
Limitace genetického testování v onkologii
Limitations of Genetic Testing in Oncology
Genetic testing of cancer syndromes is based on the existing knowledge of monogenic causes of oncologic diseases. In cases of high risk genes, the findings concerning the carrier status of pathogenic mutation can be of clinical use in the prediction of risks and for preventive care. In non carriers in families with mutation in the high risk gene, the risk of cancer diseases may not be the same as the population risk and some preventive follow up is recommended. The clinical use of genes with mild or moderate risk of cancer is problematic and could lead to distorted conclusions about the actual cause of the familial form of the disease. Predictive testing in genes with moderate risk of cancer (2 – 3 times) is not offered, or the non carriers are followed in the same way as carriers. The use of genes with mild risk is not recommended in clinical practice. Molecular genetic testing also has its limitations and its quality depends on the methods and technology used and the existing knowledge of the significance of mutations. In some variants it is not clear yet whether they are just insignificant polymorphisms or pathogenic mutations. The interpretation of test results in the context of the whole family history is important.
Key words:
genetic testing – mutations – genes
Authors:
L. Foretová 1; M. Navratilova 1; E. Macháčková 1
Authors‘ workplace:
Oddělení epidemiologie a genetiky nádorů, Masarykův onkologický ústav, Brno
1
Published in:
Klin Onkol 2009; 22(Supplementum): 65-68
Overview
Genetické testování nádorových syndromů je založeno na dosavadních znalostech monogenních příčin onkologických onemocnění. U vysoce rizikových genů je možné klinické využití zjištění nosičství patogenní mutace v predikci rizika a v preventivní péči. U ne-nosičů mutace v rodině s prokázanou mutací nemusí být riziko na úrovni běžné populace a doporučuje se preventivní sledování. U genů s předpokládaným středním a nízkým rizikem je klinické využití dosud sporné a mohlo by vést ke zkresleným závěrům o skutečné příčině familiární formy nemoci. U genů s předpokládaným středním rizikem nádoru (2 – 3krát) se prediktivní testování nenabízí nebo se doporučuje ne-nosiče mutace preventivně sledovat stejným způsobem jako nosiče. U genů s nízkým rizikem se klinické využití vůbec nedoporučuje. Molekulárně genetické testování má také své limitace a jeho kvalita závisí na použitých metodách, přístrojové technice a dosavadních znalostech o významu mutací. U některých změn není dosud jasné, zda se jedná o nevýznamný polymorfizmus nebo patogenní mutaci. Interpretace výsledků testování v kontextu rodinné anamnézy je důležitá.
Klíčová slova:
genetické testování – mutace – geny
Úvod
Každé onkologické onemocnění může být zapříčiněno zděděnou mutací v jednom nebo více genech, která způsobuje vyšší náchylnost ke vzniku nádorů. Pravděpodobnost, že se nádorové onemocnění skutečně během života objeví, záleží na mnoha dalších genetických i negenetických faktorech.
U mnoha genů bylo prokázáno, že zděděná patogenní mutace může způsobovat velmi vysoké riziko nádorového onemocnění, mnohdy přesahující desetinásobek populačního rizika (např. mutace v genech BRCA1/ 2, TP 53, MLH1, MSH2). I v těchto genech však různé typy mutací mohou způsobovat různě vysoká rizika dle uložení mutace a typu poruchy tvorby proteinu.
Postupně jsou zkoumány nové a nové geny a je zjišťováno, že mnohé hrají také důležitou úlohu v etiologii nádorových onemocnění, nicméně ne tak významnou jako geny vysokého rizika. Předpokládá se, že porucha jejich funkce ovlivňuje nárůst rizika jen mírně nebo středně závažně (např. heterozygotní mutace v genech CHEK2, BRIP1, NBS1, PALB2 a ATM pro nádory prsu aj.) [1 – 3].
