
Castlemanova choroba
Castleman Disease
Backgrounds:
Castleman disease is a rare non-clonal lymphoproliferative disorder with the etiopathogenesis not yet thoroughly clarified. Clinically, either unicentric (localized) or multicentric (generalized) forms are recognized while, histopathologically, hyaline-vascular, plasma-cell and mixed variants of the disease exist. These types vary one from another in their clinical courses and, importantly, in methods of therapeutic management. While the unicentric hyaline-vascular form usually manifests as benign growth of a single lymph node and treatment response to complete surgical excision reaches up to 100%, the multicentric plasmocellular variant is an aggressive disease with generalized symptoms, laboratory abnormalities and the need for systemic therapy.
Aim:
The paper provides an overview of information on Castleman disease from its clinical and histopathological signs to diagnostic and therapeutic options. It deals with the role of cytokines and HHV-8 virus infection in the disease pathophysiology and is supplied with ample pictorial documentation of radiographic findings including ultrasonography, computed tomography and hybrid imaging by positron emission tomography (PET) in combination with simultaneously taken full-body computed tomography (CT) scans, the so called PET/CT. We also present photographs of histological specimens taken from an HIV and HHV-8 negative patient with the plasmocellular multicentric form.
Conclusions:
Consequent to its low incidence, Castleman disease is often misdiagnosed or diagnosed with a delay. Therefore, it is always necessary to include this rare condition in differential diagnostics of lymphadenopathy, microcytic anemia as well as B-symptoms (night sweats, fevers and weight loss). In conclusion, we also stress the significance of full-body PET/CT scanning during staging and treatment response evaluation.
Key words:
Castleman disease – lymphadenopathy – tocilizumab – interleukin – HHV-8 – ultrasonography – computed tomography – PET scan
This publication was prepared as part of the Internal Grant Agency of the Czech Republic grants NT 12215-4, the Ministry of Education, Youth and Sports of the CR grants MSM0021622434, LC06027 and the Ministry of Health of the Czech Republic’s Internal Grant Agency grants IGA MZd NT11154, NT12130, NT12215 and NS10408.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Submitted:
4. 5. 2011
Accepted:
27. 5. 2011
Authors:
P. Szturz 1; M. Moulis 2; Z. Adam 1; R. Šlaisová 3; R. Koukalová 4; Z. Řehák 4; P. Volfová 1; R. Hájek 1; J. Mayer 1
Authors‘ workplace:
Interní hematoonkologická klinika, LF MU a FN Brno
1; Ústav patologie, LF MU a FN Brno
2; Radiologická klinika, LF MU a FN Brno
3; Oddělení nukleární medicíny, PET centrum Masarykova onkologického ústavu, Brno
4
Published in:
Klin Onkol 2011; 24(6): 424-434
Category:
Reviews
Overview
Východiska:
Castlemanova choroba je vzácné neklonální lymfoproliferativní onemocnění s dosud ne zcela jasnou etiopatogenezí. Klinicky rozlišujeme unicentrickou (lokalizovanou) a multicentrickou (generalizovanou) formu, histopatologicky pak hyalinně-vaskulární, plazmocelulární a smíšenou variantu nemoci. Tyto typy se navzájem liší nejen klinickým průběhem onemocnění, ale především způsobem léčebného ovlivnění. Zatímco unicentrická hyalinně-vaskulární forma se obvykle manifestuje ve formě benigního zvětšování lymfatické uzliny a kompletní chirurgická excize dosahuje 100 % léčebných odpovědí, tak multicentrická plazmocelulární varianta je agresivní choroba s celkovými příznaky, laboratorními odchylkami a nutností systémové léčby.
Cíl:
Tato práce přináší přehled informací o Castlemanově chorobě od klinických a histopatologických znaků nemoci až po možnosti diagnostiky a léčby. Věnuje se úloze cytokinů a infekce HHV-8 virusem v patofyziologii nemoci a je doplněna bohatou obrazovou dokumentací radiografických nálezů, zahrnujících zobrazení pomocí ultrasonografie, výpočetní tomografie a hybridního zobrazování pozitronovou emisní tomografií (PET) v kombinaci se současně snímanými celotělovými skeny výpočetní tomografie (CT), tzv. PET/CT vyšetření. Prezentujeme zde i fotografie histologických preparátů HIV a HHV-8 negativního pacienta s plazmocelulární multicentrickou formou.
Závěr:
Vzhledem k nízké incidenci bývá Castlemanova choroba špatně nebo opožděně diagnostikována. Je třeba ji proto vždy zahrnout do diferenciální diagnostiky lymfadenopatie, mikrocytární anémie, ale i B-symptomů (noční poty, febrilie, hubnutí). Závěrem rovněž zdůrazňujeme význam celotělového PET/CT skenování při stážovacích vyšetřeních a hodnocení léčebné odpovědi u pacientů.
Klíčová slova:
Castlemanova choroba – lymfadenopatie – tocilizumab – interleukin – HHV-8 – ultrasonografie – výpočetní tomografie – PET skener
Úvod
V roce 1956 popsal dr. Benjamin Castleman soubor pacientů s hyperplazií mediastinálních lymfatických uzlin připomínající thymom [1]. Během dalších 50 let se v anglické literatuře objevily pojmy jako angiofollicular (mediastinal) lymph node hyperplasia, giant lymph node hyperplasia [2], lymph node or lymphoid hamartoma [3], angiomatous lymphoid hyperplasia, benign (giant) lymphoma [3,4] či follicular lymphoreticuloma [5]. Zastřešující se pro ně stalo eponymum Castlemanova choroba (Castleman disease – CD).
V roce 1972 byl klinicky a histopatologicky vymezen hyalinně-vaskulární a méně častý plazmocelulární typ. Nemoc navíc může postihovat pouze jednu uzlinovou lokalitu anebo se může šířit dále pod obrazem generalizované lymfadenomegalie. U pacientů s CD se tedy můžeme setkat se 3 histomorfologickými (hyalinně-vaskulární, plazmocelulární a smíšený) a 2 klinickými (unicentrický a multicentrický) typy [5]. Tyto typy se navzájem liší nejen klinickým průběhem onemocnění, ale i prognózou a především pak způsobem léčebného ovlivnění. Zatímco unicentrická lokalizovaná forma CD je vyléčitelná excizí postižených lymfatických uzlin, multicentrický typ je často refrakterní k léčbě, a to i přes použití kortikoidů nebo chemoterapie [6].
Nemoc většinou postihuje oblast krku (42 %), mediastina (31 %) a břicha (23 %), častý je však výskyt i v oblasti axil [7,8]. Prevalence tohoto onemocnění není známa, odhadovaný počet případů v USA sahá od 30 000 k 100 000 [9]. Popsána byla vyšší incidence CD u mužů [10]. Stěžejní roli v patofyziologii Castlemanovy choroby hraje dysregulace cytokinové sítě, ale jak uvádíme dále v textu, je tato problematika složitější a souvisí i s herpesvirovými infekcemi. Cytokiny jsou solubilní proteiny a peptidy regulující prostřednictvím receptorů různé procesy svého mikroprostředí [11]. Jako klíčový cytokin u tohoto chorobného stavu byl označen interleukin-6 [12], což, jak uvádíme dále v textu, má praktický dopad na léčbu CD.
CD je v české literatuře opomíjeným tématem, pomocí databáze Bibliographia medica Čechoslovaca jsme v české literatuře nalezli pouze 11 článků zabývajících se touto tematikou. Tato přehledná práce předkládá čtenářům souhrnný pohled na tuto chorobu, zabývá se jak klinickými a histopatologickými znaky, tak i diagnostikou a léčbou CD. Text je doplněn bohatou obrazovou dokumentací zahrnující nálezy z výpočetní tomografie a ultrasonografie a dále i fotografie histologických preparátů. Uvedeny jsou rovněž výsledky získané pomocí PET/CT vyšetření, tedy hybridního zobrazování pozitronovou emisní tomografií (PET) v kombinaci se současně snímanými celotělovými skeny pomocí výpočetní tomografie (CT), které byly u jednoho pacienta korelovány s nálezem in vivo.
Klinické rozdělení
CD může být limitována na jednu uzlinovou oblast, nebo ji může onemocnění přesahovat. Na základě toho pak rozlišujeme unicentrickou (lokalizovanou) a multicentrickou (generalizovanou) formu CD.
Unicentrická forma
Lokalizovaná forma se klinicky manifestuje ve formě benigního, nebolestivého, pomalého zvětšování lymfatické uzliny. Příznaky však může působit při kompresi přilehlých struktur nebo je její nález náhodný při rutinní lékařské prohlídce [13]. Podle různých studií mělo 31–50 % pacientů asymptomatickou masu, u 33–69 % se vyskytly příznaky [13–15].
Unicentrická forma CD postihuje v 46–70 % případů mediastinum, v 3–39 % břicho a v 10–15 % periferní uzlinové oblasti [13,16–19]. Úplné chirurgické odstranění má kurativní potenciál [13]. Medián nejčastějšího výskytu nemoci leží mezi 20 a 35 lety věku [17,20], muži i ženy jsou postiženi rovnoměrně [21]. Podle jedné studie 23 nemocných (48 %) byly ženy a 25 (52 %) byli muži [13]. U této formy onemocnění nebyla popsána zvýšená smrtnost [21].
Multicentrická forma
Generalizovaná forma CD byla jako multicentrická označena v roce 1985 [22]. Poprvé však byla popsána již v roce 1978 s charakteristickými znaky v podobě lymfadenopatie, systémových příznaků, organomegalie, progresivního klinického průběhu s maligním potenciálem [15]. Jedná se tedy o atypické lymfoproliferativní onemocnění charakterizované systémovou lymfadenopatií a celkovými zánětlivými příznaky [23] (obr. 1).
