
Zpřesněme indikaci podávání inhibitorů kinázové aktivity EGFR
Indication of EGFR Kinase Inhibitors Should Be Refined
Even though lung cancer incidence began to decline in the majority of industrialized countries, is still belong to cancers with one of the highest incidence and mortality rates. In the Czech Republic, epidermal growth factor receptor (EGFR) kinase activity inhibitors erlotinib and gefitinib are approved for the use as the second - and third-line treatment of non-small-cell lung cancer. In a cohort of non-small-cell lung cancer patients, erlotinib administration led to tumour regression in less than 20% of patients. However, when used in patients with EGFR-activating mutations, e. g. L858R or delE746-A750, the response rate increased to 75–82% in several parallel clinical studies. Similarly, improved response rate was reported in patients bearing amplified wild-type EGFR gene. In contrary, patients with T790M, D761Y, L747S, and T854A mutations (and some other rare abberations) were found to be resistant to treatment with small-molecule inhibitors targeting the active site of the kinase domain. These mutations do not change the EGFR affinity to gefitinib or erlotinib but the mutated receptor is able to bind ATP into its active site even in the presence of erlotinib or gefitinib, similar to a wild-type receptor without an inhibitor. Besides that, when the EGFR molecule bears both the activating (e. g. L858R) and resistance-inducing mutation (e. g. T790M), the tumour acquires resistance to both erlotinib and gefitinib treatment. Currently, research focuses on a development of new strategies that would allow treatment of patients bearing mutations inducing resistance to the small-molecule inhibitors targeted on the active site of EGFR kinase domain. Contrary to the current guidelines for Czech oncologists, identification of EGFR with any of the above mentioned resistance-inducing somatic mutations should be considered as an explicit contraindication for non-small-cell cancer treatment using small-molecule EGFR kinase activity inhibitors erlotinib or gefitinib. This should also include patients in whom a resistance-inducing mutation is detected together with any of the activating mutations or deletions.
Key words:
adenocarcinoma – lung cancer – protein kinase inhibitors – antitumor agents – genetic predisposition to disease – neoplastic process – genetic markers – pharmacological biomarkers
Authors:
P. Heneberg
Authors‘ workplace:
3. lékařská fakulta Univerzity Karlovy, Praha
Published in:
Klin Onkol 2011; 24(2): 87-93
Category:
Reviews
Overview
I přes počínající pokles v posledních letech je v industrializovaných zemích karcinom plic nádorovým onemocněním s jednou z nejvyšších incidencí i úmrtností. Pro léčbu jeho nejčastější, nemalobuněčné formy je v druhé a třetí linii léčby v České republice mimo jiné doporučeno použití erlotinibu (případně gefitinibu), tj. inhibitorů receptorové tyrozinové kinázy EGFR. V běžné populaci však při léčbě erlotinibem dochází k regresi tumoru jen u méně než 20 % pacientů. V posledních letech se ukazuje, že podávání těchto inhibitorů dosahuje výrazně vyšší účinnosti u kohort pacientů s aktivačními mutacemi EGFR, např. L858R či delE746-A750, kdy úspěšnost léčby v několika paralelních studiích dosáhla 75–82 %. O něco nižší odpověď na podávání nízkomolekulárních inhibitorů kinázové aktivity je též u pacientů s amplifikací normálního genu pro tento receptor. Naopak pacienti s mutacemi T790M, D761Y, L747S a T854A (a některými jinými vzácnějšími aberacemi) jsou k léčbě nízkomolekulárními inhibitory cílícími na aktivní místo kinázové domény rezistentní. Tyto mutace sice nemění afinitu EGFR ke gefitinibu či erlotinibu, ale způsobují, že mutovaný receptor i s navázaným inhibitorem je schopen vázat ATP do aktivního místa kinázové domény podobně jako normální receptor bez inhibitoru. Navíc pokud se v rámci molekuly EGFR vyskytne u pacienta kombinace mutace aktivující (např. L858R) s mutací způsobující rezistenci (např. T790M), léčba erlotinibem či gefitinibem je zcela neúčinná. V současnosti jsou proto vyvíjeny nové terapeutické strategie, které by umožnily léčbu pacientů s mutacemi způsobujícími rezistenci k nízkomolekulárním inhibitorům EGFR cíleným na aktivní místo kinázové domény. Oproti současnému stavu by identifikace EGFR s některou ze somatických mutací způsobujících rezistenci měla být považována za jednoznačnou kontraindikaci léčby nemalobuněčného karcinomu plic nízkomolekulárními inhibitory kinázové aktivity EGFR, tj. erlotinibem či gefitinibem, a to i v případě, že současně je s některou z těchto mutací v nádorových buňkách detekována i mutace či delece aktivující.
Klíčová slova:
adenokarcinom – karcinom plic – inhibitory proteinkinázy – antitumorózní látky – genetická predispozice k nemoci – nádorové procesy – genetické markery – biomarkery farmakologické
Východiska
Incidence maligních nádorů plic, kdysi raritních onemocnění, se v České republice začala razantně zvyšovat v průběhu 50. a 60. let, především u mužů. U těch již v posledních dvou desítkách let dochází k pomalému poklesu incidence tohoto onemocnění. U žen došlo k nárůstu počtu onemocnění později, v některých industrializovaných zemích je již viditelné plateau, popřípadě počínající pokles (obr. 1). I přesto zůstávají nádory plic jedním z nejčastěji se vyskytujících zhoubných nádorů s velmi vysokou úmrtností.