V roce 1994 a 1995 byly objeveny geny BRCA1 a BRCA2, které jsou zodpovědné za velkou část familiární predispozice k nádorovému onemocnění prsu a ovaria [4 – 6]. V současné době jsou hledány další geny, které by byly schopny vyvolat podobně vysoká rizika onemocnění a které by vysvětlovaly hereditární predispozici k nádorovému onemocnění u vysoce rizikových rodin (4 a více případů v rodině, většinou v mladém věku), kde byla vazebnou analýzou vyloučena příčinná souvislost s geny BRCA1 nebo BRCA2.
Tento výzkum však dosud nebyl úspěšný a spekuluje se, že u mnoha rodin se může jednat o polygenní model rizika s aditivním efektem více (i mnoha) genů s mírnějším rizikem.
Mutace a jejich význam v genetickém testování nádorových onemocnění
Pokud mluvíme o testování suspektních hereditárních forem nádorů, provádí se testování zárodečných mutací z DNA izolované z krve pacientů.
Vyhledávání změn (mutací) v sekvenci DNA se provádí různými způsoby. Pokud nejde o detekci již známé konkrétní mutace a cílem je zachytit jakoukoli změnu, lze použít k testování různé screeningové metody, které umožní rychlé vyhledání těch úseků genu (část sekvence DNA), které jsou „odlišné“ od standardního typu. Konkrétní mutace je pak charakterizována sekvenováním. Screeningové metody se neustále vyvíjejí, zvyšuje se jejich senzitivita i specifita a také dochází k automatizaci práce, a tím k redukci chyby způsobené lidským faktorem.
Záměny bazí – substituce
Nukleotidové substituce jsou nejčastější genetickou variací, která představuje až 90 % z množství detekovaných variant v DNA sekvenci [7]. Převážná většina z nich jsou neškodné polymorfní varianty bez fenotypového projevu. Průměrná frekvence běžných nukleotidových substitucí v lidském genomu se pohybuje kolem jedné na 1 000 bazí [8].
Tzv. „nonsense“ substituce způsobují v místě záměny vznik terminačního kodonu a jsou patogenní. Mnohdy však záměna baze vede ke změně jedné aminokyseliny kódované daným tripletem a je sporné, zda vůbec ovlivňuje riziko onemocnění, nebo nikoliv. Tyto varianty jsou označovány jako varianty s neznámým klinickým účinkem a genetici nemohou tyto varianty použít pro klinické testování příbuzných osob, pokud není dokázáno některým z funkčních testů, zda je nebo není skutečně patogenní. Pro mnoho genů však neexistují validní funkční testy a varianty zůstávají na dlouhou dobu klasifikovány jako „Unknown Variants“ (UV) [9].
Pokud se jedná o rodinu s vysoce rizikovou rodinnou anamnézou (obr. 1), potom je možné několik vysvětlení tohoto sporného (neinformativního) výsledku: a) jedná se o variantu, která je patogenní, ale nelze to v současné době potvrdit, b) příčina hereditární etiologie je v jiném dosud neznámém nebo netestovaném genu, c) příčina je ve více genetických faktorech (polygenní), které v současné době nelze identifikovat, d) nebo se nejedná o hereditární onemocnění, ale to je možné jen v rodinách, kde není familiární kumulace příliš závažná (obr. 2).