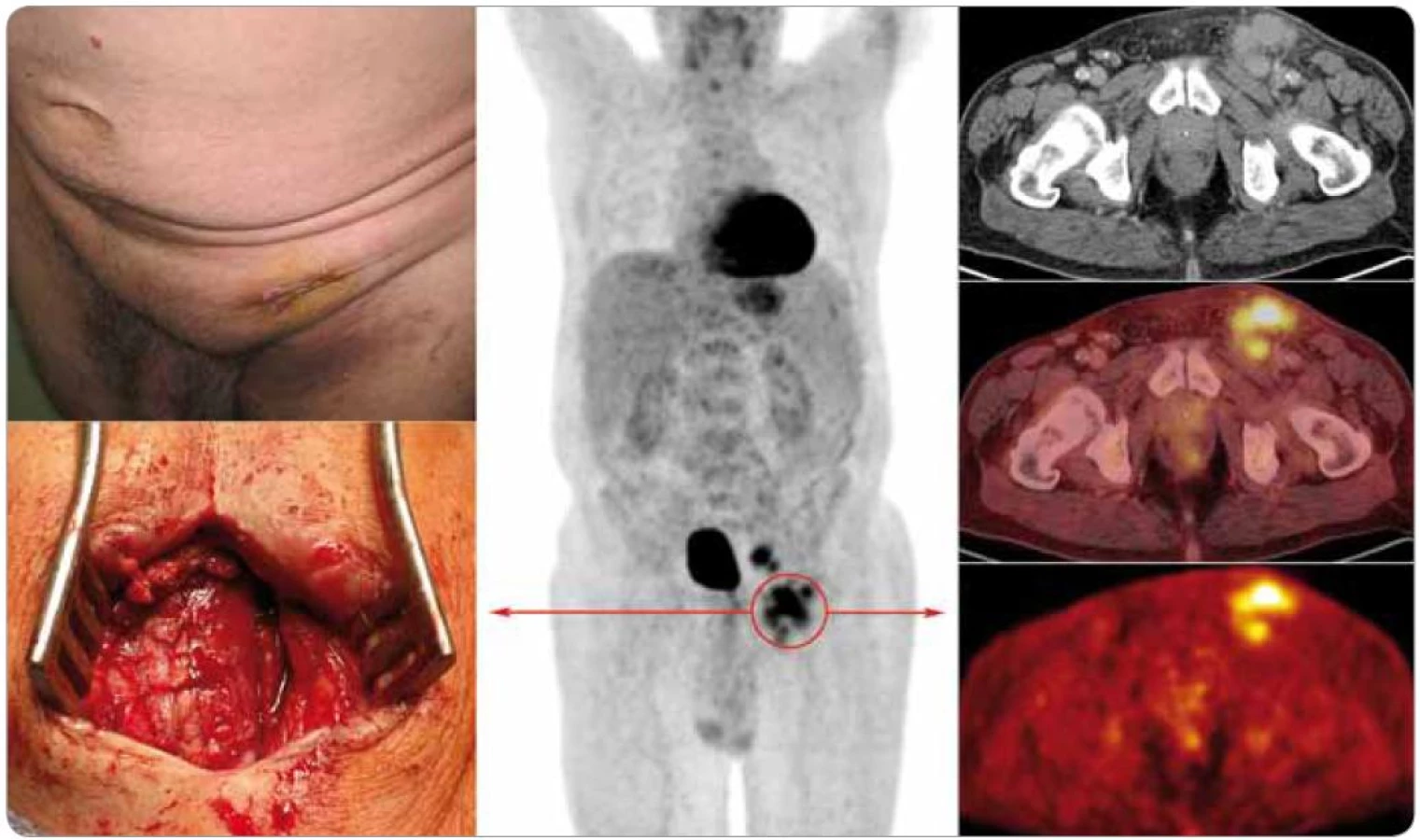
Multicentrická forma je zastoupena pouze v 10 % případů CD, nemocní jsou většinou starší než v případě unicentrické formy, pohlaví jsou zastoupena stejně [21]. Medián věku pacientů leží podle různých autorů mezi 48 a 57 lety [13,17,20]. Ve 13 % je asociovaná s Kaposiho sarkomem [24].
Histopatologické rozdělení
Na základě histopatologického nálezu lze Castlemanovu chorobu rozdělit na hyalinně-vaskulární, plazmocelulární a smíšený typ. Uvážíme-li navíc existenci 2 klinických forem onemocnění, můžeme se setkat minimálně s 6 typy onemocnění s více či méně podobnými projevy. Na pomyslné škále agresivity onemocnění by se na straně benigních lézí nacházela hyalinně-vaskulární unicentrická CD a na protilehlé straně torpidní plazmocelulární multicentrická varianta s typickými systémovými příznaky. Hyalinně-vaskulární varianta zodpovídá za většinu, tedy 80–90 % všech případů CD, u zbývajících pacientů se jedná o plazmocelulární nebo smíšenou formu [3,4].
Hyalinně-vaskulární varianta
Hyalinně-vaskulární, v typických případech asymptomatická varianta CD bývá nalézána u 74–91 % pacientů s lokalizovanou formou CD, vzácný je výskyt u multicentrické choroby [8,13,16,25,26]. Klinicky se hyalinně-vaskulární varianta projevuje jako pomalu rostoucí masa. Muži a ženy jsou postiženi rovnoměrně. Medián výskytu leží ve 4. dekádě [8,16,21,26–28]. Postižené uzliny dosahují velikosti asi 6–7 cm (rozptyl 1–25 cm) [21].
Hyalinně-vaskulární typ, někdy také označovaný jako angiofolikulární typ, je morfologicky charakterizován folikuly a malými hyalinizovanými germinálními centry s intrafolikulární proliferací kapilár [29]. Při histologickém vyšetření nalézáme v uzlině zmnožené folikuly, které jsou rozložené v kůře i ve dřeni uzliny. Folikuly mohou být velké nebo malé, v jednotlivých případech však mají přibližně stejnou velikost a často obsahují dvě i více germinálních center. Hyalinně změněná germinální centra vykazují depleci malých lymfocytů a zmnožení folikulárních dentritických buněk. Plášťová zóna těchto folikulů je rozšířená a je tvořená typicky koncentricky uspořádanými prsteny malých lymfocytů (toto uspořádání připomíná slupku cibule). Nápadná je radiálně penetrující vaskularizace germinálních center, často s jednou prominující sklerotickou cévou. Koncentricky uspořádaná plášťová zóna společně s prominující sklerotickou cévou připomíná vzhledem lízátko. Další důležitou známkou je vaskulární proliferace mezi folikuly, často s perivaskulární hyalinizací [2,13,30].
Plazmocelulární varianta
Agresivnější, plazmocelulární forma CD se vyskytuje nejčastěji ve 3. dekádě, muži a ženy jsou postiženi rovnoměrně [21]. Tento subtyp bývá nalézán asi u 9–33 % pacientů s lokalizovanou formou CD, dominantní je totiž výskyt u multicentrické CD [3,4,8,13,16,21,25,26,29]. U lokalizované formy se tvoří agregát lymfatických uzlin, na rozdíl od hyalinně-vaskulární lokalizované varianty se solitární zvětšenou uzlinou [16].
V literatuře je uváděno, že plazmocelulární varianta je často doprovázena imunodeficiencí, infekcemi, Kaposiho sarkomem (13 %), non-Hodgkinskými lymfomy (18 %), hemangiomy, plazmocytomy, karcinomy kolon, ledvin, štítné žlázy, dále i smíšenou chorobou pojiva, ale i POEMS syndromem [31–35]. Plazmocelulární a smíšená varianta je častěji sdružena se systémovými příznaky a abnormálními laboratorními nálezy než varianta hyalinně-vaskulární [8,16,27]. Pacienti mají často systémové projevy, jako je horečka, únavnost, nechutenství, hubnutí a noční poty, hepatomegalii, splenomegalii, kožní vyrážku [23,24,36].
U plazmocelulární varianty, unicentrické i multicentrické, byla nalezena zvýšená sérová hladina interleukinu-6 zodpovídající za průvodní systémové příznaky [37–42]. Na rozdíl od multicentrické formy vyžadující systémovou léčbu má chirurgická excize u unicentrické plazmocelulární varianty kurativní potenciál [2].
Na rozdíl od hyalinně-vaskulární varianty má plazmocelulární CD méně charakteristické rozlišovací histologické znaky. Pro plazmocelulární variantu CD je typické rozšíření interfolikulárních oblastí, ve kterých jsou velmi četné polyklonální plazmatické buňky tvořící souvislá pole. Plazmatické buňky bývají obvykle zralé a lze je identifikovat podle jejich excentricky uloženého jádra s loukoťovitě uspořádaným chromatinem a podle perinukleárního projasnění. V některých případech, zejména u pacientů HHV-8 pozitivních, jsou přítomny i nezralé a atypické plazmatické buňky, včetně plazmablastů. Plazmatické buňky se v hojné míře nalézají také ve dřeni uzliny. Folikuly mohou být hyperplastické, se zvětšenými polarizovanými germinálními centry a rozšířenou plášťovou zónou, ve které se obvykle nachází populace větších plazmablastů (plazmocytoidních imunoblastů). Dále bývají interfolikulárně zmnožené drobné epiteloidní venuly. Hyalinně-vaskulární změny, pokud jsou přítomny, bývají vyjádřeny v malé míře [2,13,30].
Jelikož tyto znaky nejsou specifické, je nezbytné vyloučení dalších jednotek, které mohou napodobit plazmocelulární CD, tedy B-lymfomy, vzácné plazmocytomy, reaktivní lymfadenopatie asociované s infekcí, autoimunitní choroby, jako je revmatoidní artritida, nebo reaktivní lymfadenopatie asociované s jinými imunodeficity [2,25,43]. Histologické nálezy plazmocelulární varianty u HIV a HHV-8 negativního pacienta jsou zachyceny na obr. 2 a 3.