![(A) Úmrtnost na nádory plic, průdušnice, průdušek a pleury u mužů (•) a žen (º) v USA v letech 1930–1998 [50]. Data jsou přepočítána na standardizovanou populaci roku 2000, zobrazena je roční úmrtnost na 100 tisíc obyvatel. (B) Věkově standardizovaná časová křivka incidence (•) a mortality (º) zhoubného nádoru průdušnice, průdušky a plíce (C33, C34) v České republice v letech 1977–2007 [51].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/4458929643e19fc9c8d9d6e93eff089e.jpg)
Česká onkologická společnost ČLS JEP na svých webových stránkách pravidelně aktualizuje „Zásady cytostatické léčby maligních onkologických onemocnění“ vytvářené pro práci lékařů léčících onkologické diagnózy. V jejich poslední aktualizaci platné k době psaní tohoto příspěvku [1] bylo pro léčbu nemalobuněčného karcinomu plic ve druhé a třetí linii léčby doporučeno použití erlotinibu (případně gefitinibu), tj. inhibitorů receptorové tyrozinové kinázy EGFR (epidermal growth factor receptor). V rámci kontraindikací je uvedeno, že „v žádné prospektivní studii dosud nebyla definována podskupina nemocných, která by z léčby erlotinibem neměla prospěch. Při retrospektivní analýze studie BR.21 nebyl nalezen rozdíl mezi nemocnými s mutacemi EGFR a nemocnými bez mutací; výhoda léčby erlotinibem byla dokumentována i v podskupině nemocných mužů – kuřáků s dlaždicobuněčným karcinomem“.
První inhibitory EGFR
Erlotinib a gefitinib jsou dnes již klasikou biologické léčby. Jsou jedněmi z prvních inhibitorů tyrozinových kináz, které úspěšně prošly pokročilejšími fázemi klinických testů. Prvním, který byl zahrnut do pokročilejších fází klinických testů, byl gefitinib (Iressa, Astra-Zeneca Pharmaceuticals, Wilmington, BE), kdy slibné výsledky randomizované klinické studie fáze II [2,3] vedly v USA k jeho rychlému schválení pro terapii nemalobuněčného nádoru plic ve třetí linii. Nicméně následná studie fáze III u pacientů s pokročilým nemalobuněčným karcinomem plic (kteří již absolvovali 1–2 chemoterapie) předpoklady nenaplnila, podávání orální dávky 250 mg gefitinibu nezpůsobilo statisticky významný rozdíl oproti podávání placeba [4], a FDA proto přestal doporučovat gefitinib pro výše zmíněnou indikaci. Později se ukázal být účinným při léčbě pacientů s prokázanou aktivační mutací EGFR; pro tyto pacienty je nyní registrován i v EU, v době psaní článku však ještě v ČR nebyla pro tento inhibitor stanovena úhrada.
Druhý z inhibitorů EGFR, erlotinib (Tarceva, Genentech, Inc., South San Francisco, CA a OSI Pharmaceuticals, Inc., Melville, NY), si vedl lépe. BR.21, klinická studie fáze III zaměřená na randomizované pacienty s pokročilým nemalobuněčným karcinomem plic, kteří předtím byli podrobeni 1–2 cyklům chemoterapie, vedla ke statisticky významnému zlepšení přežívání pacientů (průměr 6,7 vs 4,7 měsíce; přežití ≥ 1 rok 31 % vs 21 %) a ke zlepšení kvality života [5]. V době psaní tohoto příspěvku byl erlotinib registrován i v České republice, jeho užití pro léčbu nemalobuněčného karcinomu plic bylo hrazeno deseti českým pneumoonkologickým centrům.
Současný stav poznání
Již během časných fází vývoje obou zmíněných inhibitorů se objevilo hned několik prediktorů zvýšené úspěšnosti léčby pomocí těchto látek. Mezi ně patřily nekuřáctví, východoasijský původ, histologická klasifikace nádoru jako adenokarcinom či bronchoalveolární karcinom a ženské pohlaví. V běžné populaci léčba gefitinibem či erlotinibem vede k regresi nádoru u méně než 20 % pacientů s pokročilým nemalobuněčným karcinomem plic, kteří byli podrobeni 1 či 2 cyklům chemoterapie [5]. Naproti tomu když byla provedena studie na pacientech korejské národnosti, nekuřácích, s diagnostikovanými adenokarcinomy, převážně ženách (92 %), bez předchozí léčby (72 %), u těchto pacientů vyústilo podávání erlotinibu v 56% úspěšnost léčby, s mediánem přežití 19,7 měsíce a s přežitím ≥ 1 rok 76 % [6]. V tu chvíli nebylo příliš zřejmé, co za zjištěnými rozdíly stojí, jednotlivé klinické studie nelze navzájem bezvýhradně srovnávat. Diametrálně odlišné výsledky ovšem naznačovaly genetickou podmíněnost zjištěných rozdílů; v populaci s výše uvedenými charakteristikami jsou zároveň častější histologicky odlišné typy karcinomů, což ve zmíněných studiích nebylo příliš zohledněno.
Retrospektivní analýzou studie BR.21, jakož i dalších studií probíhajících v posledních letech byly skutečně zjištěny významné rozdíly v úspěšnosti léčby u pacientů s odlišným genetickým pozadím. Jak již proběhlo i českým tiskem [7–9], ukázalo se, že léčba je úspěšná zejména u pacientů s aktivačními mutacemi v EGFR, popřípadě s amplifikací genu pro tento receptor [10,11]. Mezi nejčastější patří ≥ 12-nukleotidové delece v exonu 19 neměnící čtecí rámec a zahrnující konzervovaný motiv LREA (45 % aktivačních mutací) a mutace L858R v exonu 21 (40 % aktivačních mutací). Tyto skutečnosti byly reflektovány v sérii klinických studií fáze II [12–16], které ve všech pěti případech dosáhly úspěšnosti léčby 75–82 %. Z výsledků zmíněných studií lze vyzdvihnout především skutečnost, že u pacientů s delecí v exonu 19 byla léčba úspěšná u 95 % případů, u pacientů s mutací v exonu 21 u 67 % případů (ve srovnání s průměrnou < 20% úspěšností léčby u kohorty náhodně zvolených pacientů s nemalobuněčným karcinomem plic). Nicméně zůstává ještě k objasnění, zda přítomnost samotných mutací neovlivňuje přežívání pacientů. Někteří kliničtí onkologové spekulují o možnosti, že předmětné mutace jsou spíše prognostickými markery přežití pacienta nezávisle na probíhající léčbě [17]. Jako negativní prediktor bývá vesměs využívána přítomnost aktivujících mutací v genu KRAS [18], většinou však nedochází k jejich simultánnímu výskytu spolu s mutacemi EGFR.
