocných. Bohužel resistence k léčbě bývá u řady nemocných na oba léky současně. Proto záměna jednoho léku za druhý či jejich kombinace problém zpravidla nevyřeší.
U akutních stavů, kdy potřebujeme zasáhnout rychle, je problémem několikahodinové prodlení do nástupu účinku u klopidogrelu. Inhibitory receptorů IIb/ IIIa, tedy v pravém smyslu slova antiagregancia, mají nevýhodu nutnosti parenterálního podání, vyššího krvácivého potenciálu a vyššího rizika protrombotického stavu po jejich vysazení při rebound fenoménu.
Jaké jsou perspektivy v jednotlivých skupinách protidestičkových léků?
Mechanizmem účinku protidestičkových léků je inhibovat primární, tj. destičkovou hemostázu ve fázi adheze, aktivace, degranulace či vlastní agregace trombocytu. Řada postupů je zatím v preklinických fázích, některé již pokročily do klinického hodnocení, jiné jsou dokonce uvolněny ke klinickému užití. Tento přehled je věnován inovacím směřujícím k brzkému rozšíření našich léčebných možností.
Inhibice destičkových funkcí ve fázi adheze
První fázi, tj. adhezi destiček k subendoteliálním strukturám, zejména ke kolagenu, umíme ovlivnit na několika místech – inhibicí destičkového receptoru Ib v místě vazby na von Willebrandův faktor (vWF), blokádou vazné domény von Willebrandova faktoru či snížením aktivity vWF obsazením vazných míst specifickými protilátkami (obr. 1A).
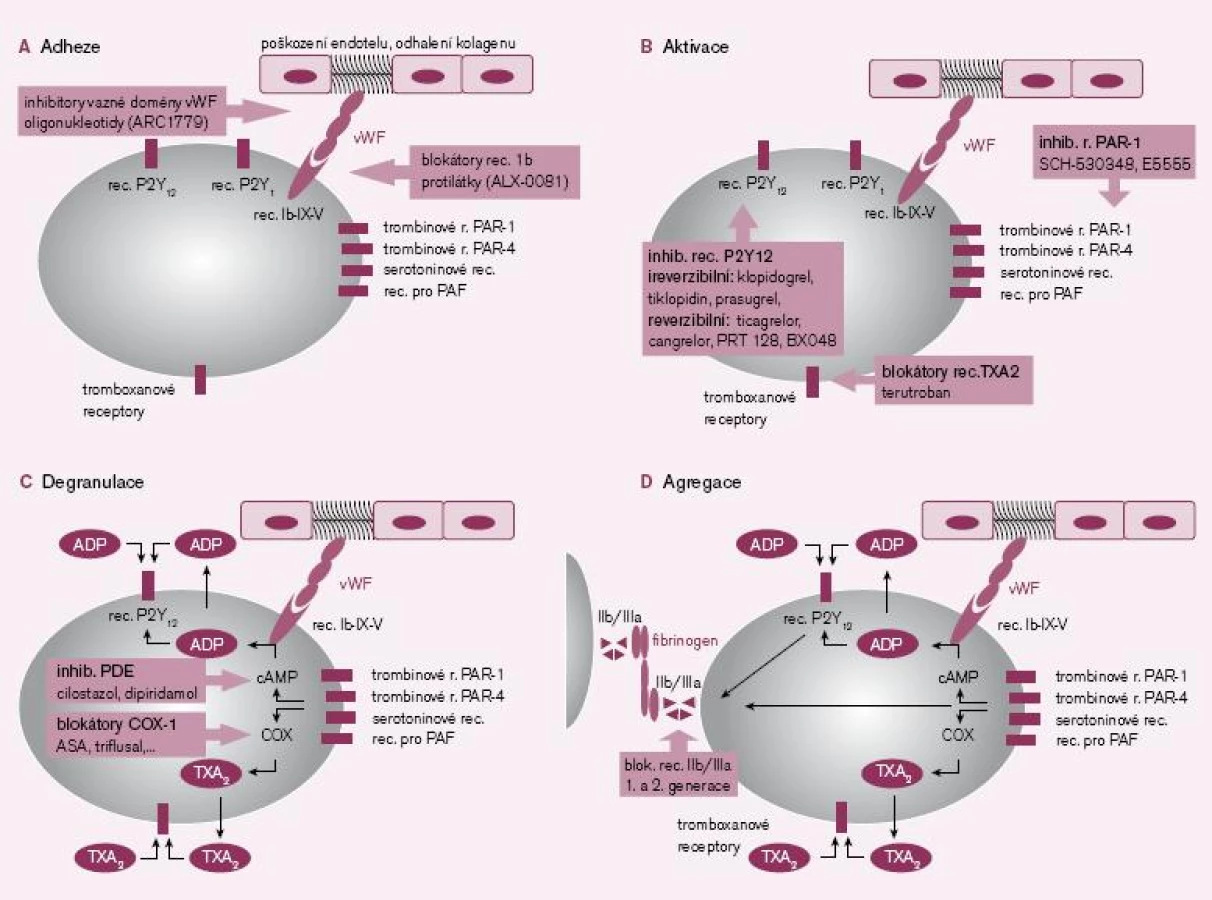
Prvním typem blokátorů adheze jsou inhibitory destičkových receptorů Ib (resp. komplexu glykoproteinových receptorů Ib-IX-V) typu specifických fragmentů protilátek. Nanoprotilátka – ALX-0081 – je velmi zajímavou inovací. Jedná se o ultramalý fragment přirozeného řetězce imunoglobulinu, který si ponechal bivalentní afinitu k vaznému místu destičkového receptoru GPIb s doménou A1 von Willebrandovu faktoru. Obsazením vazby nedojde ani k adhezi, ani k aktivaci trombocytu. Přípravek ALX-0081 je ve II. fázi hodnocení u nemocných s koronárními příhodami typu non‑STEMI léčenými PCI, účinnost a bezpečnost je porovnávána s abciximabem [1 – 2]. Druhým směrem testování je inhibice zvýšené aktivity vWF při trombofilních mutacích u nemocných s trombotickými mikroangiopatiemi, tj. v profylaxi atak trombotické trombocytopenické purpury.
Druhou možností přístupu je blokáda funkce von Willebrandova faktoru. Tento vazný protein na jedné straně váže trombocyt k obnaženým kolagenním vláknům, na straně druhé váže další aktivované trombocyty a přispívá k narůstání a ke stabilizaci destičkového trombu. Von Willebrandův faktor můžeme inhibovat monoklonálními protilátkami proti vWF (např. IgG protilátka AJW200) či inhibitory vazných receptorů (např. blokátor vazné domény vWF k destičkovému receptoru Ib – ARC1779).