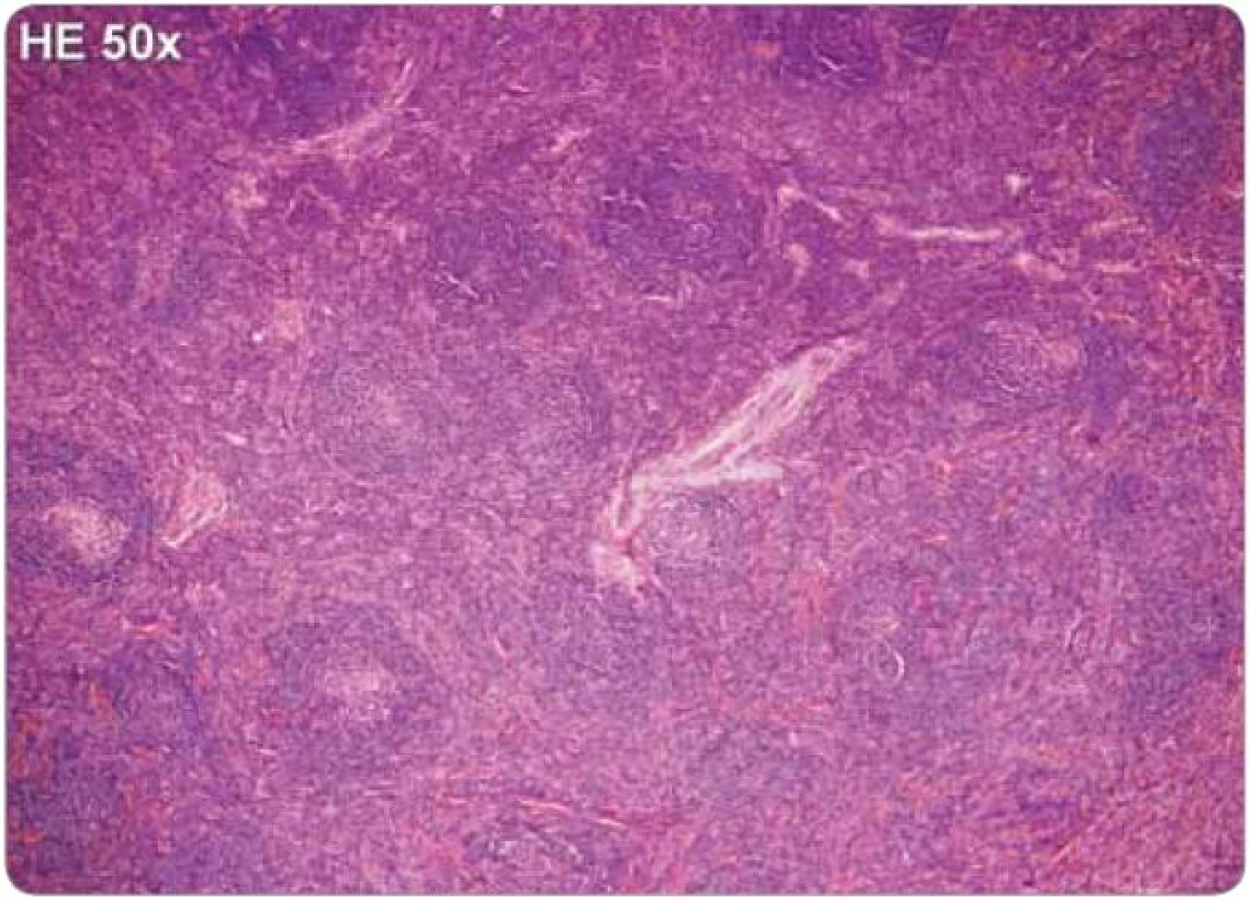
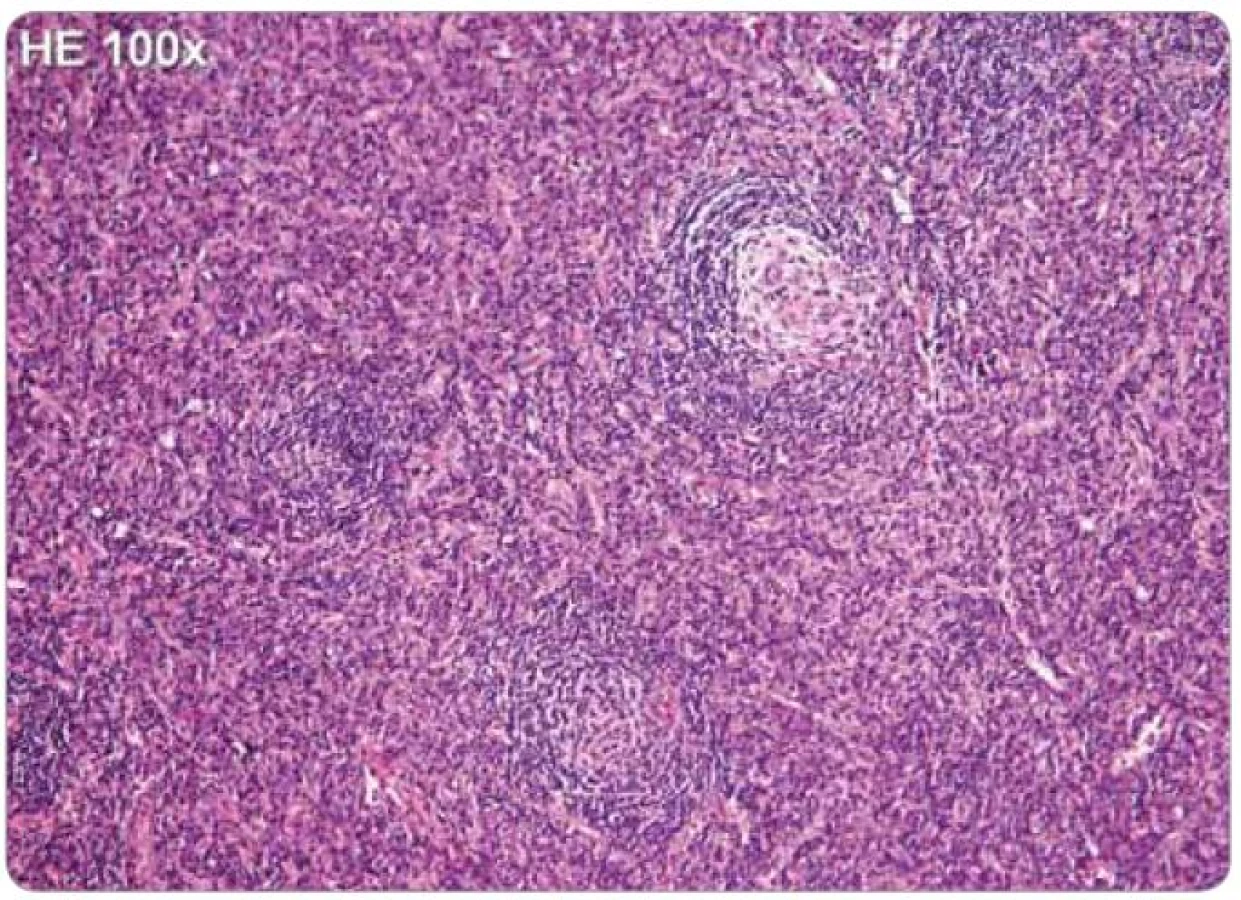
Příznaky onemocnění
Pacient, kterého popsal před více než 50 lety dr. Castleman, trpěl dlouhou dobu horečkami a slabostí a až s odstupem mnoha let mu byla zjištěna mediastinální expanze na skiagramu [44]. Příznaky CD vyplývají jednak z místního růstu patologické rezistence projevujícího se různými kompresivními syndromy a jednak z aktivace cytokinových signalizačních drah vedoucí k projevům systémovým. Zatímco unicentrické formy s nejčastějším histopatologickým korelátem ve formě hyalinně-vaskulárních lézí bývají často asymptomatické či oligosymptomatické s příznaky komprese okolních struktur (bolest, dušnost, kašel, chrapot, porucha polykání, průjmy a další), tak u plazmocelulární varianty multicentrického CD jsou celkové příznaky téměř konstantním jevem. Hyalinně-vaskulární varianta je asociována s celkovými příznaky pouze v méně než 10 % případů [21].
U multicentrické CD je nejčastějším příznakem horečka a asi polovina nemocných si stěžuje na hubnutí a noční poty [24,45]. Za klinické projevy je zodpovědná dysregulovaná trvalá nadprodukce interleukinu-6 B-buňkami zárodečných center hyperplastických lymfatických uzlin [23,39]. Bylo zjištěno, že hladina interleukinu-6 nejvíce koreluje právě s aktivitou multicentrické CD [29]. Nadprodukce interleukinu-6 je zřejmě rovněž zodpovědná za špatný nutriční stav pacientů s multicentrickou CD [23] a má snad význam i při rozvoji kachexie a chronického zánětu [23,46]. Splenomegalie je u multicentrické CD vyjádřena asi u 33–79 %, hepatomegalie se vyskytuje téměř výlučně se splenomegalií a pozorujeme ji asi u 63 % pacientů [21]. Popsán však byl i případ unicentrické hyalinní varianty CD, která se klinicky chovala jako multicentrická se systémovými příznaky (anémie, hubnutí, elevace CRP, zvýšená sedimentace erytrocytů, POEMS syndrom, plazmocytóza ve dřeni) [29]. V retrospektivní studii 52 pacientů s CD však Ye et al popsali 4 pacienty s multicentrickou CD (1 hyalinně-vaskulární a 3 plazmocelulární varianty), z nichž žádný netrpěl systémovými příznaky [13].
Laboratorní nálezy
U pacientů s plazmocelulární nebo smíšenou formou CD bývají přítomny abnormální laboratorní výsledky zahrnující anémii, hypoalbuminemii, hypocholesterolemii, hypergamaglobulinemii a elevaci proteinů akutní fáze [23,24,36]. U všech nemocných s multicentrickou CD byla zjištěna zvýšená sedimentace, v 90 % anémie, trombocytopenie nebo elevace transamináz u 2/3 vyšetřených pacientů [24]. V případě unicentrické plazmocelulární varianty se anémie vyskytuje asi v 90 % případů a zvýšená sedimentace asi u 80 % nemocných [21].
Mikrocytární anémie zřejmě souvisí s interleukinem-6 a hepcidinem, tedy stejně jako u ostatních anémií typu chronických chorob, a má vztah k probíhajícímu chronickému zánětu v organizmu [3]. Interleukin-6 (ale ne interleukin-1 nebo tumor necrosis factor alpha) přímo zvyšuje jaterní produkci hepcidinu, klíčového regulátoru v metabolizmu železa [47,48]. Hepcidin blokuje uvolňování železa z jaterních makrofágů a snižuje absorpci železa ve střevě, čímž omezuje dodávku železa do vyvíjejících se erytrocytů v kostní dřeni [3,49]. Nadměrná produkce hepcidinu je zřejmě zodpovědná za mikrocytární anémii u pacientů s CD a tato produkce může být tlumena blokováním signalizační cesty interleukinu-6 pomocí tocilizumabu [50]. U multicentrické plazmocelulární varianty se může objevit i autoimunitní hemolytická anémie [3].
Patogeneze
CD se vyznačuje hyperproliferací specifických B-buněk produkujících cytokin interleukin-6. Ačkoliv bylo navrhováno několik imunologických mechanizmů zahrnujících nadprodukci interleukinu-6 a infekci virusem HHV-8, zůstává etiopatogeneze onemocnění neznámá [45]. Nalezena byla dokonce i spojitost mezi CD a infekcí HIV a virusem hepatitidy C [51–53]. Jiní autoři považují hyalinně-vaskulární CD za reaktivní folikulární hyperplazii, která se vyvinula jako odpověď na neznámý antigenní stimulus [54].
Vztah mezi systémovými příznaky plazmocelulární varianty CD a faktorem secernovaným postiženými uzlinami byl rozpoznán v roce 1989 [42]. Tento B-buňky stimulující cytokin dostal později označení interleukin-6 a jeho role v systémové manifestaci CD se opakovaně potvrdila [35,37,39,41]. Interleukin-6 je cytokin s širokým rozsahem biologických účinků. Reguluje imunologické reakce, zánětlivé odpovědi a hematopoézu. Dysregulovaná nadprodukce interleukinu-6 B-buňkami zárodečných center je zahrnuta do patogeneze plazmocelulární CD [6,39,55]. Ukázalo se, že interleukin-6 indukuje u myší stav podobný CD [38]. Má stimulační účinek v procesu vyzrávání B-buněk a rovněž je dáván do souvislosti s maligními lymfoproliferacemi, jako je mnohočetný myelom a jiné lymfomy [29,56].