Kombinovaná terapie
Protože se objevují případy současné aplikace chemoterapie a inhibitorů kinázové aktivity EGFR, je na místě zdůraznit, že proběhlo hned několik klinických studií fáze III, které zjišťovaly výhodnost tohoto postupu [19–22]. Obecně lze říci, že ani jedna z těchto studií nezjistila statisticky průkazné zlepšení stavu zkoumaných kohort pacientů s pokročilým nemalobuněčným karcinomem, kteří byli léčeni gefitinibem nebo erlotinibem v kombinaci se standardní chemoterapií oproti pacientům, kterým byla podávána jen cisplatina, carboplatina, paklitaxel či gemcitabin. Spekuluje se dokonce o protichůdném efektu těchto léčebných postupů. Na druhou stranu u některých podskupin pacientů byly zjištěny pozitivní efekty kombinované léčby [22], tyto podskupiny ale zatím nebyly dostatečně charakterizovány a zpřesnění jejich definice je předmětem několika v současnosti probíhajících klinických studií.
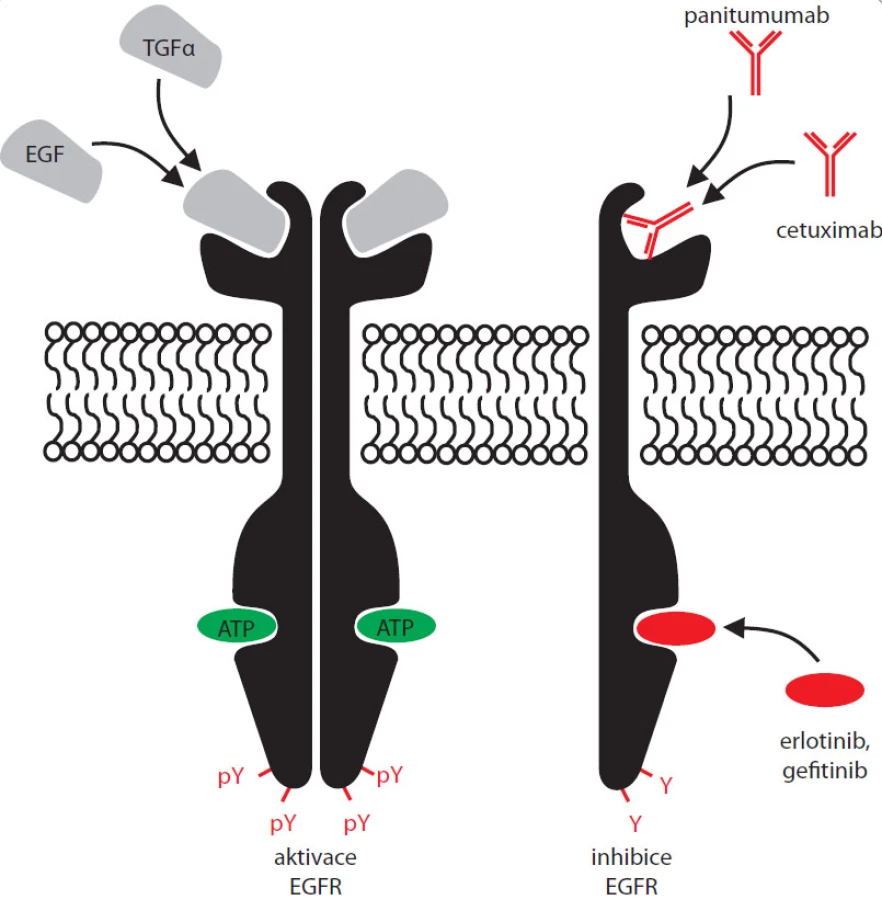
Dalším druhem kombinované terapie je aplikace inhibitorů kinázové aktivity EGFR spolu s monoklonálními protilátkami cílenými do vazebného místa pro ligand tohoto receptoru (EGF) – viz obr. 2. Nejznámějšími z dnes užívaných monoklonálních protilátek proti EGFR je chimerická humanizovaná myší IgG1 monoklonální protilátka cetuximab (Erbitux) a lidská monoklonální IgG2 protilátka panitumumab (Vectibix). Z pohledu molekulárních mechanizmů signalizace přes EGFR se zdá být nepravděpodobné, že by použití blokačních protilátek proti vazebnému místu pro EGF vedlo k výraznějšímu zlepšení stavu u pacientů s aktivačními mutacemi EGFR. Takto zmutovaný receptor je totiž většinou konstitutivně aktivní, je schopen aktivovat sám sebe bez nutnosti vazby extracelulárního ligandu, jehož navázání protilátka blokuje. Naopak u pacientů, jejichž nádor nevykazuje žádné mutace v EGFR, popřípadě vykazuje pouze zmnožení lokusu pro EGFR, by kombinovaná terapie mohla vést k výraznější regresi nádoru oproti léčbě pouze jedním z těchto typů léčiv. Efekt tohoto typu léčby byl však zatím přesvědčivě dokumentován pouze na myších xenotransplantátech [23] a v klinických studiích fáze I [24–26]. Výsledky následných fází klinických testů by měly být známy v nejbližších letech.