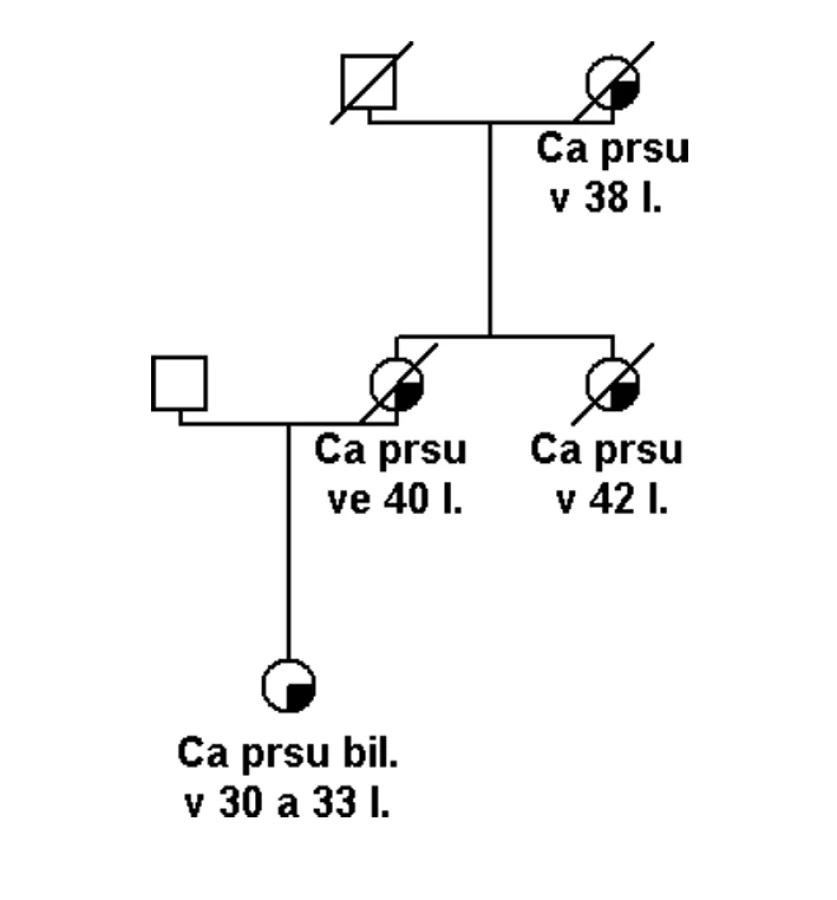
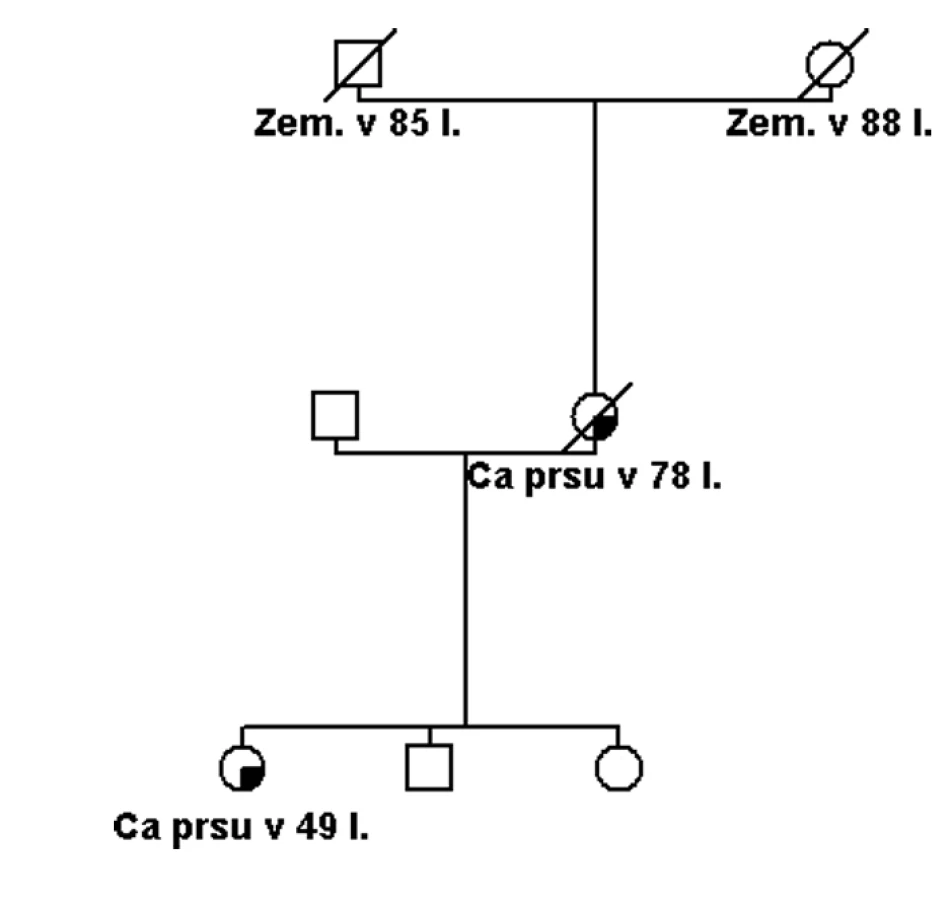
Záměna nukle otidu v kódující sekvenci, která způsobí záměnu aminokyseliny v polypeptidovém řetězci, může být příčinou onemocnění pouze tehdy, narušuje li důležitou strukturní nebo přímo funkční doménu proteinu. Obecně jsou mezidruhově konzervativní aminokyseliny v polypeptidovém řetězci s větší pravděpodobností funkčně významné. Zároveň mutace, které zaměňují původníaminokyselinu za aminokyselinu odlišných fyzikálních vlastností, s větší pravděpodobností mohou mít vliv na strukturu, a tím následně i stabilitu a funkci polypeptidu [10].