Virus HHV-8 má úzký vztah k CD, zejména k multicentrické CD u HIV pozitivních pacientů [21]. HHV-8 produkuje virusový analog interleukinu-6 (50% podobnost genu), který má přímý efekt, ale také zvyšuje hladinu lidského interleukinu-6 u infikovaného hostitele [57,58]. Virusový interleukin-6 sdílí funkční vlastnosti s lidským korelátem, a aniž by tvořil komplex s receptorem, může se vázat s glykoproteinem gp130, což je vlastní přenašeč receptorového signálu interleukinu-6. Tocilizumab (monoklonální protilátka proti receptoru pro interleukin-6, viz dále) není schopen blokovat tuto formu nereceptorové aktivace gp130. Přesto léčba s tocilizumabem byla s to zlepšit příznaky a laboratorní parametry HHV-8 pozitivních pacientů s CD. Lze tedy usuzovat, že i když virusový interleukin-6 může být u těchto pacientů exprimován, je to stále lidský interleukin-6, který je dominantně zodpovědný za systémové příznaky u HHV-8 pozitivních pacientů s multicentrickou CD [23]. Virusový interleukin-6 má hematopoetický i angiogenní efekt [59], přesto je ještě třeba přesnou roli virusového interleukinu-6 v patogenezi HHV-8 příbuzných nemocí objasnit [60].
V patogenezi hyalinně-vaskulárního typu hraje důležitou roli vaskulární endoteliální růstový faktor (vascular endothelial growth factor, VEGF), který přispívá k výrazné vaskulární proliferaci tohoto subtypu onemocnění [43].
Diagnostika
Pro diagnostiku onemocnění je rozhodující histologický nález [4]. Na CD by se mělo myslet vždy u pacientů s Kaposiho sarkomem a POEMS syndromem, tedy ještě před biopsií uzlin, které jinak bývají tím prvním, co nás navede ke správné diagnóze [21].
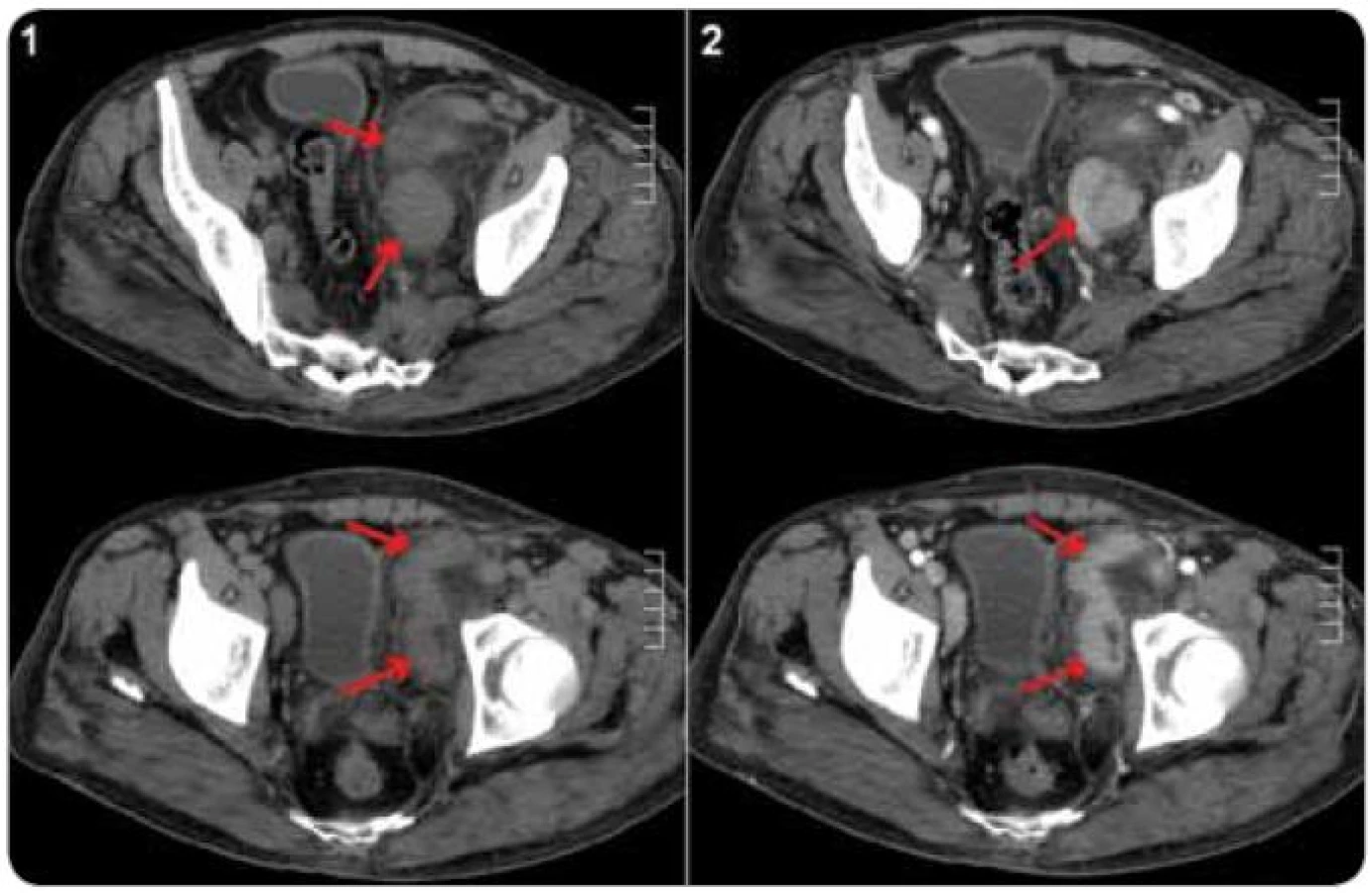
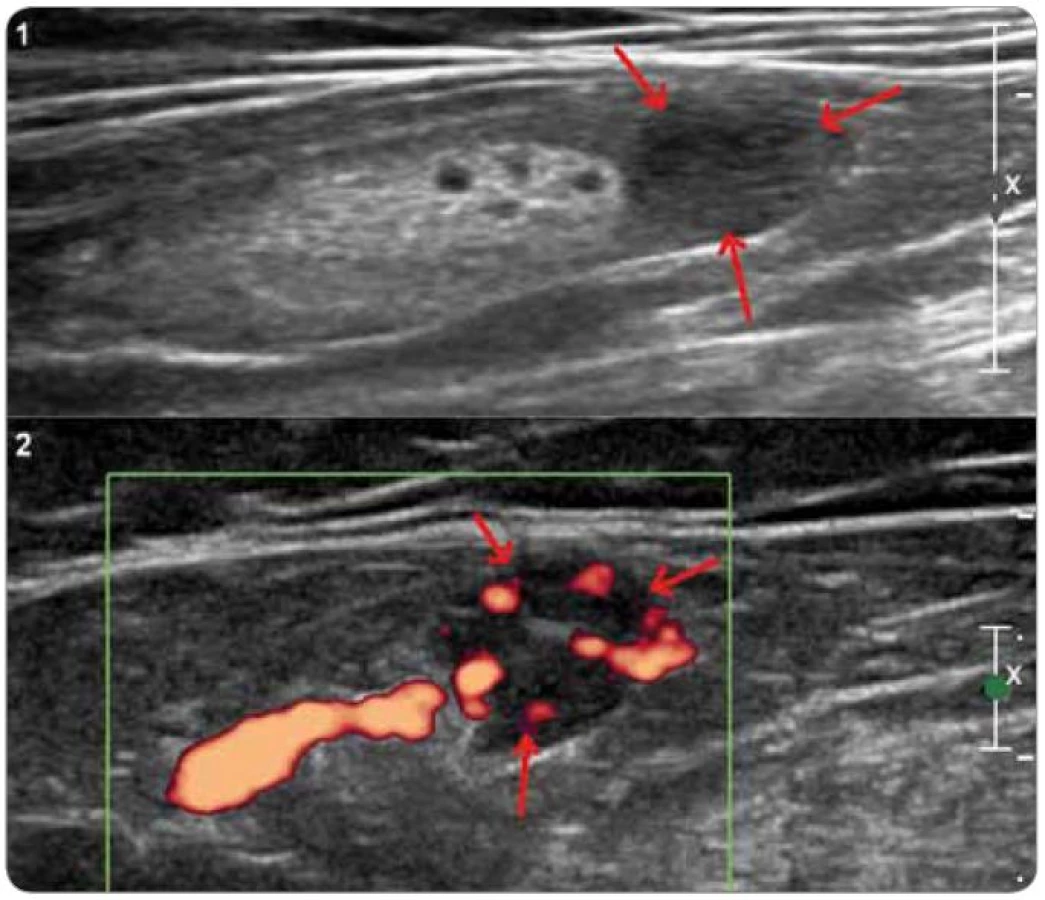
Pro odlišení unicentrické CD od multicentrické CD je nutné vyšetření krevního obrazu, CRP a jaterních testů a dále radiologické metody pro zjištění případné krční, hrudní, břišní a pánevní lymfadenopatie [21,61] (obr. 4 a 5). Testování interleukinu-6 a případné pozitivity virusu HHV-8 sice nejsou rutinně prováděna ve všech laboratořích, jejich stanovení má však význam u pacientů se systémovými projevy, aby mohlo být rozhodnuto o optimální léčbě [21].
Vychytávání galium citrátu na scintigrafii je nekonzistentní, jeho využití je proto omezeno [21,62,63]. Objevily se zprávy o použití galiové scintigrafie k odlišení CD od lymfomu, kdy akumulace radiofarmaka byla v lézích CD menší než v případě lymfomových infiltrátů [64]. Dnes je však od tohoto vyšetření upuštěno a je nahrazeno zobrazováním pozitronovou emisní tomografií (PET), případně v kombinaci se současně snímanými celotělovými skeny výpočetní tomografie (CT), tzv. PET/CT vyšetřením. Aplikováno je radiofarmakum 2-18F-fluoro-2-deoxy-D-glukóza (18F-FDG).
18F-FDG-PET a 18F-FDG-PET/CT přes svou vysokou senzitivitu pro diagnostiku neoplazií nejsou vhodné k odlišení CD od jiných benigních nebo maligních lymfoproliferací [65]. CD jakožto benigní choroba vykazuje mírnou až střední akumulaci radiofarmaka nad lymfatickými uzlinami [65,66]. Lymfomy s nižším stupněm malignity však mohou někdy také vykazovat nízké vychytávání značené glukózy na PET skenech [67]. PET/CT je cenné vyšetření při rozlišování unicentrické CD od multicentrické CD [65] a pro sledování léčebné odpovědi, jak rovněž ukazují naše výsledky (obr. 6).