Posledním druhem kombinované terapie, o kterém se zde zmíníme, je podávání inhibitorů kinázové aktivity EGFR spolu s inhibitory VEGFR (vascular endothelial growth factor receptor). Signální dráhy vedoucí přes tyto dva receptory sdílejí některé efektorové molekuly, blokace jedné z drah částečně potlačuje aktivitu dráhy druhé prostřednictvím autokrinní signalizace. Na myších xenotransplantátech se prokázalo, že inhibice obou drah zároveň přináší lepší výsledky při potlačování růstu nádorů než inhibice každé z těchto drah samostatně [27]. V rámci klinických studií byla aplikována kombinace erlotinibu a bevacizumabu (Avastin). V klinické studii fáze II byla aplikována standardní chemoterapie (docetaxel nebo pemetrexed), identická chemoterapie v kombinaci s bevacizumabem anebo léčba erlotinibem v kombinaci s bevacizumabem. Došlo k mírnému zlepšení, úspěšnost léčby byla 12,2 % (chemoterapie), 12,5 % (chemoterapie + bevacizumab) a 17,9 % (erlotinib + bevacizumab); mírně se prodloužilo i přežití a oproti chemoterapii vykazovala léčba kombinací erlotinibu a bevacizumabu méně vedlejších příznaků [28]. V současnosti jsou dokončovány studie využívající kombinovanou léčbu erlotinibem a bevacizumabem u pacientů, kteří již podstoupili chemoterapii. Blokády obou receptorů při podávání 300 mg denně je schopen také ZD6474 (vandetanib). V klinické studii fáze II byly zjištěny lepší výsledky v porovnání s podáváním erlotinibu, ZD6474 má však poměrně silné vedlejší účinky [29]. Nízké dávky ZD6474 (100 mg denně) se ale ukazují být účinné v kombinaci s docetaxelem [30], v současnosti proto probíhá klinická studie fáze III. Jde prozatím o jediný inhibitor EGFR, u nějž byly prokázány pozitivní účinky při současné aplikaci s chemoterapií.
Kontraindikace podávání inhibitorů kinázové aktivity EGFR
I když ještě v březnu 2010 P. Zatloukal v Medical Tribune uvádí, že „není k dispozici žádný spolehlivý marker, na jehož základě by před zahájením biologické léčby bylo možno identifikovat nemocné, u nichž biologická léčba bude neúčinná a neměla by být podána“ [7], opak je pravdou. Už roku 2005 byly v PLoS Medicine a v New England Journal of Medicine zveřejněny dvě studie [31,32] potvrzující, že rezistence k inhibitorům kinázové aktivity EGFR je asociována s některými mutacemi v tomto genu. Těmto mutacím jsou připisovány také často pozorované relapsy zhoubného bujení, ke kterým často dochází 6–12 měsíců po zahájení terapie erlotinibem či gefitinibem. Mezi nejrozšířenější a nejvíce studované mutace potlačující účinek nízkomolekulárních inhibitorů kinázové aktivity EGFR patří T790M v exonu 20, tj. v kinázové doméně předmětného receptoru. Je třeba podotknout, že i mutace T790M má aktivující účinek podstatně zvyšující kinázovou aktivitu EGFR. Jde o mutační hotspot známý i u ostatních receptorových kináz – T315I v ABL, T674I v PDGFRA a T670I v kináze KIT. Na rozdíl od analogické mutace T315I v kináze ABL, která působí jako sterická zábrana pro vazbu imatinibu, EGFR T790M příliš nemění afinitu molekuly EGFR ke gefitinibu či erlotinibu. Způsobuje ale, že EGFR s navázaným gefitinibem nebo erlotinibem je opět schopen vázat ATP podobně jako normální receptor bez asociovaného inhibitoru. Receptor s mutací T790M je tedy schopen kinázové aktivity i s navázaným inhibitorem (erlotinibem či gefitinibem) [33]. Sekundární mutace T790M se objevuje u plných 50% pacientů s rezistencí k podávaným inhibitorům kinázové aktivity EGFR, typický případ z České republiky popisují např. Jančaříková et al [34]. Bylo zjištěno, že řada nemalobuněčných karcinomů obsahuje už před léčbou malé procento buněk nesoucích tuto mutaci. Během léčby erlotinibem či gefitinibem dochází k pozitivní selekci buněčných populací nesoucích mutaci T790M, a ty proto zodpovídají za brzký relaps nemoci. Podobně se chovající, ale výrazně vzácnější jsou mutace D761Y, L747S a T854A. V současné době jsou proto ve vývoji látky zaměřené na inhibici EGFR nesoucího tyto problematické mutace, zejména pak nejrozšířenější z nich, T790M. Většina nově vyvíjených inhibitorů nahradila vratnou inhibici (jakou poskytuje například gefitinib či erlotinib) inhibicí nevratnou, kdy dochází k Michaelově adiční reakci na klíčový cysteinový zbytek C797 v kinázové doméně EGFR. Tato vlastnost umožňuje efektivnější kompetici o ATP vazebné místo v porovnání s již klinicky používanými reverzibilními inhibitory. Mezi látkami úspěšně testovanými in vitro jsou neratinib (HKI-272), XL647, BIBW 2992 a PF-00299804. Výsledky jejich testování in vivo však lze prozatím označit spíše za problematické, zejména s ohledem na množství nežádoucích účinků v důsledku silné paralelní inhibice normálního nemutovaného EGFR ve zdravých tkáních [35,36], a zčásti dokonce s ohledem na poměrně překvapivou rezistenci k některým z nich v tumorech s amplifikovaným mutantním EGFR [37]. Dosud vyvíjené inhibitory kinázové aktivity EGFR byly založeny pouze na strukturách podobných 4-anilonoquinazolinovému jádru. Je možné, že lepších výsledků bude do budoucna dosaženo s kovalentním ireverzibilním inhibitorem založeným na struktuře pyrimidinu. Knihovnu tohoto typu inhibitorů recentně testovala laboratoř P. A. Jänneho s velmi nadějnými výsledky, kdy se jim podařilo identifikovat hned několik látek se specifitou o tři řády vyšší k mutantnímu EGFR T790M oproti normální molekule. Tyto látky byly velmi účinné na myších modelech plicních nádorů způsobených mutací EGFR T790M [33]. Je naděje, že zmíněné inhibitory nebudou doprovázeny dnes častými vedlejšími účinky pozorovanými při léčbě karcinomů erlotinibem či gefitinibem. Svým zaměřením jen na somaticky mutovanou molekulu nebudou působit na EGFR receptor nacházející se v tělních epitelech nesoucích normální nemutovaný EGFR. Alternativně se pro nádory s rezistencí k gefitinibu a erlotinibu testuje užití inhibitorů tyrozinové kinázy MET, inzulinového receptoru IGF-1R a heat shock proteinu Hsp90.