Přípravek ARC1779, syntetický oligonukleotid je představitelem inhibitorů vazné domény vWF. Malé molekuly oligonukleotidů, zvané aptamery, mají unikátní schopnost inhibovat bioaktivní molekuly, v případě ARC1779 blokují vaznou doménu von Willebrandova faktoru. Inhibicí kontaktního místa je znemožněna vazba k destičkovému glykoproteinovému receptoru Ib a adheze trombocytu k obnaženému kolagenu. Díky stabilitě, nízké toxicitě a výraznému efektu aptameru na inhibici adheze trombocytů dospěl vývoj do pokročilejších fází klinického hodnocení. Prověřována je účinnost a bezpečnost v indikaci prevence komplikací při intervenci příhod typu non‑STEMI či v profylaxi recidivy TIA. Dále byl efekt tohoto inhibitoru vazné domény von Willebrandovu faktoru prověřen v pilotních studiích u nemocných s trombotickými mikroangiopatiemi, zejména typu trombotické trombocytopenické purpury [3 – 4]. Stejně jako u předchozího přípravku je tato indikace zcela logická, nemocných s tímto typem je ale málo a doufejme, že oba přípravky nezůstanou na úrovni „orphan drugs“. Důvodem pro mírnou skepsi je skutečnost, že vWF není jediným ligandem destičkového receptoru Ib a při blokádě vWF může tento bivalentní protein zastoupit ligand jiný, např. a-trombin či P-selectin.
Méně nadějné – za více než deset let vývoje totiž nepokročily do fáze klinického hodnocení – jsou humanizované monoklonální protilátky IgG4 proti von Willebrandovu faktoru, které jsou testované pod kódem AJW200.
Inhibice aktivace destiček
Aktivace trombocytu je kontrolována řadou receptorů. Je klíčovým krokem, který vede ke změně tvaru (umožňující optimální adhezi ke kolagenu či jiným povrchům), aktivaci enzymů (zejm. fosfolipázy C či A2 a adenyl-cyklázy) vedoucí k degranulaci a uvolnění řady protrombotických a vazokonstrikčních substancí, k aktivaci povrchových receptorů (konkrétně vazných glykoproteinových receptorů IIb/ IIIa) a ke změně orientace membránových fosfolipidů (dovolující vazbu s protrombinázovým komplexem a akcelerující sekundární hemostázu).
Nejdůležitějšími aktivačními receptory jsou receptory tromboxanové, adenozin‑difosfátové (typu P2Y12 nebo P2Y1), trombinové
(typu PAR-1 či PAR-4), serotoninové, a‑adrenergní či receptory pro faktor aktivující destičky (platelet activating factor – PAF). Receptor kolagenový typu Ib-IX-V je nejen aktivační, ale zprostředkovává též adhezi trombocytů k subendoteliálním vrstvám kolagenu. Tyto aktivační receptory jsou za fyziologického stavu v rovnováze se stabilizačními inhibičními receptory: adenozinovými (typ A2A), β‑adrenergními (typu β2), prostacyklinovými (pro PGI2) či prostaglandinovými (pro PGE2).
Specifické jsou adenozin‑difosfátové (ADP) receptory typu P2Y12, jejichž stimulace iniciuje dlouhodobou aktivaci trombocytu a expresi agregačních glykoproteinových receptorů IIb/ IIIa. Je proto logické, že nejvíce inovativních molekul působí ve fázi aktivace destičky, především aktivace indukované ADP (obr. 1B).
Blokátory receptorů ADP typu P2Y12
Blokáda aktivace trombocytu cestou receptorů ADP nabízí řadu velmi perspektivních novinek. Na povrchu trombocytu jsou dva typy ADP receptorů, které kontrolují funkci trombocytu. Prvními jsou rychle a krátce působící receptory P2Y1, které zatím ovlivnit neumíme. Druhými jsou ADP receptory typu P2Y12, které jsou aktivovány pomaleji, zato působí dlouhodobě – zprostředkují aktivaci a jejich zapojení je klíčovým krokem vlastní agregace. I když tyto receptory můžeme inhibovat například tiklopidinem či klopidogrelem, ve vývoji je řada inovativních molekul.
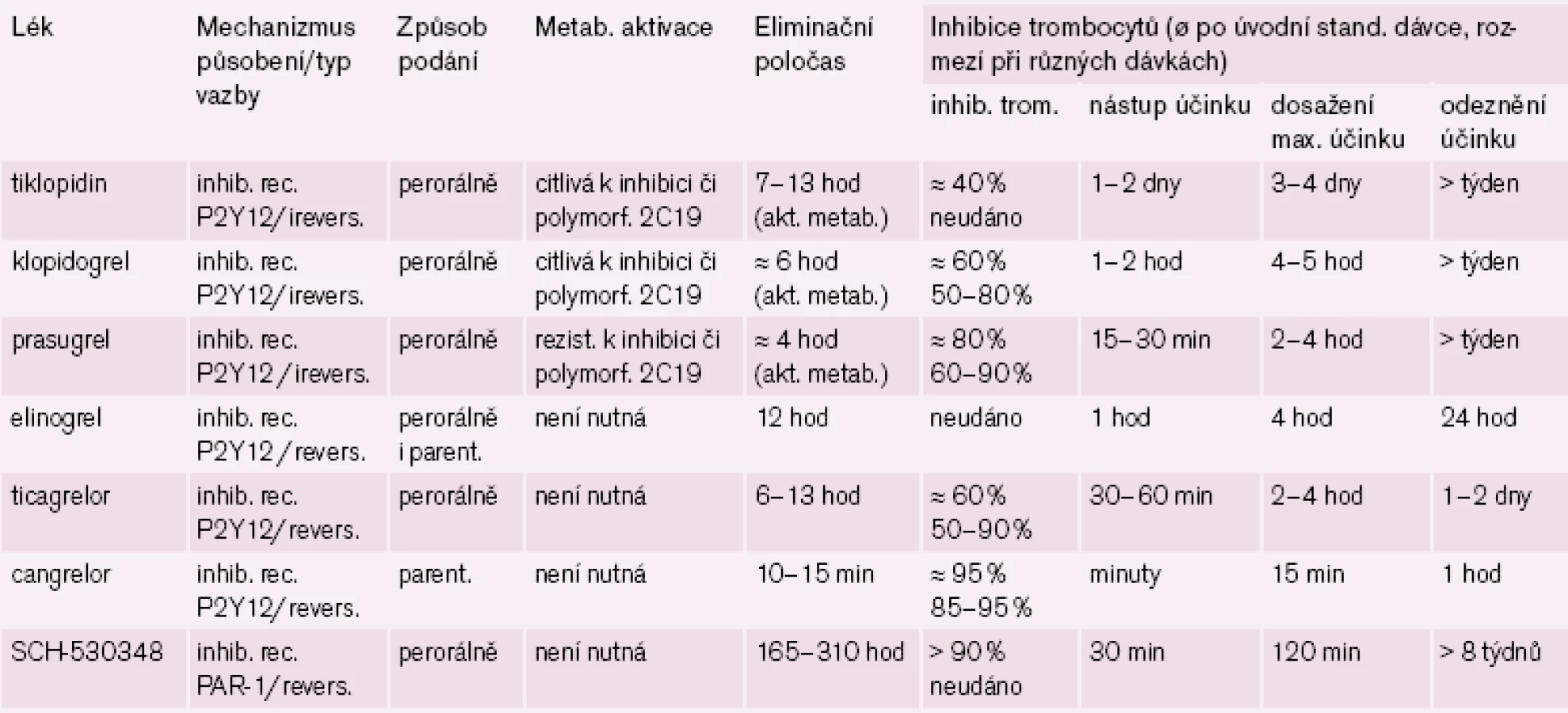
Inhibitory receptorů P2Y12 máme dvojího typu: nepřímo působící, tj. vyžadující bioaktivaci – thienopyridiny (klopidogrel, tiklopidin a prasugrel) a přímé – většinou typu analog ATP (ticagrelor, cangrelor či elinogrel). První skupina má výhodu ireverzibilní vazby na receptor s protidestičkovým účinkem po celou dobu cirkulace trombocytu, daní však je nutnost bioaktivace a pomalejší nástup účinku. Naopak předností přímých inhibitorů receptorů P2Y12 je rychlejší nástup účinku, krátká doba působení však vyžaduje přesnější spolupráci, při krvácení ale odeznění efektu může být přínosem.