Delece, inzerce a velké přestavby genů
Delece nebo inzerce jedné a více bazí (způsobující narušení čtecího rámce) a velké intragenové přestavby (delece nebo duplikace oblastí zahrnující celé exony) jsou většinou změny, které významně mění strukturu a funkci proteinu a je možné je jednoznačně označit za patogenní. Některé delece nebo inzerce, které způsobují ztrátu jedné nebo více aminokyselin a nevedou k poruše čtecího rámce, mohou ale nemusí být patogenní.
Závěry molekulárně genetického vyšetření vyžadují znalost funkce a lokalizace jednotlivých funkčních domén proteinu. Mutace lokalizované na terminálním 3’ konci proteinu již nemusí být funkčně významné, ale mohou mít vliv na stabilitu proteinu.
Mutace měnící místo sestřihu
Speciální skupinou jsou mutace měnící místo sestřihu – tzv. „splice site“ mutace. Nejsnadněji rozpoznatelné se vyskytují ve 100% konzervativních místech sestřihu dinukleotidu GT lokalizovaného na 5’ počátku intronu (donorové místo sestřihu) a dinukleotidu AG lokalizovaného na 3’ konci intronu (akceptorové místo sestřihu). Důležitou úlohu při správném sestřihu hrají také sekvence přilehlé k AG a GT sekvenci a část konzervativní intronické sekvence (větvící místo – „branch site“) lokalizované ve vzdálenosti ≈ 20 – 40 nukleotidů před terminálním AG dinukleotidem [11].
Vzácně se mohou „splice site“ mutace nacházet vzdáleně od správného místa sestřihu v intronické sekvenci nebo kdekoli v kódující sekvenci, kde by na první pohled nevýznamná záměna třetí baze kodonu mohla aktivovat kryptické místo sestřihu. K odhalení mutací měnících místo sestřihu mohou přispět počítačové programy vyhledávající v zadané DNA sekvenci lokalizaci pravděpodobného místa sestřihu.
Pomocí predikčního programu lze zjistit, s jakou pravděpodobností by mohlastudovaná varianta ovlivnit správný sestřih mRNA. Konečný aberantní sestřih však musí být u pacienta prokázán experimentálně [12]. V laboratorních podmínkách je nutno provést mRNA analýzu současně s negativní kontrolou, aby nedošlo k chybné interpretaci přirozeného alternativního sestřihu jako důsledku mutace. Jako alternativní sestřih je označován jev, kdy z jednoho genu vzniká více různých forem mRNA a proteinu. Alternativní sestřih je v buňce regulován a vzniklé izoformy bývají často tkáňově specifické nebo mohou mít rozdílnou lokalizaci v buňce. Jednotlivé izoformy tak mohou mít modifikovanou, až odlišnou funkci [11].