Kontraindikace při změnách v expresi/aktivitě jiných molekul
Již výše jsme se zmínili o mutacích v genu KRAS, které jsou negativním prediktorem odpovědi na terapii erlotinibem či gefitinibem [18]. Tyto somatické mutace se však většinou nevyskytují u pacientů s aktivačními mutacemi EGFR. Výrazně závažnějším problémem bývá získaná rezistence na erlotinib či genitinib, ke které dochází buďto mutacemi přímo v EGFR popsanými výše, anebo amplifikací kinázy MET, popřípadě zvýšením hladiny ligandu této kinázy zvaného HGF (hepatocyte growth factor) – viz obr. 3. Amplifikace kinázy MET byla nedávno opakovaně zjištěna u ~20 % pacientů s plicním karcinomem rezistentních k léčbě gefitinibem [38–40]. Jde o patrně druhý nejčastější mechanizmus rezistence, ~50 % pacientů z identických kohort neslo již výše zmíněnou mutaci T790M, tyto dvě aberace tedy vysvětlují tři čtvrtiny zjištěných rezistencí.
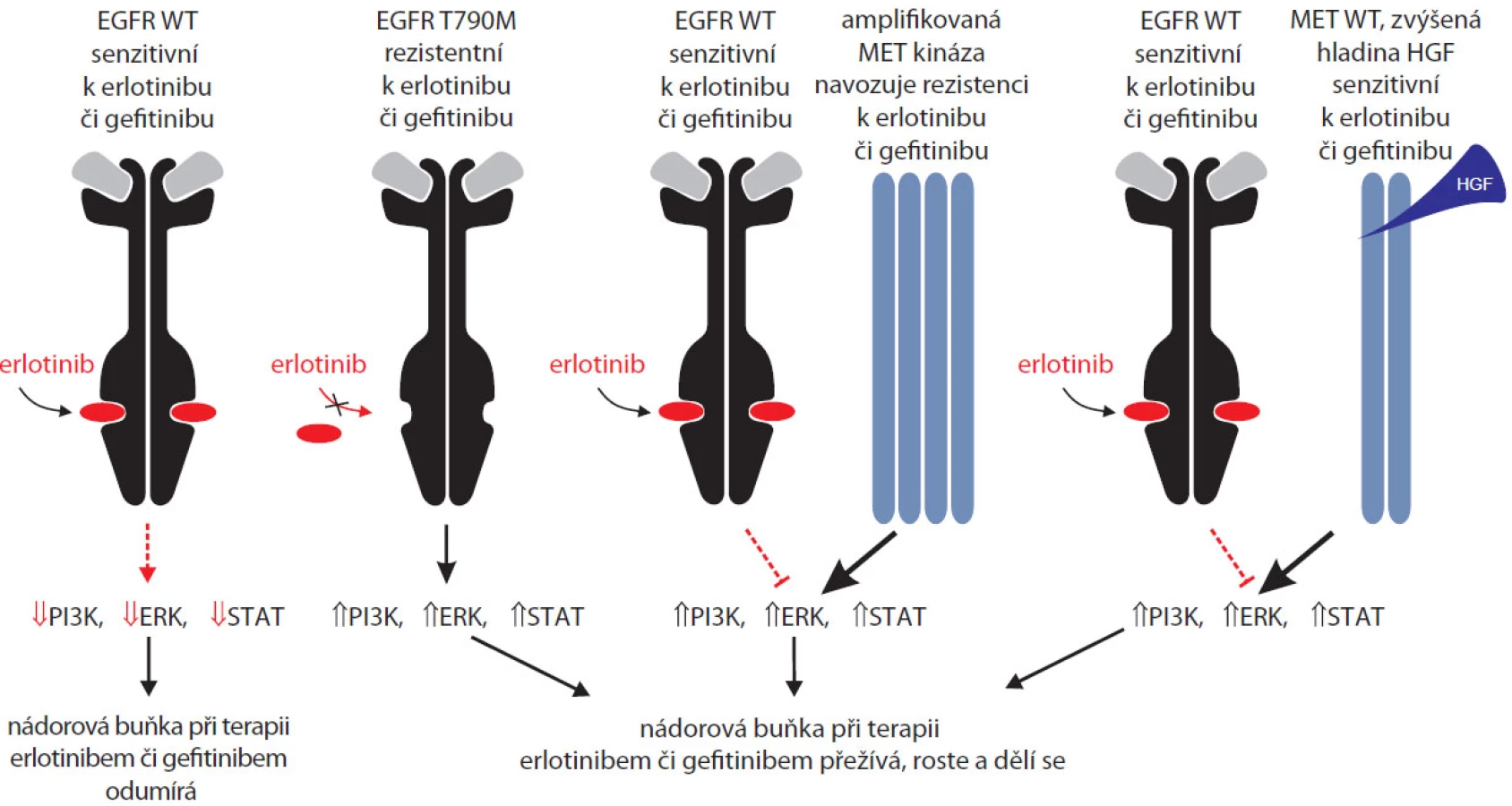
Signalizace přes kinázu MET vede k podobným efektorovým molekulám jako v případě EGFR (PI3K, ERK1/2, rodina STAT), zvýšení aktivity kinázy MET je proto schopno kompenzovat farmakologickou inhibici EGFR. Amplifikace genu pro MET kinázu vede k aktivaci Src kinázy, podobně jako k ní dochází při aktivaci kinázy EGFR prostřednictvím mutace T790M. Hyperaktivní MET má ale větší schopnost fosforylovat a aktivovat Src než hyperaktivní EGFR [41]. Využití inhibitorů MET kinázy v kombinaci s gefitinibem vede k velmi slibným výsledkům u pacientů s nádory rezistentními k samotnému gefitinibu. Na základě popsaného mechanizmu někteří autoři zároveň argumentují pro využití již známých inhibitorů Src kinázy, například dasatinibu [41]. Klinická studie účinnosti dasatinibu u pacientů s nemalobuněčným karcinomem plic neodpovídajícím na gefitinib či erlotinib právě probíhá. Variantní léčbou u pacientů s rezistencí ke gefitinibu a s vysokou hladinou HGF, ligandu MET kinázy, je aplikace nově vyvinutého inhibitoru TAK-701 v kombinaci s gefitinibem [42]. Zjištění amplifikace genu MET by v současné době měla být kontraindikací k monoterapii gefitinibem či erlotinibem.