Zlatým standardem protidestičkové léčby byla až doposud duální léčba klopidogrelem a kyselinou acetylsalicylovou. Nicméně, jak bylo řečeno, část nemocných nereaguje na klopidogrel optimálně či je dokonce k léčbě rezistentní. Příčin je více, hlavním důvodem však je nedostatečná konverze klopidogrelu (podávaného pro lepší resorpci jako proléčivo) na vlastní aktivní metabolit (obr. 2). Za fyziologické situace je asi 85 % proléčiva degradováno esterázami a pouze 15 % je aktivováno na účinný metabolit. V této skutečnosti je kámen úrazu, přeměna na aktivní metabolit neproběhne vždy tak, jak bychom očekávali – podkladem může být polymorfizmus konvertujících enzymů či inhibice konverze některými léky.
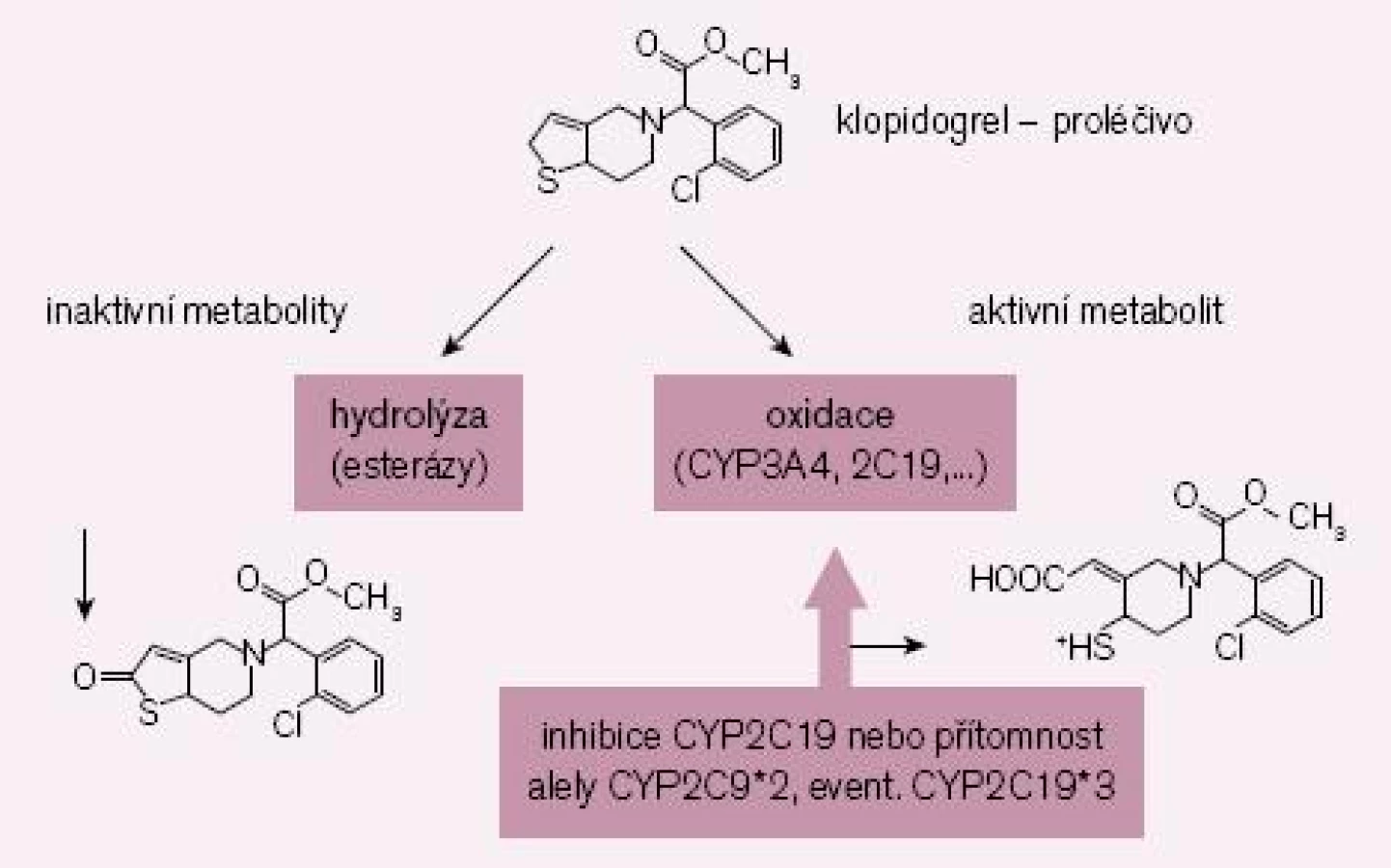
Enzymů nutných k bioaktivaci je více, klíčovým však je polymorfní oxidáza CYP 2C19. U nositelů alely CYP2C19*2 nebo CYP2C19*3 probíhá konverze velmi pomalu, významně větší část proléčiva je degradována na neúčinné metabolity a efekt klopidogrelu selhává. Jak ukázala farmakogenetická analýza studie TRITON-TIMI-38, u nemocných s alespoň jednou defektní alelou pro tento izoenzym, vyskytující se asi u 30 % populace, se objevilo o polovinu více závažných kardiovaskulárních příhod (KV úmrtí, infarkt či iktus); trombóz po implantaci stentu bylo dokonce třikrát více než u zbytku populace s funkčním izoenzymem [5]. Nález byl potvrzen recentní analýzou registru infarktů ve Francii [6].