Jaký je tedy výsledný efekt mutace měnící místo sestřihu? Frekvenčně nejčastěji popisovaným důsledkem je inaktivace místa sestřihu a následné nerozeznání exonu při sestřihu RNA probíhajícím v jádře, který vede ke konečné deleci celého exonu v mRNA sekvenci – tzv. „exon skipping“. Častým důsledkem je také aktivace kryptického místa sestřihu, pokud se nachází v blízkosti přirozeného místa sestřihu. Poměrně vzácně pak dochází k vytvoření pseudo-exonu v intronické sekvenci, případně k retenci celého intronu v případě velmi krátkých nebo terminálních intronů. U některých mutací narušujících místo sestřihu byla jako důsledek popsána i kombinace více variant aberantního sestřihu a reziduální správný sestřih [13 – 14]. Konečným důsledkem aberantního sestřihu je delece nebo inzerce úseků mRNA sekvence, což vede při translaci k deleci nebo inzerci části proteinové sekvence, nebo frekvenčně častěji dochází k posunu čtecího rámce a k předčasné terminaci translace.
Klinická hodnocení výsledků testování
Laboratorní zpráva by měla být pro klinického genetika jednoznačně srozumitelná a měla by popisovat i dosavadní znalosti o uvedené mutaci. Na základě těchto poznatků je možné využití výsledků testování v praxi.
Neinformativní výsledek u pacienta
Pokud je v rizikové rodině testován pacient s typickým nádorovým onemocněním syndromu a není nalezena kauzální mutace, může se jednat o: a) příčinu hereditární etiologie v jiném dosud neznámém nebo netestovaném genu (monogenní), b) příčinu ve více genetických faktorech (polygenní), které v současné době nelze identifikovat, c) nebo se nejedná o hereditární onemocnění, ale o tom je možné spekulovat jen v případě málo závažné rodinné anamnézy a u typů nádorových onemocnění, které mohou mít i silnou negenetickou etiologickou příčinu (např. nádory kolorekta).
Genetik vždy musí při interpretaci posuzovat všechny rizikové faktory, které mohly hrát důležitou úlohu. Jedná se i o předchozí léčbu, dlouhodobé chronické onemocnění, rizikové chování nebo životní a pracovní prostředí atd.
Vždy je důležité, aby si vyšetřovaná rodina uvědomila, že negativní výsledek je neinformativní a není vyloučením možnosti dědičné příčiny.
Genetik ve zprávě odhaduje empirická rizika pokud možno pro širší rodinu a doporučuje vhodná preventivní opatření.
Pozitivní výsledek a prediktivní testování
Pokud je testování u pacienta (probanda) pozitivní a je zjištěna kauzální mutace, je vhodné pozvat rodinné příslušníky k prediktivnímu testování. Prediktivní testování má určitou výpovědní hodnotu, která může být dourčité míry limitovaná.
Pokud se jedná o přímé příbuzné testovaného probanda nebo probandky a výsledek prediktivního testu je negativní, je možné říci, že s největší pravděpodobností bylo vysoké riziko nádorového syndromu vyloučeno. S největší pravděpodobností z toho důvodu, že nemůžeme říci na sto procent, zda u nemocné osoby byla zjištěná kauzální mutace jedinou genetickou změnou, která určovala riziko onemocnění. V naprosté většině rodin se skutečně bude jednat o monogenní příčinu, ale existují vzácně i rodiny, kde je možné zjistit homozygoty (identické mutované alely stejného genu, např. dvě stejné mutace v genu BRCA1), složené heterozygoty (různé alely stejného genu, např. dvě různé mutace v genu BRCA1). Tyto je možné zjistit při dokončení kompletního testování všech exonů danéhogenu (i po zachycení patogenní mutace). Nebo se může jednat o dvojité heterozygoty (mutace ve dvou různých genech, např. BRCA1 a CHEK2 aj.), což by mohlo být nejčastější variantou, i když dosud málo prozkoumanou (obr. 3). Kombinace genů vysokého rizika a mnoha genů nižšího a středního rizika, které budou modulovat výši rizika onemocnění, je v onkologii pravděpodobná. Toto musíme mít na paměti při uzavírání každého genetického vyšetření a indikování preventivní péče v onkologii.