Mezi další alternativní dráhy aktivované v buňkách nemalobuněčného karcinomu plic s rezistencí ke gefitinibu či erlotinibu patří aktivace signalizace přes IgF-1Rb/IRS-1 prostřednictvím ztráty IGF-vazebných proteinů. K těmto mutacím dochází v buňkách s normálním EGFR, rezistence k léčbě gefitinibem či erlotinibem je tedy způsobena využitím jiné signální dráhy [43], nikoliv amplifikací či mutací EGFR.
Detekce mutací v EGFR
Senzitivita k erlotinibu či gefitinibu je ovlivňována především mutacemi EGFR, screening počtu kopií EGFR pomocí FISH (fluorescence in situ hybridization) či CISH (chromogenic in situ hybridization) je sice možný, ale detekci počtu kopií daného receptoru nelze na rozdíl od mutačního screeningu použít jako samostatný prediktor odpovědi na plánovanou terapii nízkomolekulárními inhibitory kinázové aktivity EGFR [44]. V naprosté většině dosud zaznamenaných případů jde o mutace somatické, běžnými technikami je tedy možno je identifikovat pouze z biopsie plicního karcinomu. Navíc by měla být na minimum omezena kontaminace biopsie buňkami obklopujícími tumor a buňkami imunitního systému, které samozřejmě danou somatickou mutaci v naprosté většině případů nenesou a jejich zvýšené zastoupení ve vzorku určeném k izolaci DNA může výrazně zkreslit výstup v podobě klasického Sangerova sekvencování.
S ohledem na problematický odběr biopsií u pacientů s plicními karcinomy a i na komplikace s následnou detekcí [44,45] je žádoucí zásadní změna dosud používaných metod. Zlepšení v podobě cílené selekce nádorových buněk představuje na některých českých pracovištích již používaná laserová mikrodisekce [46,47]. Přesto zůstává problémem načasování odběru vzorků, které by v ideálním případě měly být dostupné jednak před zahájením léčby a jednak při relapsech. V současnosti se ale všude ve světě potýká léčba těchto nemocí se třemi zásadními problémy:
- a) nedostupnost biopsie karcinomu při relapsu onemocnění,
- b) i když je materiál dostupný, dochází často k efektu alelického vyředění (allelic dilution) a s tím spojeným dramatickým snížením senzitivity tradičních sekvenačních technik,
- c) přítomnost mutací mimo molekulu EGFR, například v již zmíněné kináze MET, což dále zvyšuje finanční nároky na provedení analýzy.
Alternativou k biopsii je do budoucna snad použití qPCR DxS analýzy z pacientovy plazmy. Zároveň existují snahy o zvýšení senzitivity používaných metod, které by předešly falešně negativním výsledkům způsobeným efektem alelického vyředění. Jako alternativy zatím přicházejí do úvahy HPLC (high performance liquid chromatography), tandemová hmotnostní spektrometrie anebo vysokodenzitní pikolitrové reaktory [47]. Bohužel, na rutinní využití zmíněných metod si v klinické praxi budeme muset ještě chvíli počkat.
Závěr
V České republice se v posledních letech rozšířil screening nemalobuněčného karcinomu plic na aktivující mutace v genu EGFR a navazující podávání kinázového inhibitoru erlotinib [9,46–49]. V této souvislosti je nutno podotknout, že mutace T790M je schopna zcela eliminovat hypersenzitivitu k erlotinibu či gefitinibu způsobenou aktivujícími mutacemi (např. L858R či delE746-A750). Podávání kinázových inhibitorů erlotinib či gefitinib pacientům, jejichž nádor exprimuje EGFR nesoucí zároveň oba typy mutací, nemá žádný účinek na dobu přežití. Podobně podávání kinázových inhibitorů erlotinib či gefitinib nemá žádný účinek na dobu přežití pacientů, u nichž se objevila mutace T790M až sekundárně, v průběhu léčby. Mutace T790M (popřípadě i D761Y, L747S a T854A) jsou zcela jednoznačným, spolehlivým prediktivním diagnostickým markerem, na základě jehož přítomnosti je možno před zahájením biologické léčby identifikovat nemocné, u nichž bude biologická léčba erlotinibem či gefitinibem neúčinná a neměla by být podána.
Tato práce byla podpořena výzkumným záměrem 3. LF UK v Praze MSM0021620814.
This research was supported by a research goal of the 3rd Faculty of Medicine, Charles University in Prague No. MSM0021620814.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do bi omedicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
RNDr. Petr Heneberg, Ph.D.
3. lékařská fakulta UK
Ruská 87
100 00 Praha 10
e-mail: Petr.Heneberg@lf3.cuni.cz
Obdrženo/Submitted: 7. 10. 2010
Přijato/Accepted: 2. 2. 2011
Sources
1. Zásady cytostatické léčby maligních onkologických onemocnění. Česká onkologická společnost ČLS JEP, Česká republika. 11. vydání [aktualizováno 1. srpna 2010; citováno 4. října 2010]. Dostupné z: http://www.linkos.cz/odbornici/info_praxe/standardy.php? t=1.