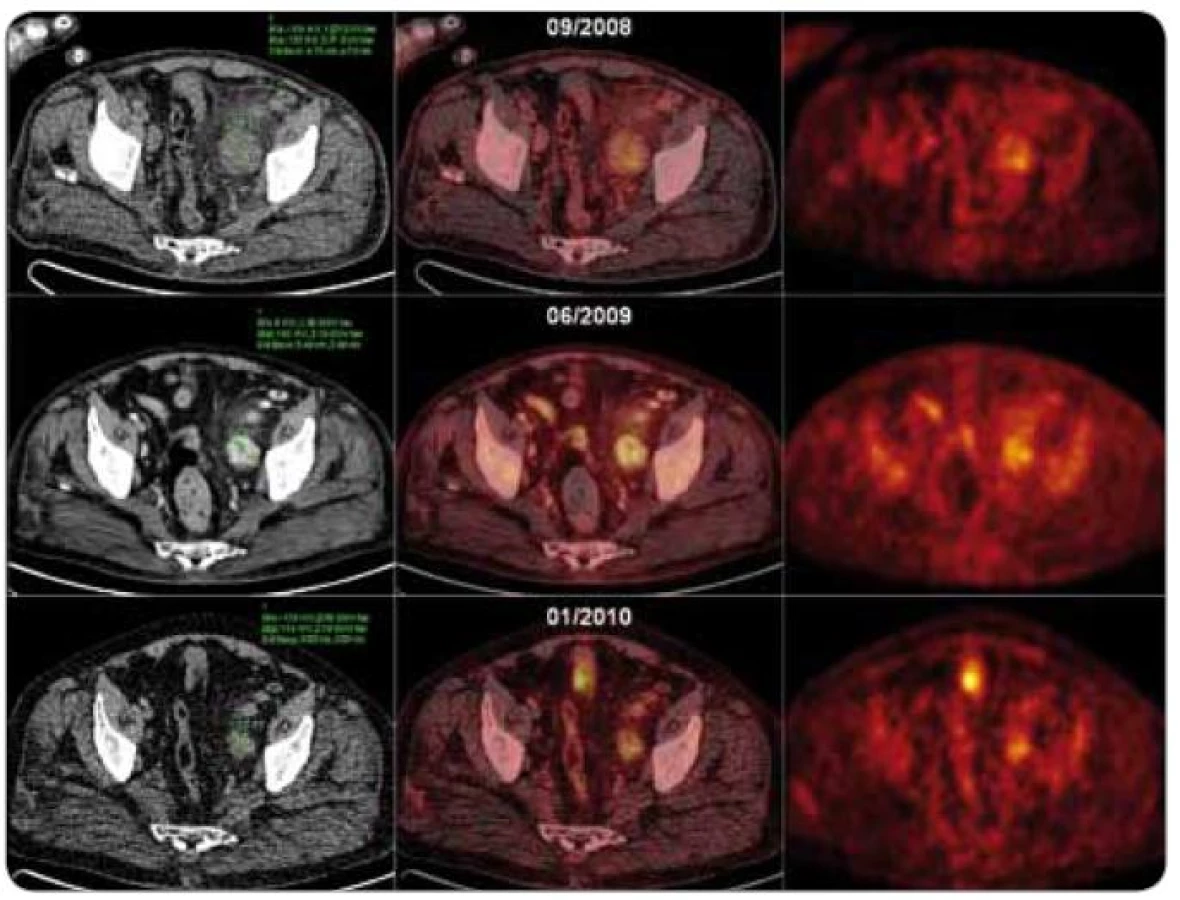
CD je důležitou součástí diferenciální diagnostiky příčiny nenádorové lymfadenopatie [2] a mikrocytární anémie, kdy je po negativní endoskopii doporučováno provedení screeningového CT hrudníku a břicha [2,3].
Terapie
Doporučení k léčbě CD lze jen obtížně formulovat, neboť neexistuje žádná standardní terapie. Literatura je tvořena popisy jednotlivých případů a malých souborů a až na výjimky (viz níže) nejsou k dispozici výsledky randomizovaných studií [6,13,40,68–72].
Chirurgická léčba
Kompletní chirurgická excize patologické masy je v případě unicentrické choroby téměř vždy kurativní, a to jak u hyalinně-vaskulární, tak plazmocelulární varianty [36,73–79]. Pětiletého přežití pak dosahuje 100 % pacientů [16,80]. K recidivě však může dojít po subtotální nebo parciální resekci. V jedné studii podstoupilo všech 48 pacientů s lokalizovanou CD kompletní chirurgickou resekci a všichni přežili s excelentní prognózou; po dobu sledování (22–115 měsíců) se neobjevily žádné příznaky nemoci [13].
Yoshizaki et al představili ve své práci 2 pacienty se smíšenou formou CD, jednoho s unicentrickou a druhého s multicentrickou variantou. U obou pacientů byla zvýšená sérová hladina interleukinu-6. Zatímco chirurgická resekce lokalizované formy vedla k úpravě klinického stavu pacienta a k normalizaci hladiny interleukinu-6, v případě multicentrické choroby neměl invazivní výkon na největší lymfatické uzlině žádný terapeutický efekt s přetrvávající elevací interleukinu-6 [39].
Výsledky chirurgického debulkingu u multicentrické CD nedosáhly ani dle další dostupné literatury významnějších léčebných odpovědí [20]. Jeden pacient s hyalinně-vaskulární formou multicentrické CD však přežil dlouhodobě (sledování 82 měsíců) po cervikální a axilární disekci lymfatických uzlin [13]. U operativy hyalinně-vaskulární CD je navíc třeba velké opatrnosti pro přítomnost rozvinuté vaskularizace tumoru, kdy resekce bývá často spojena s velkými perioperačními ztrátami krve [73].
Radioterapie
Radioterapie byla použita s různou mírou úspěchu u neresekabilních nádorů [45,81]. Může být rovněž alternativou u unicentrické CD pro pacienty, kteří nejsou schopni podstoupit chirurgický výkon [21]. Chronowski et al uvádí soubor 18 případů s CD, kteří byli léčeni radioterapií. U 13 pacientů (72 %) bylo dosaženo kompletní nebo parciální remise. Pouze 3 z těchto 13 pacientů (23 %) měli multicentrickou CD, účinnost u těchto pacientů však mohla být ovlivněna současně podanou adjuvantní chemoterapií nebo steroidy [45].
Chemoterapie
Vzorem pro cytoredukční léčbu u multicentrické choroby se staly režimy určené pro terapii non-Hodgkinských lymfomů [21]. Mezi nejčastěji používané režimy se řadí CHOP (cyklofosfamid, vinkristin, doxorubicin, prednison) a CVAD (cyklofosfamid, vinkristin, doxorubicin, dexametazon), oba využívané se smíšenými úspěchy. Publikované malé studie udávají počet léčebných odpovědí mezi 50 a 67 % [20,45]. Formou kazuistik bylo popsáno i použití dalších režimů, nejednalo se však o kontrolované studie, a je tedy obtížné z těchto popisů dělat širší závěry [82].
Glukokortikoidy
Dle publikovaných popisů případů mají kortikosteroidy potenciál navodit u některých nemocných remisi onemocnění [82]. 6 z 15 pacientů (40 %) bylo v jedné studii léčeno glukokortikoidy, u 2 došlo ke kontrole nemoci při dlouhodobém podávání kortikoidů, u 4 však nebylo dosaženo žádné trvající odpovědi [22]. Parciální remise byla dokumentována u 3 z 5 pacientů (60 %) s multicentrickou CD léčených výlučně prednisonem [83]. Prolongovaná léčba nízkodávkovaným prednisonem (10 mg denně) byla úspěšná u 2 pacientů s multicentrickou hyalinně-vaskulární formou [84], ale selhala u pacienta s agresivní multicentrickou plazmocelulární variantou [85].
Dlouhodobé užívání kortikosteroidů může být spojeno s vysokým rizikem bakteriální infekce u pacientů s multicentrickou formou, kdy bylo popsáno mnoho případů úmrtí na sepsi při terapii [14,22].
Interferon alfa
Interferon alfa má antivirové a imunomodulační vlastnosti [21,86]. V literatuře lze nalézt popisy případů, u nichž měl interferon alfa výborný léčebný efekt [82]. U dvou pacientů s multicentrickou CD asociovanou s HIV infekcí byla popsána dlouhodobá remise při dlouhodobé terapii interferonem alfa. U prvního pacienta byl rovněž pozitivní záchyt HHV-8 virusu, byl léčen dávkou 5 mil jednotek 3krát týdně [87]. HHV-8 status druhého pacienta není znám, léčen byl dávkou 5 mil jednotek týdně [88].
All-trans retinová kyselina (ATRA)
ATRA má antiproliferativní účinky a snižuje intenzitu interleukin-6-dependentní signalizace [82]. Předpokládá se, že tyto vlastnosti se spolupodílely na léčebném efektu v popsaném případu úspěšné léčby u HIV a HHV-8 negativní ženy [89].
Thalidomid
Stejně jako interferon alfa a ATRA má i thalidomid imunomodulační účinky, navíc může snižovat produkci interleukinu-6 a má rovněž antiangiogenní vlastnosti [21]. Výhoda podávání thalidomidu spočívá v dobrém bezpečnostním poměru léku, absenci významné myelotoxicity, a to i při několikaletém podávání, jak dokládá případ pacienta s Crohnovou chorobou, který toleroval tento lék po dobu 5 let při zachování dobré účinnosti [90]. Limitující však může být rozvoj periferní neuropatie [91].
Vynikající účinky thalidomidu jsou doloženy několika kazuistikami. U jednoho HIV a HHV-8 pozitivního muže s Kaposiho sarkomem došlo ke zlepšení celkového stavu, zvýšení počtu destiček a negativnímu restagingovému vyšetření z kostní dřeně po 38 týdnech léčby s 200 mg thalidomidu denně; léčba byla na začátku doplněna o etoposid [92]. Jedna HIV negativní žena, jejíž HHV-8 status neznáme, měla kompletní remisi cytopenie, ascitu a perikardiálního výpotku a pouze přetrvávající lymfadenopatii po dobu 40 měsíců na monoterapii thalidomidem (iniciální dávka 300 mg byla redukována na 200 mg pro rozvoj mírné periferní neuropatie). Po tuto dobu byla pacientka asymptomatická, začala opět pracovat, aniž by ji neuropatie limitovala [93,94].
Stary et al dosáhli kombinovaným režimem s thalidomidem (200 mg, později přechod na 100 mg) a rituximabem kompletní klinické a radiologické remise plazmocelulární CD u HIV a HHV-8 pozitivního muže [95]. Ke zlepšení klinického i laboratorního statusu a regresi lymfadenomegalie a hepatosplenomegalie došlo dále u pacientky s POEMS syndromem asociovaným s hyalinně-vaskulární CD, u které byla popsána 20měsíční léčba 200 mg thalidomidu denně doplněná v prvních 8 měsících o dexametazon [96]. Thalidomidem bylo dosaženo remise nefrotického syndromu v jednom případu CD [97] a úspěchu bylo dosaženo i u HIV a HHV-8 negativní pacientky se smíšenou formou multicentrické CD asociované s paraneoplastickým pemfigem [98].
Biologická léčba
Cílená biologická léčba CD se odvíjí od úlohy interleukinu-6 v patofyziologii nemoci, je zastoupena monoklonálními protilátkami proti receptoru pro interleukin-6 nebo proti samotnému cytokinu a u HIV negativních pacientů dosahuje excelentních terapeutických výsledků [12]. Tyto látky výrazně zmírňují příznaky a upravují biochemické abnormality multicentrické CD, ačkoliv se projevy většinou po vysazení léčby vrátí [6,23,40,68].