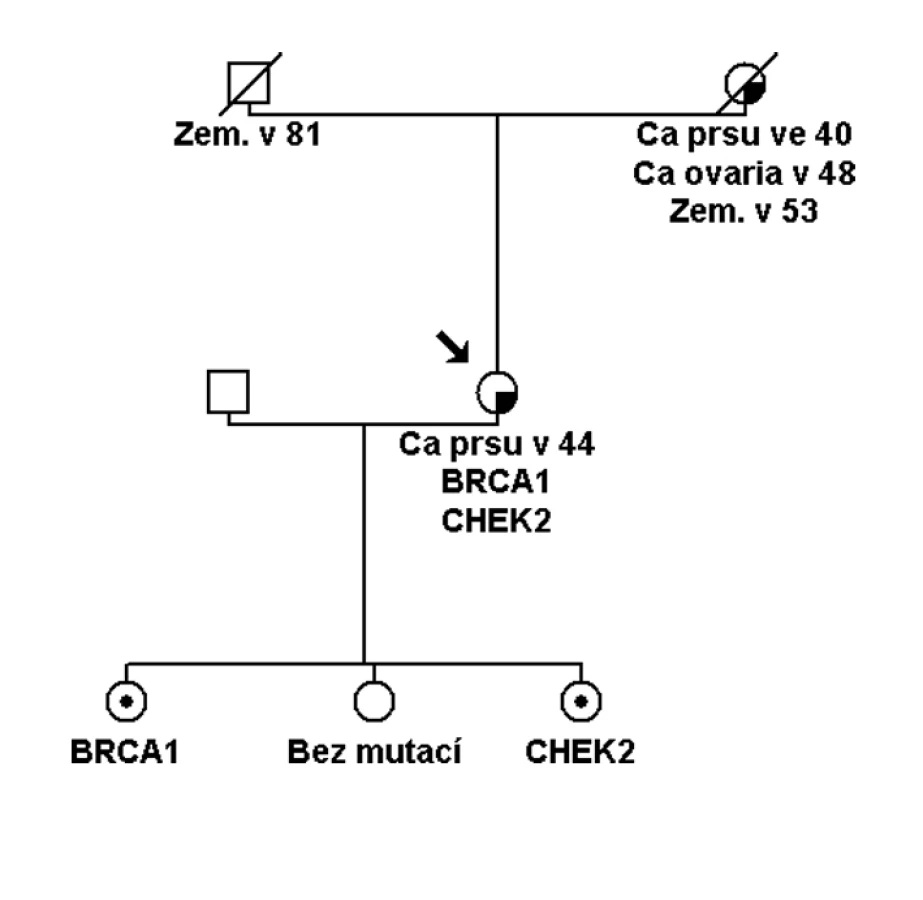
U osob z rodiny s prokázanou kauzální vysoce penetrantní mutací, kde prediktivní test u příbuzné osoby tuto mutaci vyloučil, nemůžeme vyloučit další možné genetické modifikátory rizika, které nejsme schopni v současné době identifikovat. Tuto skutečnost potvrzuje i epidemiologická studie, která prokázala velký počet fenokopi í (24 % žen s nádorem prsu bez BRCA1/ 2 mutace) v rodinách s prokázanou BRCA1/ 2 mutací [15]. Studie upozorňuje, že při kalkulaci rizik onemocnění u ne - nosičů BRCA1 nebo BRCA2 mutací v rodinách s prokázanou patogenní mutací u probanda je riziko onemocnění nádorem prsu vyšší než riziko běžné populace, RR je uváděno 5,0. U dalších studi í je odhad tohoto reziduálního familiárního rizika v pásmu středních rizik (2 – 3krát) [16]. Proto v současné době indikujeme i u zdravých ne - nosiček mutací v BRCA1 a BRCA2 genech preventivní sledování tak jakou žen s odhadovaným středním rizikem zátěže (většinou roční kontroly se začátkem o deset let dříve, než byl nejčasnější výskyt nádoru prsu v rodině, kombinace UZ a MMG dle věku). Stejně tak bychom měli postupovat u ne - nosičů mutací pro Lynchův syndrom a další syndromy a doporučit preventivní sledování jakou středního rizika onemocnění.