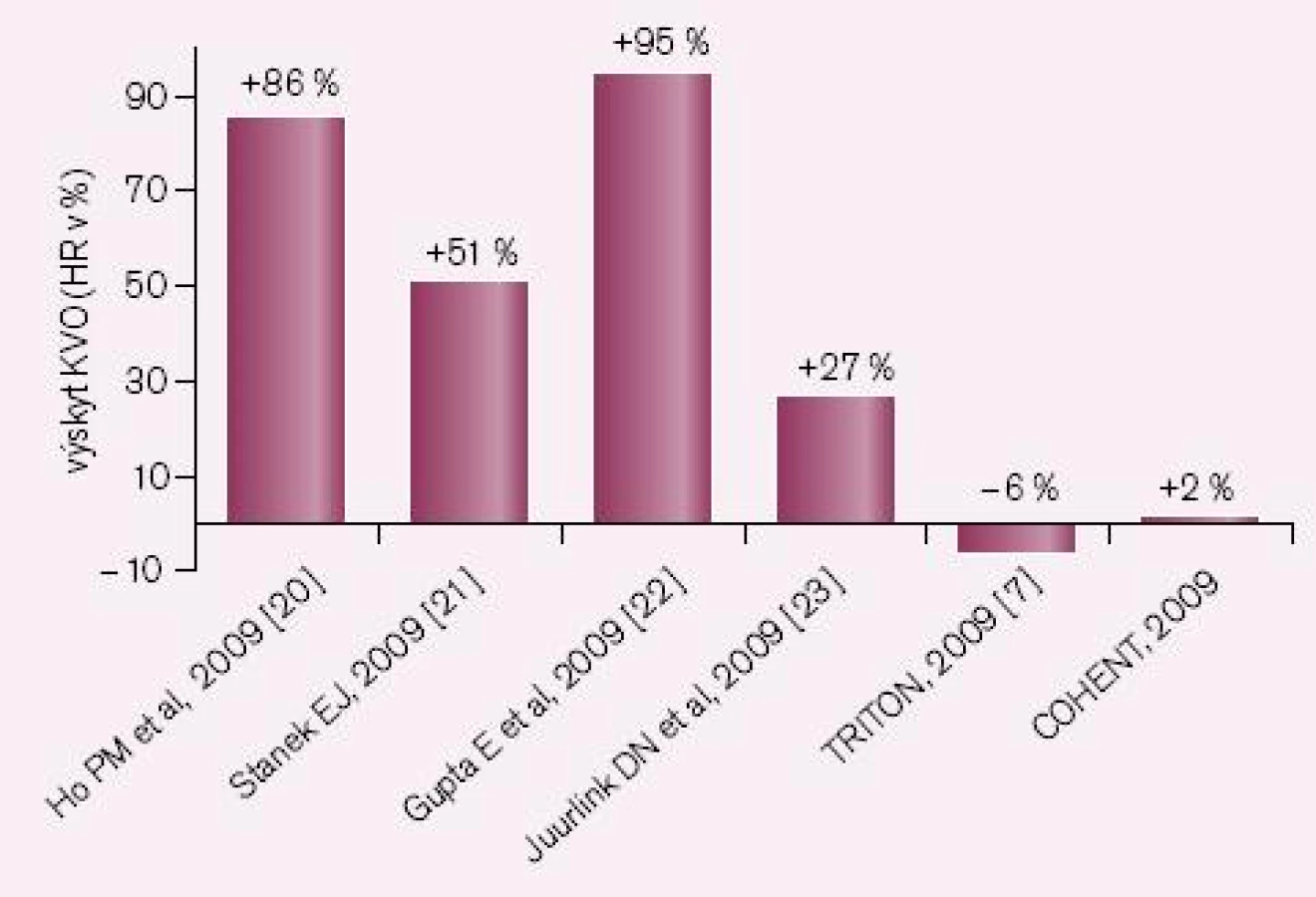
Druhou příčinou nedostatečné bioaktivace je inhibice CYP2C19 léky, zejména však některými inhibitory protonové pumpy (IPP). Nejsilnějším inhibitorem je omeprazol, nejslabšími pantoprazol a rabeprazol. Jak ukazuje přehled dostupných studií, zvýšení kardiovaskulárních příhod u nemocných po akutní koronární příhodě či po implantaci stentu je klinicky významné (obr. 3). První pětice studií jsou studie retrospektivní, rozdílné výsledky jsou diskutovány, nacházena je řada metodických výhrad. Nejvíce námitek je proti jediné prospektivní studii COHEN, jejich rozbor však je mimo rámec článku. Skutečností však zůstává, že důsledná analýza interakcí provedené FDA podpořila klinickou závažnost interakce omeprazolu s klopidogrelem. O významu nedostatečné inhibice destičkových funkcí při nižší nabídce účinného metabolitu svědčí i výsledek studie CURRENT-OASIS 7, kdy zdvojnásobení dávky klopidogrelu vedlo ke zvýšené léčebné odpovědi (obr. 4).
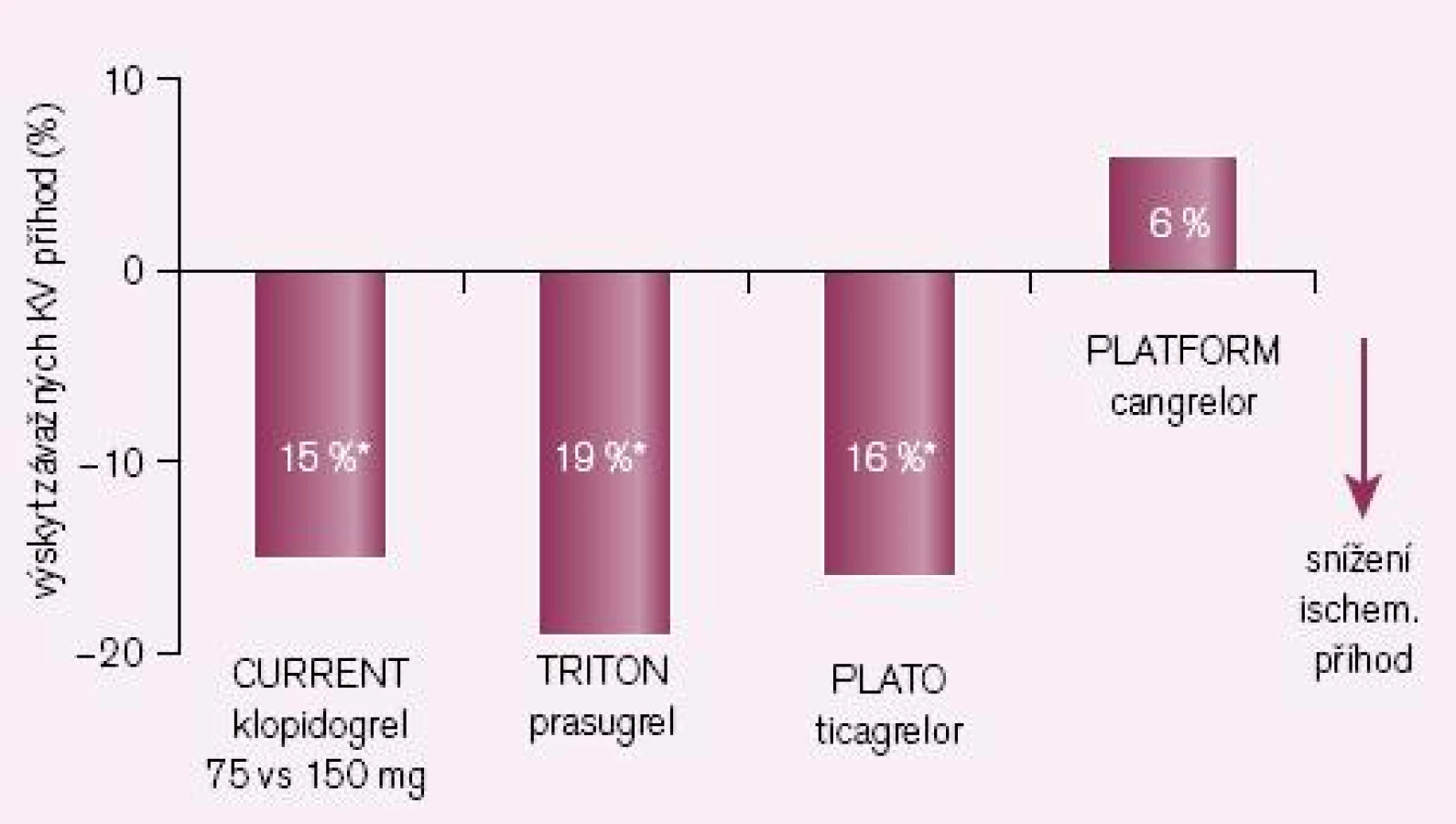
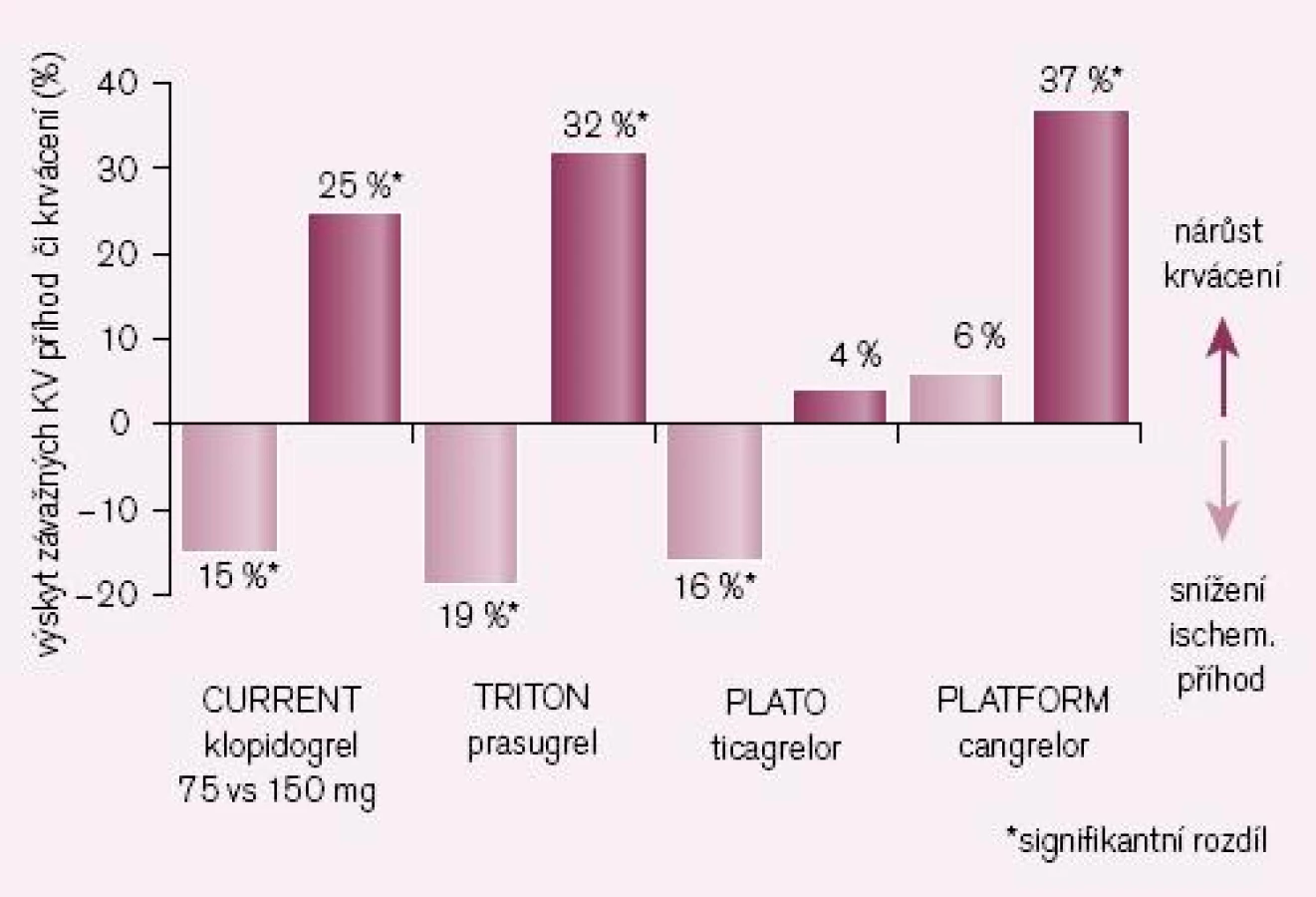
Logickým řešením tohoto handicapu je vývoj inhibitorů receptoru P2Y12, jejichž účinek není závislý na bioaktivaci. Cesta jak toho dosáhnout je dvojí: buď vývojem proléčiva rezistentního k inhibici konverze, nebo zavedením léků metabolickou konverzi nevyžadujících. První cestu reprezentuje prasugrel – ireverzibilní blokátor dlouhodobě působících receptorů P2Y12, který je představitelem dnes již třetí generace tienopyridinů. Jeho konverze na aktivní metabolit je daleko méně ovlivnitelná lékovými interakcemi či polymorfizmy izoenzymů CYP, efekt je proto spolehlivější a není omezen pouze na populaci s funkčním CYP 2C19. Při perorálním podání je nástup účinku patrný již za 30 min a efekt trvá nejméně 72 hod po ukončení léčby (tab. 1). Studie TRITON-TIMI 38 prokázala „superioritu“ prasugrelu proti klopidogrelu v léčbě akutních koronárních příhod (obr. 4) [7]. Kombinovaný ukazatel efektu (úmrtí, infarkt či iktus) poklesl proti klopidogrelu z 12,1 % na 9,9 % (tj. o absolutní 2,2 %, resp. relativních 19 %). Cenou za tento účinek byl vyšší výskyt významného krvácení o absolutních 0,6 %, resp. relativních 32 %. Tento vzestup odráží skutečnost, že prasugrel je účinný také asi u 30 % nemocných s rezistencí na klopidogrel. Zvýšení rizika krvácení není významné, neboť pravděpodobnost, že zabráníme závažné kardiovaskulární příhodě, je asi čtyřikrát větší ve srovnání s klopidogrelem nežli riziko, že pacienta poškodíme významným krvácením. Nejvýraznější účinek byl dokumentován u nemocných po implantaci stentu (trombózy v tepně v oblasti stentu klesly při léčbě prasugrelem ve srovnání s klopidogrelem na polovinu, resp. u stentu lékového poklesly o dvě třetiny).
Shrneme‑li pak v prasugrelu, který již byl uvolněn ke klinickému užití u nemocných po akutních koronárních příhodách ošetřených PCI, můžeme očekávat perspektivní lék se spolehlivějším účinkem a rychlejším nástupem efektu – hlavní místo bude jistě v léčbě akutních koronárních stavů a intervencí v tepenném systému.
Druhou větví vývoje se vydaly non‑thienopyridinové inhibitory ADP receptorů P2Y12 ticagrelor, canrelor či elinogrel. Hlavním rozdílem od první skupiny je reverzibilita inhibice a léky nemají charakter proléčiva, a není tak potřeba konverze na aktivní metabolit. Díky tomu, že nedochází k inhibici konverze, je protidestičkový efekt ve srovnání s klopidogrelem spolehlivější při srovnatelném, či dokonce nižším riziku krvácení (obr. 5).
Nejdále v klinickém hodnocení a současně s nejslibnějšími výsledky je ticagrelor. Nástup účinku po perorálním podání je možno pozorovat do hodiny, maximální účinek za 2 – 4 hod, vzhledem k reverzibilitě vazby na receptor je délka efektu kratší, středně dlouhý eliminační poločas (t1/ 2 je 6 – 13 hod) vede k podávání dvakrát denně (tab. 1).
V mega-studii PLATO byl prověřován efekt u akutních koronárních příhod v obdobném schématu jako u studie TRITON-TIMI 38, tedy ve srovnání s léčbou klopidogrelem (při bazální léčbě ASA). Kombinovaný ukazatel KV mortality, infarkt myokardu či iktus poklesly významně o relativních 16 % či o absolutních 1,9 % (obr. 4 – 5) [8]. Výskyt krvácení byl přitom v obou větvích srovnatelný. Nevyjasněnou příčinu má zvýšené pozorování asymptomatických AV blokád a dušnosti, nelze vyloučit jen náhodnou koincidenci.