První pacient, který byl léčen myší monoklonální protilátkou proti interleukinu-6, byl 27letý pacient s multicentrickou plazmocelulární formou CD. Tato terapie vedla k rychlému ústupu obtíží, poklesu CRP, zvýšení hematokritu, počtu trombocytů a hladiny albuminu. Po 84 dnech byla léčba přerušena, což bylo následováno recidivou příznaků a patologického laboratorního nálezu [40]. V následujících letech byla vyvinuta chimerická protilátka (hybridní myší a lidská) proti receptoru pro interleukin-6, tato hybridizace měla zabránit vzniku autoprotilátek při opakovaném podávání [99].
Tocilizumab (Ro-Actemra™) je humanizovaná monoklonální protilátka proti receptoru pro interleukin-6. První prospektivní studie popisuje zkušenosti s 60týdenní léčbou u 28 pacientů s plazmocelulární nebo smíšenou variantou multicentrické CD. Aplikováno bylo vždy 8 infuzí po 8 mg/kg tocilizumabu každé 2 týdny. Po 16 týdnech došlo ke zmenšení lymfadenomegalie, poklesu zánětlivých parametrů, vzestupu hemoglobinu, albuminu, celkového cholesterolu, HDL a BMI (body mass indexu), únava se snížila. U 8 pacientů (28,6 %) byla snížena dávka anebo byl prodloužen interval tocilizumabu, aniž došlo k exacerbaci nemoci. 11 pacientů (73,5 %), kteří užívali perorální kortikosteroidy před vstupem do studie, při biologické léčbě dobře snášelo redukci dávek kortikoidů [23].
Protilátka proti receptoru pro interleukin-6 rovněž zabránila rozvoji sekundární amyloidózy, protože léčba významně snížila hladiny sérového amyloidu A. Za první rok léčby nebyl zaznamenán výskyt protilátek proti této protilátce (je humanizovaná, má proto menší obsah cizorodého proteinu, a tedy menší potenciál ke vzniku neutralizujících protilátek) [23]. Intravenózně aplikovaný tocilizumab v dávce 8 mg/kg každé 2 týdny je schválený pro terapii multicentrické CD v Japonsku [6].
V průběhu studie fáze II se 132 zařazenými pacienty byly nejčastěji zaznamenány následující nežádoucí účinky tocilizumabu: infekce (55/132), abnormální laboratorní testy (60/132) jako hypercholesterolemie (4/132) či hypertriglycerolemie (5/132) [100]. Mezi další často se vyskytující nežádoucí účinky patří uroinfekce (14,3 % pacientů). U některých pacientů byl rovněž pozorován mírný a přechodný pokles leukocytů [23]. Správné a včasné odhalení rozvíjející se infekční komplikace u pacientů léčených tocilizumabem se musí v prvé řadě opírat o výsledky důkladného fyzikálního vyšetření a radiografické nálezy. Tocilizumab totiž může tlumit elevaci CRP při infekci, neboť inhibuje interleukin-6, což je hepatocyty stimulující faktor indukující tvorbu CRP. Jako marker infekce lze využívat leukocytózu, avšak tato může být zkreslena konkomitantním podáváním kortikoidů. Hospitalizace pacienta s podezřením na infekci léčeného tocilizumabem je proto indikována i při nízkém nebo nevzrůstajícím CRP [6].
Rituximab
Plazmatické buňky v plášťové zóně u některých pacientů s multicentrickou plazmocelulární formou exprimují povrchový znak CD20 [101]. Rituximab je monoklonální protilátka proti CD20, která cílové buňky exprimující tento antigen vybírá pro destrukci buď cestou komplementu, nebo přes cytotoxické buňky [81]. Mechanizmus účinku byl dokladován měřením hladiny cytokinů, zastoupením buněčných populací a HIV virusové nálože. Preparáty jako rituximab, které snižují zánětlivou cytokinovou odpověď organizmu, mají u pacientů s CD terapeutický potenciál [102]. Jednoleté remise je dosaženo u 70 % pacientů [103].
Největší studie s rituximabem zahrnovala 21 pacientů s plazmocelulární multicentrickou CD, z nichž u 20 bylo dosaženo remise onemocnění a u 14 měla tato remise i radiologický korelát. Celkové dvouleté přežití bylo 95 %. Po léčbě rituximabem poklesly zánětlivé markery, imunoglobuliny a HHV-8 virusová nálož v plazmě. Hlavním nežádoucím efektem léčby byla reaktivace Kaposiho sarkomu [104]. Do jiné studie bylo zařazeno 5 HIV pozitivních pacientů. Léčebné odpovědi bylo dosaženo u 3 nemocných a trvala 4–14 měsíců. Dva pacienti, kteří neodpovídali na léčbu, měli těžkou autoimunitní hematologickou manifestaci a dále u 2 ze 4 pacientů s Kaposiho sarkomem došlo ke zhoršení jeho příznaků [105].
Rovněž bylo popsáno úspěšné použití rituximabu po selhání předchozí léčby pomocí protilátky proti interleukinu-6 [106], kortikosteroidů [107], chemoterapie [106,108] a antivirové léčbě cidofovirem [106,109], ale např. i u HIV a HHV-8 negativního muže s CD asociovanou s autoimunitní hemolytickou anémií a Raynaudovým fenoménem [70]. S úspěchem jsou využívány kombinované režimy s klasickou chemoterapií [110] a možné je i opakování léčby u pacientů s relabovaným CD, jak ukázali Powles et al u 3 pacientů [111]. Potenciál může spočívat dále v použití jako neoadjuvantní léčba u primárně neresekabilních nebo pouze parciálně resekabilních případů unicentrické CD, jak dokládá případ takovéto úspěšné léčby u pacienta s hyalinně-vaskulární formou [112]. Zprávy o neúspěšném nasazení rituximabu jsou nečetné [113–115].
Antivirotika
Ačkoliv bylo prokázáno, že některá antivirotika (foscarnet, ganciklovir, cidofovir) mají in vitro potenciál přerušit replikaci virusu HHV-8, úspěchy v klinické praxi při použití těchto léků byly smíšené a léčebných odpovědí bylo dosaženo u foscarnetu a zejména pak u valgancikloviru [21].
Závěrečná doporučení pro léčbu CD
Léčbou volby pro lokalizovanou CD je kompletní chirurgické odstranění. U pacientů s multicentrickou chorobou může být valganciklovir nejlepší volbou pro pacienty s PCR pozitivitou HHV-8 a rituximab pro pacienty s imunohistochemicky prokázanou CD20 pozitivitou. Pro HHV-8 a CD20 negativní pacienty, zejména při těžké systémové manifestaci nebo kortikorezistenci, eventuálně s nežádoucími projevy kortikoterapie, jsou volbou chemoterapeutické režimy CHOP nebo CVAD [6,21].
Prognóza
Lokalizovaná forma CD většinou negativně ovlivňuje organizmus pouze tlakem na okolní tkáně a orgány a kompletní chirurgická resekce přináší konečné řešení. Toto však neplatí pro multicentrickou variantu. Ačkoliv je multicentrická forma CD nenádorové onemocnění, její prognóza je bez léčby špatná [6]. Většina pacientů s multicentrickou CD zemře na fulminantní infekce, renální selhání, progresi nemoci nebo příbuzné malignity (Kaposiho sarkom, folikulární dendritický buněčný nádor, non-Hodgkinský nebo Hodgkinský lymfom) [2,24,27]. Smrtnost při sepsi dosahuje až 50 % [4]. Ve 4 největších souborech pacientů s multicentrickou CD byl medián přežití 14–30 měsíců. Někteří pacienti však žili pouze několik týdnů po stanovení diagnózy a někteří přežívali až 20 let [20,22,83,116].
Mezi život ohrožující komplikace se však řadí i sekundární amyloidóza, k níž u některých pacientů může vést nadměrná produkce sérového amyloidu A vyvolaná stimulací jaterních buněk interleukinem-6. U lokalizované i multicentrické nemoci komplikované amyloidózou byla nejčastějším histopatologickým nálezem plazmocelulární varianta [10,117].
Formy renální dysfunkce u CD jsou heterogenní a projevují se abnormálním nálezem v močovém sedimentu, mírnou až střední proteinurií, mírnou hematurií nebo renální insuficiencí. Nejčastější příčinou renálního selhání u pacientů s CD je amyloidóza, následovaná mesangiální proliferující glomerulonefritidou a v malém počtu případů také intersticiální nefritidou. Do roku 2007 bylo popsáno 5 případů intersticiální nefritidy a 45 případů amyloidózy asociované s CD [10].
Jednou z nejvíce devastujících chorob asociovaných s multicentrickým CD je POEMS syndrom, který se vyskytuje asi v 15 % CD [24], multicentrická CD byla naopak zjištěna asi v 11–24 % případů POEMS syndromu [21].
Sledování pacientů po léčbě
Casper u unicentrické CD doporučuje radiologické zhodnocení lymfadenopatie za 6–12 měsíců po skončení terapie, u pacientů s multicentrickou formou je toto sledování podrobnější a kromě radiodiagnostiky zahrnuje i kontroly krevního obrazu, jaterních testů a CRP [21].
Závěr
CD je vzácná idiopatická ne-neoplastická lymfoproliferativní choroba, zatím bez definované přesné incidence, charakterizovaná hyperplazií jedné nebo více lymfatických uzlin. Jedná se o často poddiagnostikované nebo špatně diagnostikované onemocnění [13]. Je třeba ji proto mít na mysli nejen při diferenciální diagnostice lymfadenopatie, ale i mikrocytární anémie či horečky nejasného původu a dalších B-symptomů (hubnutí, noční poty). Na přiložené obrazové dokumentaci jsme rovněž potvrdili význam PET/CT skenování při stážovacím vyšetření a hodnocení léčebné odpovědi u pacientů s CD
Tato publikace byla připravena v rámci aktivity následujících grantů: grantu IGA ČR NT 12215-4 a dále pak grantů MŠMT MSM0021622434, LC06027 a grantů IGA MZd NT11154, NT12130, NT12215 a NS10408.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do bi omedicínských časopisů.
MUDr. Petr Szturz
Interní hematoonkologická klinika
LF MU a FN Brno
Jihlavská 20
625 00 Brno
e-mail: petr.szturz@fnbrno.cz
Obdrženo: 4. 5. 2011
Přijato: 27. 5. 2011
Sources
1. Castleman B, Iverson L, Menendez VP. Localized mediastinal lymphnode hyperplasia resembling thymoma. Cancer 1956; 9(4): 822–830.
2. Cronin DM, Warnke RA. Castleman disease: an update on classification and the spectrum of associated lesions. Adv Anat Pathol 2009; 16(4): 236–246.
3. Vinzio S, Ciarloni L, Schlienger JL et al. Isolated microcytic anemia disclosing a unicentric Castleman disease: The interleukin-6/hepcidin pathway? Eur J Intern Med 2008; 19(5): 367–369.
4. Wang SH, Ruan Z, Huang HL et al. A rare case of Castleman disease presenting as pulmonary mass mimicking central pulmonary malignancy. Chin Med J (Engl) 2009; 122(8): 990–991.
5. Chen CH, Liu HC, Hung TT et al. Possible roles of Epstein-Barr virus in Castleman disease. J Cardiothorac Surg 2009; 4 : 31.