2. Kris MG, Natale RB, Herbst RS et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA 2003; 290(16): 2149–2158.
3. Fukuoka M, Yano S, Giaccone G et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial). J Clin Oncol 2003; 21(12): 2237–2246.
4. Thatcher N, Chang A, Parikh P et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet 2005; 366(9496): 1527–1537.
5. Shepherd FA, Rodrigues Pereira J, Ciuleanu T et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med 2005; 353(2): 123–132.
6. Lee DH, Han JY, Yu SY et al. The role of gefitinib treatment for Korean never-smokers with advanced or metastatic adenocarcinoma of the lung: a prospective study. J Thorac Oncol 2006; 1(9): 965–971.
7. Zatloukal P. Cílená léčba nemalobuněčného karcinomu plic (NSCLC). Medical Tribune 2010; 6 [aktualizováno 28. března 2010; citováno 4. října 2010]. Dostupné z: http://www.tribune.cz/clanek/17123.
8. Hajdúch M. Mutace genů EGFR1 a KRAS. In: V. dny diagnostické, prediktivní a experimentální onkologie & II. sympózium o cílené biologické léčbě, 25.–27. 11. 2009, Olomouc. Onkologie 2009; 3 (Suppl B): 11–12.
9. Svoboda M, Fabian P, Slabý O et al. Cílená léčba bronchioloalveolárního plicního adenokarcinomu inhibitory tyrozinkinázové aktivity EGFR: kazuistika klinicky promptní a výrazné odpovědi a přehled literatury. Klin Onkol 2010; 23(4): 224–230.
10. Lynch TJ, Bell DW, Sordella R et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004; 350(21): 2129–2139.
11. Paez JG, Jänne PA, Lee JC et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304(5676): 1497–1500.
12. Asahina H, Yamazaki K, Kinoshita I et al. A phase II trial of gefitinib as first-line therapy for advanced non-small cell lung cancer with epidermal growth factor receptor mutations. Br J Cancer 2006; 95(8): 998–1004.
13. Sutani A, Nagai Y, Udagawa K et al. Gefitinib for non-small-cell lung cancer patients with epidermal growth factor receptor gene mutations screened by peptide nucleic acid-locked nucleic acid PCR clamp. Br J Cancer 2006; 95(11): 1483–1489.
14. Inoue A, Suzuki T, Fukuhara T et al. Prospective phase II study of gefitinib for chemotherapy-naive patients with advanced non-small-cell lung cancer with epidermal growth factor receptor gene mutations. J Clin Oncol 2006; 24(21): 3340–3346.
15. Sunaga N, Tomizawa Y, Yanagitani N et al. Phase II prospective study of the efficacy of gefitinib for the treatment of stage III/IV non-small cell lung cancer with EGFR mutations, irrespective of previous chemotherapy. Lung Cancer 2007; 56(3): 383–389.
16. Paz-Ares L, Sanchez JM, Garcia-Velasco A et al. A prospective phase II trial of erlotinib in advanced non-small cell lung cancer (NSCLC) patients (p) with mutations in the tyrosine kinase (TK) domain of the epidermal growth factor receptor (EGFR). ASCO Meeting Abstract 20. 6. 2006. J Clin Oncol 2006; 24 (Suppl 18): 7020.
17. Shepherd FA, Tsao MS. Unraveling the mystery of prognostic and predictive factors in epidermal growth factor receptor therapy. J Clin Oncol 2006; 24(7): 1219–1220.
18. Pao W, Wang TY, Riely GJ et al. KRAS mutations and primary resistance of lung adenocarcinoma to gefitinib or erlotinib. PLoS Med 2005; 2(1): e17.
19. Herbst RS, Giaccone G, Schiller JH et al. Gefitinib in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: a phase III trial-INTACT 2. J Clin Oncol 2004; 22(5): 785–794.
20. Giaccone G, Herbst RS, Manegold C et al. Gefitinib in combination with gemcitabine and cisplatin in advanced non-small-cell lung cancer: a phase III trial-INTACT 1. J Clin Oncol 2004; 22(5): 777–784.
21. Gatzemeier U, Pluzanska A, Szczesna A et al. Phase III study of erlotinib in combination with cisplatin and gemcitabine in advanced non-small-cell lung cancer: the Tarceva Lung Cancer Investigation Trial. J Clin Oncol 2007; 25(12): 15451–552.
22. Herbst RS, Prager D, Hermann R et al. TRIBUTE: a phase III trial of erlotinib hydrochloride (OSI-774) combined with carboplatin and paclitaxel chemotherapy in advanced non--small-cell lung cancer. J Clin Oncol 2005; 23(25): 5892–5899.
23. Huang S, Armstrong EA, Benavente S et al. Dual-agent molecular targeting of the epidermal growth factor receptor (EGFR): combining anti-EGFR antibody with tyrosine kinase inhibitor. Cancer Res 2004; 64(15): 5355–5362.
24. Preston GG, Calvo E, Papadopoulos K et al. Multi-targeted inhibition of the epidermal growth factor (EGFR) and vascular endothelial growth factor receptor (VEGFR) pathways: a phase I study of cetuximab (C), erlotinib (E), and bevacizumab (B) in patients with solid tumors. ASCO Meeting Abstracts 20. 6. 2006; 24 (Suppl 18): 3005.
25. Baselga J, Schoffski P, Rojo F et al. A phase I pharmacokinetic (PK) and molecular pharmacodynamic (PD) study of the combination of two anti-EGFR therapies, the monoclonal antibody (MAb) cetuximab (C) and the tyrosine kinase inhibitor (TKI) gefitinib (G), in patients (pts) with advanced colorectal (CRC), head and neck (HNC) and non-small cell lung cancer (NSCLC). ASCO Meeting Abstracts 20. 6. 2006; 24 (Suppl 18): 3006.
26. Naret CL, Ramalingam S, Beattie L et al. Total blockade of the epidermal growth factor receptor with the combination of cetuximab and gefitinib: a phase I study for patients with recurrent non-small cell lung cancer (NSCLC). ASCO Meeting Abstracts 20. 6. 2006; 24 (Suppl 18): 17045.