Příkladem dalšího reverzibilního non‑thienopyridinového inhibitoru ADP receptoru P2Y12 je cangrelor, který – obdobně jako ticagrelor – nevyžaduje bioaktivaci. Jeho předností je velmi rychlý nástup účinku (v řádu minut), účinek však je krátký, odezní během desítek minut (tab. 1). Vzhledem ke krátkému efektu a nutnosti kontinuální parenterální aplikace byl testován u akutních koronárních příhod (studie CHAMPION PLATFORM [9]) jen v akutním, periprocedurálním období. V jedné větvi byl u nemocných s akutní koronární příhodou indikovaných k provedení PCI aplikován cangrelor před výkonem (bolus + infuze) s následnou léčbou klopidogrelem, druhá větev byla léčena jen klopidogrelem. Při sledování celkové mortality, infarktu myokardu či nutnosti revaskularizace se v prvních 48 hod nepotvrdil předpoklad příznivějšího působení cangreloru na pokles mortality a morbidity, krvácivých příhod přitom významně přibylo (obr. 4 – 5) [10]. Naděje vkládané do cangreloru – vyplývající z jeho velmi rychlého nástupu účinku – se tak nepotvrdily.
Posledním přímým inhibitorem receptorů P2Y12, který je v pokročilejších fázích klinického hodnocení, je elinogrel. I zde probíhají či se připravují studie ověřující efekt v různých indikacích (GRAVITAS, 3T/ 2R, DANTE, TRIGGER-PCI a ARTIC). Výsledky však ještě nejsou k dispozici.
Blokátory trombinových receptorů PAR-1
Aktivace trombocytu vytvořeným trombinem propojuje sekundární hemostázu s primární. V současné době jsou dva přípravky inhibující trombinový receptor PAR-1 (protease‑activated receptor 1) ve fázi klinického hodnocení. Vzhledem k tomu, že trombinové receptory nemají vliv na adhezi trombocytů, je pravděpodobné, že místo inhibitorů receptoru PAR-I zůstane v kombinaci s blokátory ADP či tromboxanové cesty aktivace.
Ve třetí fázi klinického hodnocení je přípravek uváděný stále pod kódovým číslem SCH - 530348. Tento selektivní reverzibilní blokátor destičkového receptoru PAR-1 je prověřován v indikaci profylaxe cévních příhod u aterotrombotických onemocnění. V pokročilejších fázích hodnocení je testován v kombinaci s ostatními protidestičkovými léky, zejména s ASA či klopidogrelem. Tento antagonista destičkového trombinového receptoru má rychlý nástup účinku (do hodiny) a dlouhou dobu působení (t1/ 2 je 160 až 310 hod) umožňující podávat perorálně jedenkrát denně (po aplikaci nasycovací dávky). I když je blokáda receptoru reverzibilní, je zajištěn efekt po dobu několika týdnů (tab. 1).
Ze studií II. fáze hodnocení je nejvýznamnější studie TRA-PCI, v níž se ukázala bezpečnost přípravku SCH-530348 při předpokladu účinnosti u nemocných s primární PCI. Při úvodní dávce 40 mg a udržovací 2,5 mg se neobjevilo závažné krvácení, výskyt MACE sice ve srovnání s placebem klesl, nicméně čísla byla příliš malá k jakýmkoli závěrům [10 – 11]. Na základě příznivých výsledků v pilotních klinických studiích provedených zejména na japonské populaci v indikaci prevence recidivy iktu a infarktu myokardu (při léčbě ASA) či v ovlivnění periprocedurálních infarktů (na pozadí duální protidestičkové léčby) pokročily studie do III. fáze hodnocení. Ve studii TRA*CER je prověřován efekt po akutní koronární příhodě typu non‑STEMI v kombinaci se standardní, zpravidla duální
protidestičkovou léčbou. Obdobně, ve studii TRA-2(o)P-TIMI 50 u chronických kardiovaskulárních onemocnění, tj. po překonaném infarktu myokardu, po iktu či s ICHDK, je u 27 tis. nemocných hodnocen pokles řady mortalitně/ morbiditních parametrů při stávající léčbě, tedy i při ASA [12].
Druhým inhibitorem receptorů PAR-1 je E5555, který vedle blokády aktivace destičky tlumí též uvolnění molekul akutní fáze, zejména cytokinů interleukinu-6, sCD40L a P-selektinu. Bohužel, klinické hodnocení u nemocných s akutními koronárními příhodami nepřekročilo II. fázi hodnocení (studie LANCELOT). První výsledky ukazují jen středně intenzivní protidestičkový účinek, místo tohoto inhibitoru receptoru PAR-1 proto bude pravděpodobně také v kombinaci s ASA či s inhibitory ADP receptorů [1,13]. Vzhledem k tomu, že studie III. fáze hodnocení jsou plánovány pouze pro japonskou populaci, není pravděpodobné, že by tento inhibitor trombinových receptorů byl v nejbližších letech v Evropě uveden.
Destičkové receptory PAR-4 mají zřejmě jen podpůrný účinek. Po inhibici receptoru PAR-1 sice může být trombocyt aktivován stimulací receptorů PAR-4, nicméně dávka trombinu musí být mnohonásobně vyšší. O možnostech farmakologické blokády těchto receptorů nejsou publikovány žádné zprávy.
Blokátory tromboxanových receptorů a inhibitory syntézy tromboxanu A2
Novinek ve skupině inhibitorů syntézy tromboxanu A2 (TXA2) či blokátorů jeho receptorů není mnoho. Alternativou ASA je triflusal. Tento derivát ASA preferenčně a ireverzibilně inhibuje cyklo-oxygenázu 1 (COX‑1). Inhibice COX‑1 je sice slabší než po podání ASA, nicméně tento nedostatek bohatě kompenzuje skutečnost, že se jedná o blokádu semi-selektivní. To znamená, že je potlačena syntéza trombocytární cyklo-oxygenázy, tedy syntéza TXA2, ale v endoteliích COX‑1 je tlumena o řád méně, a blokáda syntézy prostacyklinu je tak nevýznamná. Navíc triflusal stimuluje NO-syntázu a zvýšenou nabídkou oxidu dusnatého funkci endotelu zlepšuje.
Potenciálem triflusalu by mohla být protidestičková léčba nemocných s intolerancí či rezistencí na ASA. Též je možno uvažovat o užití triflusalu jako alternativě ASA. V posledních letech proběhla řada studií v indikaci sekundární prevence po TIA či po iktu, ve kterých byl triflusal srovnán s ASA (TACIP, TAPIRSS). Dopad na snížení mortality/ morbidity byl stejný, krvácivé komplikace však byly při triflusalu významně nižší. Účinek v prevenci iktu u nemocných s fibrilací síní (sledována monoterapie vs kombinace s antivitaminem K) byl sice nadějný, při kombinaci poklesl výskyt trombembolických příhod, nicméně počty zařazených nemocných nesplňovaly současné nároky na prognostickou studii [14]. Recentně byl triflusal úspěšně testován v pilotní studii SNUH-DES v rámci triální protidestičkové léčby (spolu s kombinací ASA + klopidogrel) u nemocných po implantaci lékového stentu [15]. Ač je triflusal pravděpodobně stejně účinný jako ASA a stran krvácení je bezpečnější, úspěšnému zavedení, zdá se, brání vyšší cena.