6. Matsuyama M, Suzuki T, Tsuboi H et al. Anti-interleukin-6 receptor antibody (tocilizumab) treatment of multicentric Castleman‘s disease. Intern Med 2007; 46(11): 771–774.
7. Danon AD, Krishnan J, Frizzera G. Morpho-immunophenotypic diversity of Castleman‘s disease, hyaline-vascular type: with emphasis on a stroma-rich variant and a new pathogenetic hypothesis. Virchows Arch A Pathol Anat Histopathol 1993; 423(5): 369–382.
8. Frizzera G. Castleman‘s disease and related disorders. Semin Diagn Pathol 1988; 5(4): 346–364.
9. Moore DF, Preti A, Tran SM. Prognostic implications following an indeterminate diagnostic work-up of lymphoma. Blood 1996; 88 (Suppl 1): 229a.
10. Morita-Hoshi Y, Tohda S, Miura O et al. An autopsy case of multicentric Castleman‘s disease associated with interstitial nephritis and secondary AA amyloidosis. Int J Hematol 2008; 87(1): 69–74.
11. Klener P et al. Cytokiny ve vnitřním lékařství. Praha: Grada Publishing 1997.
12. van Rhee F, Stone K, Szmania S et al. Castleman disease in the 21st century: an update on diagnosis, assessment, and therapy. Clin Adv Hematol Oncol 2010; 8(7): 486–498.
13. Ye B, Gao SG, Li W et al. A retrospective study of unicentric and multicentric Castleman‘s disease: a report of 52 patients. Med Oncol 2010; 27(4): 1171–1178.
14. Bowne WB, Lewis JJ, Filippa DA et al. The management of unicentric and multicentric Castleman‘s disease: a report of 16 cases and a review of the literature. Cancer 1999; 85(3): 706–717.
15. Gaba AR, Stein RS, Sweet DL et al. Multicentric giant lymph node hyperplasia. Am J Clin Pathol 1978; 69(1): 86–90.
16. Keller AR, Hochholzer L, Castleman B. Hyaline-vascular and plasma-cell types of giant lymph node hyperplasia of the mediastinum and other locations. Cancer 1972; 29(3): 670–683.
17. Ko SF, Wan YL, Ng SH et al. Imaging features of atypical thoracic Castleman disease. Clin Imaging 2004; 28(4): 280–285.
18. Gangopadhyay K, Mahasin ZZ, Kfoury H. Pathologic quiz case 2. Castleman disease (giant lymph node hyperplasia). Arch Otolaryngol Head Neck Surg 1997; 123(10): 1137, 1139.
19. Johkoh T, Müller NL, Ichikado K et al. Intrathoracic multicentric Castleman disease: CT findings in 12 patients. Radiology 1998; 209(2): 477–481.
20. Herrada J, Cabanillas F, Rice L et al. The clinical behavior of localized and multicentric Castleman disease. Ann Intern Med 1998; 128(8): 657–662.
21. Casper C. The aetiology and management of Castleman disease at 50 years: translating pathophysiology to patient care. Br J Haematol 2005; 129(1): 3–17.
22. Frizzera G, Peterson BA, Bayrd ED et al. A systemic lymphoproliferative disorder with morphologic features of Castleman‘s disease: clinical findings and clinicopathologic correlations in 15 patients. J Clin Oncol 1985; 3(9): 1202–1216.
23. Nishimoto N, Kanakura Y, Aozasa K et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood 2005; 106(8): 2627–2632.
24. Peterson BA, Frizzera G. Multicentric Castleman‘s disease. Semin Oncol 1993; 20(6): 636–647.
25. McCarty MJ, Vukelja SJ, Banks PM et al. Angiofollicular lymph node hyperplasia (Castleman‘s disease). Cancer Treat Rev 1995; 21(4): 291–310.
26. Menke DM, Camoriano JK, Banks PM. Angiofollicular lymph node hyperplasia: a comparison of unicentric, multicentric, hyaline vascular, and plasma cell types of disease by morphometric and clinical analysis. Mod Pathol 1992; 5(5): 525–530.
27. Flendrig JA, Schillings PM. Benign giant lymphoma: the clinical signs and symptoms. Folia Medica Neerlandica 1969; 12 : 119–120.
28. Martin JM, Bell B, Ruether BA. Giant lymph node hyperplasia (Castleman‘s disease) of hyaline vascular type. Clinical heterogeneity with immunohistologic uniformity. Am J Clin Pathol 1985; 84(4): 439–446.
29. Choi JH, Jo YJ, Gong SJ et al. Unicentric castleman disease is not clearly distinguished from multicentric type: a case report. Clin Lymphoma Myeloma 2008; 8(4): 256–259.
30. Ioachim HL, Medeiros LJ. Ioachim‘s lymph node pathology. Philadelphia: Lippincott Williams & Wilkins 2008.
31. Feigert JM, Sweet DL, Coleman M et al. Multicentric angiofollicular lymph node hyperplasia with peripheral neuropathy, pseudotumor cerebri, IgA dysproteinemia, and thrombocytosis in women. A distinct syndrome. Ann Intern Med 1990; 113(5): 362–367.
32. Bitter MA, Komaiko W, Franklin WA. Giant lymph node hyperplasia with osteoblastic bone lesions and the POEMS (Takatsuki’s) syndrome. Cancer 1985; 56(1): 188–194.
33. Mandler RN, Kerrigan DP, Smart J et al. Castleman’s disease in POEMS syndrome with elevated interleukin-6. Cancer 1992; 69(11): 2697–2703.
34. Bélec L, Mohamed AS, Authier FJ et al. Human herpesvirus 8 infection in patients with POEMS syndrome-associated multicentric Castleman’s disease. Blood 1999; 93(11): 3643–3653.
35. Dispenzieri A, Kyle RA, Lacy MQ et al. POEMS syndrome: definitions and long-term outcome. Blood 2003; 101(7): 2496–2506.
36. Seco JL, Velasco F, Manuel JS et al. Retroperitoneal Castleman’s disease. Surgery 1992; 112(5): 850–855.
37. Leger-Ravet MB, Peuchmaur M, Devergne O et al. Interleukin-6 gene expression in Castleman’s disease. Blood 1991; 78(11): 2923–2930.
38. Brandt SJ, Bodine DM, Dunbar CE et al. Dysregulated interleukin 6 expression produces a syndrome resembling Castleman’s disease in mice. J Clin Invest 1990; 86(2): 592–599.
39. Yoshizaki K, Matsuda T, Nishimoto N et al. Pathogenic significance of interleukin-6 (IL-6/BSF-2) in Castleman’s disease. Blood 1989; 74(4): 1360–1367.
40. Beck JT, Hsu SM, Wijdenes J et al. Brief report: alleviation of systemic manifestations of Castleman’s disease by monoclonal anti-interleukin-6 antibody. N Engl J Med 1994; 330(9): 602–605.
41. Hsu SM, Waldron JA, Xie SS et al. Expression of interleukin-6 in Castleman’s disease. Hum Pathol 1993; 24(8): 833–839.
42. Yabuhara A, Yanagisawa M, Murata T et al. Giant lymph node hyperplasia (Castleman’s disease) with spontaneous production of high levels of B-cell differentiation factor activity. Cancer 1989; 63(2): 260–265.
43. McClain KL, Natkunam Y, Swerdlow SH. Atypical cellular disorders. Hematology Am Soc Hematol Educ Program 2004 : 283–296.
44. Castleman B, Towne VW. Case records of the Massachusetts General Hospital; weekly clinicopathological exercises; founded by Richard C. Cabot. N Engl J Med 1954; 251(10): 396–400.
45. Chronowski GM, Ha CS, Wilder RB et al. Treatment of unicentric and multicentric Castleman disease and the role of radiotherapy. Cancer 2001; 92(3): 670–676.
46. Argilés JM, López-Soriano FJ. The role of cytokines in cancer cachexia. Med Res Rev 1999; 19(3): 223–248.
47. Nemeth E, Valore EV, Territo M et al. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood 2003; 101(7): 2461–2463.
48. Nemeth E, Rivera S, Gabayan V et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest 2004; 113(9): 1271–1276.
49. Ganz T. Hepcidin - a regulator of intestinal iron absorption and iron recycling by macrophages. Best Pract Res Clin Haematol 2005; 18(2): 171–182.
50. Kawabata H, Tomosugi N, Kanda J et al. Anti-interleukin 6 receptor antibody tocilizumab reduces the level of serum hepcidin in patients with multicentric Castleman’s disease. Haematologica 2007; 92(6): 857–858.
51. Roca B. Castleman’s Disease. A Review. AIDS Rev 2009; 11(1): 3–7.
52. Wang H, Wieczorek RL, Zenilman ME et al. Castleman’s disease in the head of the pancreas: report of a rare clinical entity and current perspective on diagnosis, treatment, and outcome. World J Surg Oncol 2007; 5 : 133.
53. Collins LS, Fowler A, Tong CY et al. Multicentric Castleman’s disease in HIV infection. Int J STD AIDS 2006; 17(1): 19–24, quiz 25.
54. Kojima M, Nakamura S, Miyawaki S et al. Progressive transformation of germinal center presenting with histological features of hyaline-vascular type of Castleman’s disease. APMIS 2005; 113(4): 288–295.
55. Nishimoto N. Clinical studies in patients with Castleman’s disease, Crohn’s disease, and rheumatoid arthritis in Japan. Clin Rev Allergy Immunol 2005; 28(3): 221–230.
56. van Kooten C, Rensink I, Aarden L et al. Effect of IL-4 and IL-6 on the proliferation and differentiation of B-chronic lymphocytic leukemia cells. Leukemia 1993; 7(4): 618–624.
57. Moore PS, Boshoff C, Weiss RA et al. Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science 1996; 274(5293): 1739–1744.
58. Veldhuis GJ, van der Leest AH, de Wolf JT et al. A case of localized Castleman’s disease with systemic involvement: treatment and pathogenetic aspects. Ann Hematol 1996; 73(1): 47–50.
59. Aoki Y, Jaffe ES, Chang Y et al. Angiogenesis and hematopoiesis induced by Kaposi’s sarcoma-associated herpesvirus-encoded interleukin-6. Blood 1999; 93(12): 4034–4043.
60. Waterston A, Bower M. Fifty years of multicentric Castleman’s disease. Acta Oncol 2004; 43(8): 698–704.
61. Chvojka J. Angiografický obraz retroperitoneální formy castlemanovy choroby. Česk Radiol 1984; 38(1): 55–60.
62. Stansby G, Hilson A, Hamilton G. Gallium scintigraphy in the diagnosis and management of multifocal Castleman’s disease. Br J Radiol 1991; 64(758): 165–167.
63. Okamoto I, Iyonaga K, Fujii K et al. Absence of gallium uptake in unicentric and multicentric Castleman’s disease. Intern Med 2003; 42(8): 735–739.