Závěr
Možnosti genetického testování v onkologii se neustále rozšiřují. Zlepšuje se i záchytnost mutací díky novým přístrojům a metodám. Klinické testování má význam především u genů s vysokým rizikem onemocnění. Geny středního a nízkého rizika jsou stále ve většině případů používány ve výzkumu.
Vyloučení zvýšeného rizika onemocnění i u ne-nosičů vysoce penetrantních mutací v pozitivně testované rodině je problematické. S velkou pravděpodobností je možné jim vyloučit vysoká rizikaonemocnění, která existují u nosičů, nicméně je důležité na ně pohlížet z mnoha důvodů jako na osoby s rodinnou zátěží s možností mírně až středně zvýšených rizik nádorů daného syndromu a doporučit vhodnou prevenci.
MUDr. Lenka Foretová, Ph.D.
Oddělení epidemiologie
a genetiky nádorů
Masarykův onkologický ústav
Žlutý kopec 7
656 53 Brno
e-mail: foretova@mou.cz
Sources
1. Claes K, Poppe B, Machackova E et al. Differentiating pathogenic mutations from polymorphic alterations in the splice sites of BRCA1 and BRCA2. Gen Chromos Cancer 2003; 37 : 314 – 320.
2. Collins FS, Brooks LD, Charkravarti A. A DNA polymorphism discovery resource for research on human genetic variation. Genome Res 1998; 8 : 1229 – 1231.
3. Cotton RGH, Scriver CR. Proof of “disease causing” mutation. Hum Mut 1998; 12 : 1 – 3.
4. Katki HA , Gail MH, Greene MH. Breast-cancer risk in BRCA-mutation-negative women from BRCA-mutation-positive families. Lance Oncol 2007; 8(12): 1042 – 1043.
5. Kleibl Z, Novotny J, Bezdickova D et al. The CHEK2 c. 1100delC germline mutation rarely contributes to breast cancer development in the Czech Republic. Breast Cancer Res Treat. 2005; 90(2): 165 – 167.
6. Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base - pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet 1992; 90 : 41 – 54.
7. Miki Y, Swensen J, Shattuck - Eidens D et al. A strong candidate for the Breast and ovarian cancer susceptibility gene BRCA1. Science 1994; 266 : 66 – 71.
8. Rogan PK, Faux BM, Schneider TD. Information analysis of human splice site mutations. Hum Mut 1998; 12 : 153 – 171.
9. Smith A, Moran A, Boyd MC et al. Phenocopies in BRCA1 and BRCA2 families: evidence for modifier genes and implications for screening. J Med Genet 2007; 44(1): 10 – 15.
10. Strachan T, Read AP. Human Molecular Genetics. 2nd ed. BIOS Scientific Publishers Ltd. 1999.
11. Tailon - Miller P, Gu Z, Li Q et al. Overlapping genomic sequences: a treasure trove of single nucleotide polymorphisms. Genome Res 1998; 8 : 748 – 754.
12. Vašíčková P, Macháčková E, Lukešová M et al. Varianty neznámého významu a intragenová přeskupení v genech BRCA1 a BRCA2. Klin Onkol 2006; 19(Suppl): 58 – 62.
13. Walsh T, King MC. Ten genes for inherited breast cancer. Cancer Cell 2007; 11(2): 103 – 105.
14. Weischer M, Bojesen SE, Ellervirk Ch et al. CHEK2 1100delC genotyping for clinical assesment of breast cancer risk: Metaanalysis of 26 000 patient cases and 27 000 controls. J Clin Oncol 2008; 26(4): 542 – 548.
15. Wooster R, Bignell G, Lancaster J et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995; 378 : 789 – 792.
16. Wooster R, Neuhausen SL, Mangion J et al. Localisation of a breast cancer susceptibility gene, BRCA2, to chromosome13q12 –
13. Science 1994; 265 : 2088-2090.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2009 Issue Supplementum
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Safety and Tolerance of Metamizole in Postoperative Analgesia in Children
Most read in this issue
- Hereditární pankreatitida
- Gorlinův syndrom
- Mnohočetná endokrinní neoplazie typ 2 – syndrom MEN 2
- Mnohočetná endokrinní neoplazie typ 1 – syndrom MEN 1