Vedle inhibice syntézy tromboxanu A2 inhibicí COX je možno blokovat i tromboxanové receptory. Nadějným antagonistou těchto
receptorů je terutroban. Terutroban je selektivní antagonista TP receptorů (tj. receptorů pro vazokonstrikční a proagregační prostanoidy TXA2 a PGG2 či PGH2), který má v experimentu antitrombotické, vazodilatační a antiaterosklerotické vlastnosti. Potvrdí‑li se závěry preklinických studií, které v experimentu dokumentují výraznou regresi aterosklerózy, pak by mohlo jít o velmi zajímavý lék. Prozatím je terutroban testován v indikaci snížení trombotického a trombembolizačního rizika u nemocných s cerebrovaskulárním postižením (studie PERFORM) a v progresi diabetické nefropatie, kde pilotní studie dokumentovaly příznivý efekt [16]. Příznivý účinek u diabetické nefropatie se přičítá protektivnímu účinku na endotelie a vazodilataci.
Jak je z uvedeného přehledu patrné, vývoj v této oblasti v posledních letech výrazně pokročil. Je tak reálná naděje, že nejbližší roky nám přinesou nová antitrombotika působící již ve fázi adheze, která zamezí aktivaci, degranulaci a agregaci trombocytů. Pro klinika je významné, že na rozdíl od pozdějších fází inhibice primární hemostázy nedojde k uvolnění řady destičkových působků (degranulaci) s vazospastickým a proagregačním účinkem (TXA2, serotonin či PAF).
Blokátory serotoninových receptorů a receptorů pro PAF
Aktivace trombocytů serotoninovými receptory je méně významná. Také blokáda serotoninových receptorů ketanserinem či naftidrofurylem ovlivňuje aktivaci trombocytů jen okrajově. Vzhledem k jen mírně zpomalené rychlosti agregace se protidestičkový efekt inhibitorů serotoninu na výsledném klinickém efektu významně nepodílí [17].
Obdobně se neosvědčila blokáda PAF (platelet‑activating factor) v indikaci protidestičkové léčby. Blokátory PAF – např. rupatadin či lexipafant – mající určitý efekt v léčbě alergických onemocnění či akutní pankreatitidy mají antiagregační účinek zanedbatelný.
Inhibice degranulace trombocytů
Degranulace trombocytů je důležitou složkou primární hemostázy. Bylo identifikováno téměř 200 látek, které jsou uvolněny právě destičkami. Řada z těchto působků má zásadní význam a funguje zpravidla více mechanizmy účinku. V prvé řadě aktivují další trombocyty a atrahují je do místa léze. Další důležitou funkcí je vazokonstrikce, neboť omezení průtoku zvýší lokální koncentraci hemostatických působků a proteáz koagulační kaskády. Navíc vazokonstrikce sníží ztráty krve zpomalením krevního toku. Při degranulaci je uvolněn zejména TXA2, ADP či serotonin, mající výše uvedené účinky. Navíc dochází k sekreci řady molekul akutní fáze, růstových faktorů, koagulačních faktorů nebo vazoadhezivních proteinů (obr. 1C).
Inhibice sekrece je proto důležitou součástí protidestičkové strategie. Hlavní léčebnou strategií není však inhibice syntézy jednotlivých faktorů, ale zabránění aktivace trombocytu a tlumení děje, který degranulaci iniciuje. Výjimkou je léčba kyselinou acetylsalicylovou, triflusalem či obecně inhibitory cyklo-oxygenázy 1. Blokádou COX‑1 tlumíme syntézu tromboxanu A2 a dalších vazokonstrikčních
prostanoidů – prostaglandinů G2 a H2. Tato léčebná strategie byla probrána již výše.
Druhou strategií je stabilizace trombocytu zvýšením nabídky cyklického adenozin monofosfátu (aktivací adenyl-cyklázy) či cyklického guanozin monofosfátu (cGMP). Zejména zvýšení dostupnosti cAMP má mírný klinický význam, dochází k nižšímu uvolňování adenozin‑difosfátu a k útlumu další aktivace trombocytů. Prakticky jediným farmakologickým postupem na této úrovni je blokáda degradace cAMP inhibicí fosfodiesterázy (PDE). Určitý, byť klinicky nevelký efekt má blokáda PDE dipyridamolem či cilostazolem. Nutno však zdůraznit, že tento postup má vysloveně jen podpůrný účinek, základem zůstává působení na mediátory aktivace destiček. Zvýšení nabídky cGMP dosáhneme donátory NO, konkrétně molsidominem. Klinický dopad tohoto postupu nebyl testován, v experimentu je inhibice destiček jen hraničně významná.
Inhibice agregace trombocytů
Agregace trombocytů je posledním krokem destičkové hemostázy. Spočívá ve vzájemné vazbě trombocytů fibrinovými můstky. Po aktivaci glykoproteinových receptorů IIb/ IIIa je vazná sekvence receptorů obsazena bivalentními vaznými proteiny (fibrinem, vWF, vitronectinem aj.) a vzniká primární trombus. Inhibice v tomto místě je výhodná, neboť zde se spojují všechny podněty iniciující agregaci trombocytů. Druhou významnou předností blokády agregace je i možnost desagregace destiček, tj. rozpuštění čerstvého destičkového trombu. Na druhé straně je nutno uvést, že tento krok přichází až po degranulaci trombocytů a uvolnění výše popsaných působků. Navíc snížením agregace trombocytů na všechny podněty globálně je zvýšeno riziko krvácení (obr. 1D).
Vývoj v oblasti nových nepeptidových antagonistů receptoru GP IIb/ IIIa zatím stagnuje. Velká plejáda „nadějných“, perorálně účinných molekul, byla sice testována, nicméně buď byla překážkou jejich zavedení nízká účinnost (zpravidla pro protrombotický efekt na podkladě rebound fenoménu při nízké afinitě k receptoru), či špatná tolerance pro vysoký výskyt hemoragických příhod při dlouhodobém podávání [18]. Dosavadní přípravky na bázi chimerických protilátek (abciximab) či malé molekuly inhibující vaznou sekvenci (tirofiban či eptifibatid) jsou zatím posledním slovem v této kapitole. Doufejme jen, že ne trvale.
Druhou zajímavou cestou, vedle blokády již exprimovaných receptorů IIb/ IIIa, je snížení exprese těchto receptorů, tedy vývoj inhibitorů exprese glykoproteinových receptorů IIb/ IIIa. Omezením nabídky vazných míst pro fibrinogen či jiné bivalentní proteiny by se mohla zpomalit akcelerovaná agregace u nemocných, se kterou se setkáváme u příhod na aterotrombotickém podkladě. Přirozeným proteinem z rodiny peptidů typu fibrinu je peptid FX06, který zatím v in vitro studiích významně tlumil vlastní expresi receptorů IIb/ IIIa. Do fáze klinického hodnocení peptidu FX06 je však ještě daleko [19].