27. Guy SP, Ashton S, Hughes G et al. Gefitinib (Iressa, ZD1839) enhances the activity of the novel vascular targeting agent ZD6126 in human colorectal cancer models. Clin Cancer Res 2003; 9 : 6143.
28. Fehrenbacher L, O’Neill V, Belani CP et al. A phase II, multicenter, randomized clinical trial to evaluate the efficacy and safety of bevacizumab in combination with either chemotherapy (docetaxel or pemetrexed) or erlotinib hydrochloride compared with chemotherapy alone for treatment of recurrent or refractory non-small cell lung cancer. ASCO Meeting Abstracts 20. 6. 2006; 24 (Suppl 18): 7062.
29. Natale RB, Bodkin S, Govindan R et al. ZD6474 versus gefitinib in patients with advanced NSCLC: final results from a two-part, double-blind, randomized phase II trial. ASCO Meeting Abstracts 20. 6. 2006; 24 (Suppl 18): 7000.
30. Heymach JV, Johnson BE, Prager DE et al. A phase II trial of ZD6474 plus docetaxel in patients with previously treated NSCLC: follow-up results. ASCO Meeting Abstracts 20. 6. 2006; 24 (Suppl 18): 7016.
31. Pao W, Miller WA, Politi KA et al. Acquired resiteance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005; 2(3): e73.
32. Kobayashi S, Boggon TJ, Dayram T et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med 2005; 352(8): 786–792.
33. Zhou W, Ercan D, Chen L et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009; 462(7276): 1070–1074.
34. Jančaříková D, Pešek M, Benešová L et al. Acquired resistance of pulmonary adenocarcinoma to initially successful targeted therapy due to EGFR mutation T790M. Anticancer Res 2007; 27(4A): 1879–1882.
35. Li D, Shinamura T, Ji H et al. Bronchial and peripheral murine lung carcinomas induced by T790M-L858R mutant EGFR respond to HKI-272 and rapamycin combination therapy. Cancer Cell 2007; 12(1): 81–93.
36. Besse B, Eaton KD, Soria JC et al. Neratinib (HKI-272), an irreversible pan-ErbB receptor tyrosine kinase inhibitor: preliminary results of a phase 2 trial in patients with advanced non-small cell lung cancer. Presented at the 20th Annual EORTC-NCI-AACR Symposium, Geneva, Switzerland 2008, abstract 203.
37. Ercan D, Zejnullahu K, Yonesaka K et al. Amplification of EGFR T790M causes resistance to an irreversible EGFR inhibitor. Oncogene 2010; 29(16): 2346–2356.
38. Engelman JA, Zejnullahu K, Mitsudomi T et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316(5827): 1039–1043.
39. Arteaga CL. HER3 and mutant EGFR meet MET. Nat Med 2007; 13(6): 675–677.
40. Turke AB, Zejnullahu K, Wu YL et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010; 17(1): 77–88.
41. Yoshida T, Okamoto I, Okamoto W et al. Effects of Src inhibitors on cell growth and epidermal growth factor receptor and MET signaling in gefitinib-resistant non-small cell lung cancer cells with acquired MET amplification. Cancer Sci 2010; 101(1): 167–172.
42. Okamoto W, Okamoto I, Tanaka K et al. TAK-701, a humanized monoclonal antibody to hepatocyte growth factor, reverses gefitinib resistance induced by tumor-derived HGF in non-small cell lung cancer with an EGFR mutation. Mol Cancer Ther 2010; 9(10): 2785–2792.
43. Guix M, Faber AC, Wang SE et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Clin Invest 2008; 118(7): 2609–2619.
44. Sholl LM, Xiao Y, Joshi V et al. EGFR mutation is a better predictor of response to tyrosine kinase inhibitors in non-small cell lung carcinoma than FISH, CISH, and immunohistochemistry. Am J Clin Pathol 2010; 133(6): 922–934.
45. Jänne PA. Challenges of detecting EGFR T790M in gefitinib/erlotinib-resistant tumours. Lung Cancer 2008; 60 (Suppl 2): S3–S9.
46. Dziechciarková M, Berkovcová J, Trojanec R et al. Využití laserové mikrodisekce pro přípravu komplexních vzorků z nádorové tkáně pro účely mikrogenomických analýz. Klin Onkol 2007; 20 : 323–329.
47. Dziechciarková M, Berkovcová J, Trojanec R et al. Využití laserové mikrodisekce pro přípravu komplexních vzorků z nádorové tkáně pro účely mikrogenomických analýz. Klin Onkol 2006; 19 (Suppl): 355–360.
48. Mendoza L. Targeted therapies in the treatment of advanced non-small-cell lung cancer: update. Klin Onkol 2009; 22(4): 131–138.
49. Berkovcová J, Hajdúch M, Dziechciarková M et al. Predikce účinnoti tyrozinkinázových inhibitorů EGFR1 v léčbě nemalobuněčných plicních karcinomů. Klin Onkol 2006; 19(3): 171–176.
50. Wingo PA, Cardinez CJ, Landis SH et al. Long-Term Trends in Cancer Mortality in the United States, 1930–1998. Cancer 2003; 97 (Suppl 12): 3133–3275.
51. Dušek L, Mužík J, Kubásek M et al. Epidemiologie zhoubných nádorů v České republice, verze 7.0. Masarykova univerzita, Brno, ČR 2007 [aktualizováno 14. ledna 2011; citováno 14. ledna 2011]. Dostupné z: http://www.svod.cz.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2011 Issue 2
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- Prehľad potenciálnych onkomarkerov detekcie skorých fáz rakoviny vaječníkov
- Metastázujúci choriokarcinóm u 26-ročnej ženy – kazuistika
- Adjuvantní terapie u karcinomu rekta
- Comments on the TNM Classification of Malignant Tumours – 7th Edition