64. Podzamczer D, Ricart I, Bolao F et al. Gallium-67 scan for distinguishing follicular hyperplasia from other AIDS-associated diseases in lymph nodes. AIDS 1990; 4(7): 683–685.
65. Halac M, Ergul N, Sager S et al. PET/CT findings in a multicentric form of Castleman’s disease. Hell J Nucl Med 2007; 10(3): 172–174.
66. Murphy SP, Nathan MA, Karwal MW. FDG-PET appearance of pelvic Castleman’s disease. J Nucl Med 1997; 38(8): 1211–1212.
67. Leskinen-Kallio S, Ruotsalainen U, Någren K et al. Uptake of carbon-11-methionine and fluorodeoxyglucose in non-Hodgkin’s lymphoma: a PET study. J Nucl Med 1991; 32(6): 1211–1218.
68. Nishimoto N, Sasai M, Shima Y et al. Improvement in Castleman’s disease by humanized anti-interleukin-6 receptor antibody therapy. Blood 2000; 95(1): 56–61.
69. Corbellino M, Bestetti G, Scalamogna C et al. Long-term remission of Kaposi sarcoma-associated herpesvirus-related multicentric Castleman disease with anti-CD20 monoclonal antibody therapy. Blood 2001; 98(12): 3473–3475.
70. Ocio EM, Sanchez-Guijo FM, Diez-Campelo M et al. Efficacy of rituximab in an aggressive form of multicentric Castleman disease associated with immune phenomena. Am J Hematol 2005; 78(4): 302–305.
71. Pavlidis NA, Skopouli FN, Bai MC et al. A successfully treated case of multicentric angiofollicular hyperplasia with oral chemotherapy (Castleman’s disease). Med Pediatr Oncol 1990; 18(4): 333–335.
72. Dispenzieri A, Gertz MA. Treatment of Castleman’s disease. Curr Treat Options Oncol 2005; 6(3): 255–266.
73. Mohanna S, Sanchez J, Ferrufino JC et al. Characteristics of Castleman’s disease in Peru. Eur J Intern Med 2006; 17(3): 170–174.
74. Larroche C, Cacoub P, Godeau P. Castleman’s disease. Rev Med Interne 1996; 17(12): 1003–1013.
75. Ebisuno S, Yamauchi T, Fukatani T et al. Retroperitoneal Castleman’s disease: a case report and brief review of tumors of the pararenal area. Urol Int 1989; 44(3): 169–172.
76. Takihara H, Yamakawa G, Baba Y et al. Castleman disease. Unusual retroperitoneal location indistinguishable from malignant tumor in preoperative angiographic appearance. Urology 1993; 41(2): 162–164.
77. Kiguchi H, Ishii T, Ishikawa Y et al. Castleman’s disease of the abdomen and pelvis: report of three cases and a review of the literature. J Gastroenterol 1995; 30(5): 661–666.
78. Bartkowski DP, Ferrigni RG. Castleman’s disease: an unusual retroperitoneal mass. J Urol 1988; 139(1): 118–120.
79. Skolnik G, Wiklund LM, Risberg B. Castleman’s tumor with retroperitoneal location: a malignant-appearing benign tumor. J Surg Oncol 1985; 28(2): 153–155.
80. d’Agay MF, Miclea JM, Clauvel JP et al. Castleman’s disease: a well defined histological pattern for a widely divergent clinical spectrum. Nouv Rev Fr Hematol 1989; 31(2): 145–148.
81. Nordstrom DG, Tewfik HH, Latourette HB. Giant lymph node hyperplasia: a review of literature and report of two cases of plasma cell variant responding to radiation therapy. Int J Radiat Oncol Biol Phys 1978; 4(11–12): 1045–1048.
82. Adam Z, Krejčí M, Vorlíček J. Hematologie: přehled maligních hematologických nemocí. Praha: Grada Publishing 2008.
83. Weisenburger DD, Nathwani BN, Winberg CD et al. Multicentric angiofollicular lymph node hyperplasia: a clinicopathologic study of 16 cases. Hum Pathol 1985; 16(2): 162–172.
84. Summerfield GP, Taylor W, Bellingham AJ et al. Hyaline-vascular variant of angiofollicular lymph node hyperplasia with systemic manifestations and response to corticosteroids. J Clin Pathol 1983; 36(9): 1005–1011.
85. Bertero MT, De Maestri M, Caligaris-Cappio F. Cyclophosphamide/cyclosporin-A treatment of multicentric Castleman’s disease with Kaposi’s sarcoma. Haematologica 2000; 85(2): 216–217.
86. Adam Z, Vorlíček J. Biologie, farmakologie a přehled léčebného užití interferonů. Klin Onkol 1996; 9(4): 115–120.
87. Kumari P, Schechter GP, Saini N et al. Successful treatment of human immunodeficiency virus-related Castleman’s disease with interferon-alpha. Clin Infect Dis 2000; 31(2): 602–604.
88. Nord JA, Karter D. Low dose interferon-alpha therapy for HIV-associated multicentric Castleman’s disease. Int J STD AIDS 2003; 14(1): 61–62.
89. Rieu P, Droz D, Gessain A et al. Retinoic acid for treatment of multicentric Castleman’s disease. Lancet 1999; 354(9186): 1262–1263.
90. Fishman SJ, Feins NR, D’Amato RJ et al. Long-term remission of Crohn’s disease treated with thalidomide: a seminal case report. Angiogenesis 1999; 3(3): 201–204.
91. Špička I, Hájek R, Gregora E et al. První zkušenosti s léčbou mnohočetného myelomu v České republice. Klin Onkol 2002; 15 (Suppl): 42–43.
92. Jung CP, Emmerich B, Goebel FD et al. Successful treatment of a patient with HIV-associated multicentric Castleman disease (MCD) with thalidomide. Am J Hematol 2004; 75(3): 176–177.
93. Lee FC, Merchant SH. Alleviation of systemic manifestations of multicentric Castleman’s disease by thalidomide. Am J Hematol 2003; 73(1): 48–53.
94. Starkey CR, Joste NE, Lee FC. Near-total resolution of multicentric Castleman disease by prolonged treatment with thalidomide. Am J Hematol 2006; 81(4): 303–304.
95. Stary G, Kohrgruber N, Herneth AM et al. Complete regression of HIV-associated multicentric Castleman disease treated with rituximab and thalidomide. AIDS 2008; 22(10): 1232–1234.
96. Kim SY, Lee SA, Ryoo HM et al. Thalidomide for POEMS syndrome. Ann Hematol 2006; 85(8): 545–546.
97. Menegato MA, Canelles MF, Tonutti E et al. Remission of nephrotic syndrome after thalidomide therapy in a patient with Castleman’s disease. Clin Nephrol 2004; 61(5): 352–356.
98. Miltenyi Z, Toth J, Gonda A et al. Successful immunomodulatory therapy in castleman disease with paraneoplastic pemphigus vulgaris. Pathol Oncol Res 2009; 15(3): 375–381.
99. Sato K, Tsuchiya M, Saldanha J et al. Reshaping a human antibody to inhibit the interleukin 6-dependent tumor cell growth. Cancer Res 1993; 53(4): 851–856.
100. Actemra Intravenous Infusion 200 Safety Information Tabulation Results from Approval to June 30, 2006.
101. Hall PA, Donaghy M, Cotter FE et al. An immunohistological and genotypic study of the plasma cell form of Castleman’s disease. Histopathology 1989; 14(4): 333–346, discussion 429–432.
102. Bower M, Veraitch O, Szydlo R et al. Cytokine changes during rituximab therapy in HIV-associated multicentric Castleman disease. Blood 2009; 113(19): 4521–4524.
103. Oksenhendler E. HIV-associated multicentric Castleman disease. Curr Opin HIV AIDS 2009; 4(1): 16–21.
104. Bower M, Powles T, Williams S et al. Brief communication: rituximab in HIV-associated multicentric Castleman disease. Ann Intern Med 2007; 147(12): 836–839.
105. Marcelin AG, Aaron L, Mateus C et al. Rituximab therapy for HIV-associated Castleman disease. Blood 2003; 102(8): 2786–2788.
106. Corbellino M, Bestetti G, Scalamogna C et al. Long-term remission of Kaposi sarcoma-associated herpesvirus-related multicentric Castleman disease with anti-CD20 monoclonal antibody therapy. Blood 2001; 98(12): 3473–3475.
107. Ide M, Ogawa E, Kasagi K et al. Successful treatment of multicentric Castleman’s disease with bilateral orbital tumour using rituximab. Br J Haematol 2003; 121(5): 818–819.
108. Gholam D, Vantelon JM, Al-Jijakli A et al. A case of multicentric Castleman’s disease associated with advanced systemic amyloidosis treated with chemotherapy and anti-CD20 monoclonal antibody. Ann Hematol 2003; 82(12): 766–768.
109. Marietta M, Pozzi S, Luppi M et al. Acquired haemophilia in HIV negative, HHV-8 positive multicentric Castleman’s disease: a case report. Eur J Haematol 2003; 70(3): 181–182.
110. Fragasso A, Mannarella C, Ciancio A et al. Complete remission and virologic response to combined chemoimmunotherapy (R-CVP) followed by rituximab maintenance in HIV-negative, HHV-8 positive patient with multicentric Castleman disease. Leuk Lymphoma 2008; 49(11): 2224–2226.
111. Powles T, Stebbing J, Montoto S et al. Rituximab as retreatment for rituximab pretreated HIV-associated multicentric Castleman disease. Blood 2007; 110(12): 4132–4133.
112. Bandera B, Ainsworth C, Shikle J et al. Treatment of unicentric Castleman disease with neoadjuvant rituximab. Chest 2010; 138(5): 1239–1241.
113. Galeotti C, Tran TA, Franchi-Abella S et al. IL-1RA agonist (anakinra) in the treatment of multifocal castleman disease: case report. J Pediatr Hematol Oncol 2008; 30(12): 920–924.
114. Ganti AK, Pipinos I, Culcea E et al. Successful hematopoietic stem-cell transplantation in multicentric Castleman disease complicated by POEMS syndrome. Am J Hematol 2005; 79(3): 206–210.
115. Neuville S, Agbalika F, Rabian C et al. Failure of rituximab in human immunodeficiency virus-associated multicentric Castleman disease. Am J Hematol 2005; 79(4): 337–339.
116. Oksenhendler E, Duarte M, Soulier J et al. Multicentric Castleman’s disease in HIV infection: a clinical and pathological study of 20 patients. AIDS 1996; 10(1): 61–67.
117. Šteiner I, Cerman J, Hájková P et al. Amyloidóza při Castlemanově chorobě. Lék Zpr Lék Fak Univ Karlovy Hr Králové 2004; 49(3–4): 129–139.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2011 Issue 6
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Safety and Tolerance of Metamizole in Postoperative Analgesia in Children
Most read in this issue
- Castlemanova choroba
- Dlouhodobé sledování pacienta s eozinofilním granulomem žebra
- Lékový registr – trabectedin
- Prediktivní hodnota ultrazvukových parametrů, CA-125 a indexu rizika malignity u pacientek s karcinomem ovarií