Závěr
Shrneme‑li, pak před námi stojí řada velmi nadějných molekul. Na většinu z nich, i přiúspěšném překonání III. fáze klinického hodnocení, však budeme muset ještě několik let čekat. Nicméně některé molekuly již byly uvolněny ke klinickému užití. Nejvýznamnější inovací tohoto roku bude jistě zavedení prasugrelu. Připomeňme si, že prasugrel má proti klopidogrelu výrazně menší výskyt rezistence, významně redukované nebezpečí lékových interakcí a rychlejší nástup účinku. Můžeme tak bez nadsázky říci, že se zavedením prasugrelu přestáváme v oblasti profylaxe trombotických příhod hrát ruletu, jelikož nevíme, který pacient na léčbu odpoví, ale zahajujeme promyšlenou šachovou partii s předem známými pravidly. Další perspektivu vidíme v zavedení reverzibilního inhibitoru ADP receptorů – ticagreloru. Jeho místo bude zejména u stavů s vyšším rizikem krvácení, kdy krátká doba účinku bude přínosem. Další výhodou bude rychlý nástup účinku.
V dlouhodobějším horizontu je možno spatřovat naději zejména v inhibitorech adheze trombocytů. Tato skupina by mohla mít velmi příznivý poměr snížení trombotického rizika při nízkém krvácivém potenciálu. Konečně současnou strategii duální protidestičkové léčby by mohla vystřídat terapie triální, kdy blokádu aktivace trombocytů TXA2 či ADP doplní inhibitory trombinových receptorů PAR-1.
prof. MUDr. Jan Bultas, CSc.1
doc. MUDr. Debora Karetová, CSc.2
1 Ústav farmakologie, 3. LF UK Praha
2 II. interní klinika kardiologie a angiologie, 1. LF UK Praha
Jan.Bultas@lf1.cuni.cz
Sources
1. ClinicalTrials.gov. [http:/ / clinicaltrials.gov/ ].
2. Van Bockstaele F, Holz JB, Revets H. The development of nanobodies for therapeutic applications. Curr Opin Investig Drugs 2009; 10 : 1212 – 1224.
3. De Meyer SF, Vanhoorelbeke K, Ulrichts H et al. Development of monoclonal antibodies that inhibit platelet adhesion or aggregation as potential anti‑thrombotic drugs. Cardiovasc Hematol Disord Drug Targets 2006; 6 : 191 – 207.
4. Gilbert JC, DeFeo - Fraulini T, Hutabarat RM et al. First - in‑human evaluation of anti von Willebrand factor therapeutic aptamer ARC1779 in healthy volunteers. Circulation 2007; 116 : 2678 – 2686.
5. Mega JL, Close SL, Wiviott SD et al. Cytochrome P - 450 Polymorphisms and Response to Clopidogrel. N Engl J Med 2009; 360 : 411 – 413.
6. Simon T, Verstuyft C, Mary - Krause M. French Registry of Acute ST‑Elevation and Non - ST‑Elevation Myocardial Infarction (FAST‑MI) Investigators. Genetic Determinants of Response to Clopidogrel and Cardiovascular Events. N Engl J Med 2009; 360 : 363 – 375.
7. Montalescot G, Wiviott SD, Braunwald E. TRITON - TIMI 38 investigators. Prasugrel compared with clopidogrel in patients undergoing percutaneous coronary intervention for ST‑elevation myocardial infarction (TRITON - TIMI 38): double‑blind, randomised controlled trial. Lancet 2009; 373 : 723 – 731.
8. HOTLINE I, PLATO favours ticagrelor over clopidogrel in ACS. Eur Heart J 2009; 30 : 2541.
9. Bhatt DL, Lincoff AM, Gibson CM. CHAMPION PLATFORM Investigators. Intravenous platelet blockade with cangrelor during PCI. N Engl J Med 2009; 361 : 2330 – 2341.
10. Hildemann SK, Bode C. Improving antiplatelet therapy for atherothrombotic disease: preclinical results with SCH 530348, the first oral thrombin receptor antagonist selective for PAR - 1. Hamostaseologie 2009; 29 : 349 – 355.
11. Oestreich J. SCH - 530348, a thrombin receptor (PAR - 1) antagonist for the prevention and treatment of atherothrombosis. Curr Opin Investig Drugs 2009; 10 : 988 – 996.
12. Morrow DA, Scirica BM, Fox KA et al. TRA 2(o) P - TIMI 50 Investigators. Evaluation of a novel antiplatelet agent for secondary prevention in patients with a history of atherosclerotic disease: design and rationale for the Thrombin‑Receptor Antagonist in Secondary Prevention of Atherothrombotic Ischemic Events (TRA 2 degrees P) - TIMI 50 trial. Am Heart J 2009; 158 : 335 – 341.e3.
13. Serebruany VL, Kogushi M, Dastros - Pitei D et al. The in‑vitro effects of E5555, a protease‑activated receptor (PAR) - 1 antagonist, on platelet biomarkers in healthy volunteers and patients with coronary artery disease. Thromb Haemost 2009; 102 : 111 – 119.
14. Anninos H, Andrikopoulos G, Pastromas S et al. Triflusal: an old drug in modern antiplatelet therapy. Review of its action, use, safety and effectiveness. Hellenic J Cardiol 2009; 50 : 199 – 207.
15. Suh JW, Andrikopoulos G, Pastromas S et al. Comparison of triple antiplatelet therapy including triflusal and conventional dual therapy in patients who underwent drug‑eluting stent implantation. Int Heart J 2009; 50 : 701 – 709.
16. Chamorro A. TP receptor antagonism: a new concept in atherothrombosis and stroke prevention. Cerebrovasc Dis 2009; 27 (Suppl 3): 20 – 27.
17. Kirsten R, Erdeg B, Moxter D et al. Platelet aggregation after naftidrofuryl application in vitro and ex vivo. Int J Clin Pharmacol Ther 1995; 33 : 81 – 84.
18. Zeymer U, Zahn R. Glycoprotein IIb/ IIIa antagonists: new developments. Hamostaseologie 2009; 29 : 334 – 337.
19. Hallén J, Atar D, Serebruany V. Effects of FX06 In Vitro on Platelet, Coagulation, and Fibrinolytic Biomarkers in Volunteers and Patients With Documented Coronary Artery Disease. Am J Ther 2009 [Epub ahead of print].
20. Ho PM, Maddox TM, Wang L et al. Risk of adverse outcomes associated with concomitant use of clopidogrel and proton pump inhibitors following acute coronary syndrome. JAMA 2009; 301 : 937–944.
21. Stanek EJ, Aubert RE, Flockhart DA et al. A National Study of the Effect of Individual Proton Pump Inhibitors on Cardiovascular Outcomes in Patients Treated with Clopiodogrel Following Coronary Stenting: The Clopidogrel Medco Outcomes Study [http://www.scai. org/pdf/20090506Medcoabstract.pdf] (Abstract).
22. Juurlink DN, Gomes T, Ko DT et al. A populationbased study of the drug interaction between proton pump inhibitors and clopidogrel. CMAJ 2009; 180 : 713–718.
23. Gupta E, Bansal D, Sotos J et al. Risk of Adverse Clinical Outcomes with Concomitant Use of Clopidogrel and Proton Pump Inhibitors Following Percutaneous Coronary Intervention. Dig Dis Sci 2009 [Epub ahead of print].
Labels
Paediatric cardiology Internal medicine Cardiac surgery CardiologyArticle was published in
Cardiology Review

2010 Issue 1
Most read in this issue
- Carvedilol as a treatment option for cardiovascular diseases
- Depression in patients with cardiovascular disease
- Tenecteplase in current clinical practice
- New players on the antiplatelet therapy chequerboard – what can we expect?