
Molekulární prognostické markery chronické lymfocytární leukemie a jejich klinický význam
Molecular Prognostic Markers and Their Clinical Relevance in Chronic Lymphocytic Leukemia
Chronic lymphocytic leukemia is the most common leukemia in Western countries affecting particularly elderly adults. Despite the constantly improving therapy options, chronic lymphocytic leukemia is still an incurable disease owing to considerable clinical and biological heterogeneity. Pathogenesis of chronic lymphocytic leukemia is not fully understood; however, aberrant antigenic stimulation, apoptosis deregulation and microenvironmental interactions play a crucial role in disease development. The most important molecular prognostic markers with clinical relevance include mutation status of heavy‑chain immunoglobulin genes (IGHV), presence of cytogenetic aberrations and TP53 and ATM gene mutations. Recent implementation of next generation sequencing technologies has enabled more accurate analysis of both well‑established and novel potential prognostic markers. The most relevant candidates are mutations in SF3B1, NOTCH1 and BIRC3 genes, which are now intensively studied with respect to their clinical importance. The other examined molecular mechanisms of chronic lymphocytic leukemia pathogenesis include deregulation of B ‑ cell receptor signalization and abnormal regulation of gene expression by microRNA. The precise characterization of molecular abnormalities improves the risk stratification of chronic lymphocytic leukemia patients, which could possibly benefit from new treatment approaches.
Key words:
chronic lymphocytic leukemia – biological markers – chromosome aberations – mutations – prognosis
This work was supported by the grants IGA MH CZ NT13493-4/2012, NT13576-4/2012, NT13576, AZV MZ ČR No. 15-30015A-4/2015 a 15-31834A-4/2015 a TAČR TE02000058.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted:
30. 7. 2015
Accepted:
4. 8. 2015
Authors:
V. Navrkalová 1; B. Kantorová 1; M. Jarošová 2; Š. Pospíšilová 1
Authors‘ workplace:
Interní hematologická a onkologická klinika LF MU a FN Brno2 Hemato‑ onkologická klinika LF UP a FN Olomouc
1
Published in:
Klin Onkol 2015; 28(Supplementum 3): 6-15
doi:
https://doi.org/10.14735/amko20153S6
Overview
Chronická lymfocytární leukemie je nejčastější typ leukemie v západním světě postihující zejména starší dospělé osoby. Navzdory stále se zdokonalující léčbě zůstává z důvodu značné biologické i klinické variability nadále nevyléčitelným onemocněním. Patogeneze chronické lymfocytární leukemie není dodnes plně objasněna, nicméně velkou roli hrají antigenní stimulace, narušená apoptóza a vliv mikroprostředí. Mezi nejvýznamnější molekulární prognostické faktory s jasným klinickým dopadem patří mutační stav genů pro těžký řetězec imunoglobulinů (IGHV), cytogenetické aberace a mutace genů TP53 a ATM. Zavedení nových sekvenačních technologií umožnilo v posledních letech zpřesnění analýzy stávajících i detekci nových potenciálních prognostických markerů chronické lymfocytární leukemie. Významnými kandidáty jsou mutace genů SF3B1, NOTCH1 a BIRC3, jejichž klinický dopad je předmětem intenzivního výzkumu. V neposlední řadě jsou nadále studovány také další mechanizmy patogeneze chronické lymfocytární leukemie, mezi něž patří deregulace signalizace přes B buněčný receptor a regulace genové exprese pomocí microRNA. Přesná charakterizace molekulárních abnormalit je klíčová pro lepší rozdělení rizikových skupin pacientů s chronickou lymfocytární leukemií, kteří mohou profitovat z nových terapeutických přístupů.
Klíčová slova:
chronická lymfocytární leukemie – biologické markery – chromozomální aberace – mutace – prognóza
Úvod
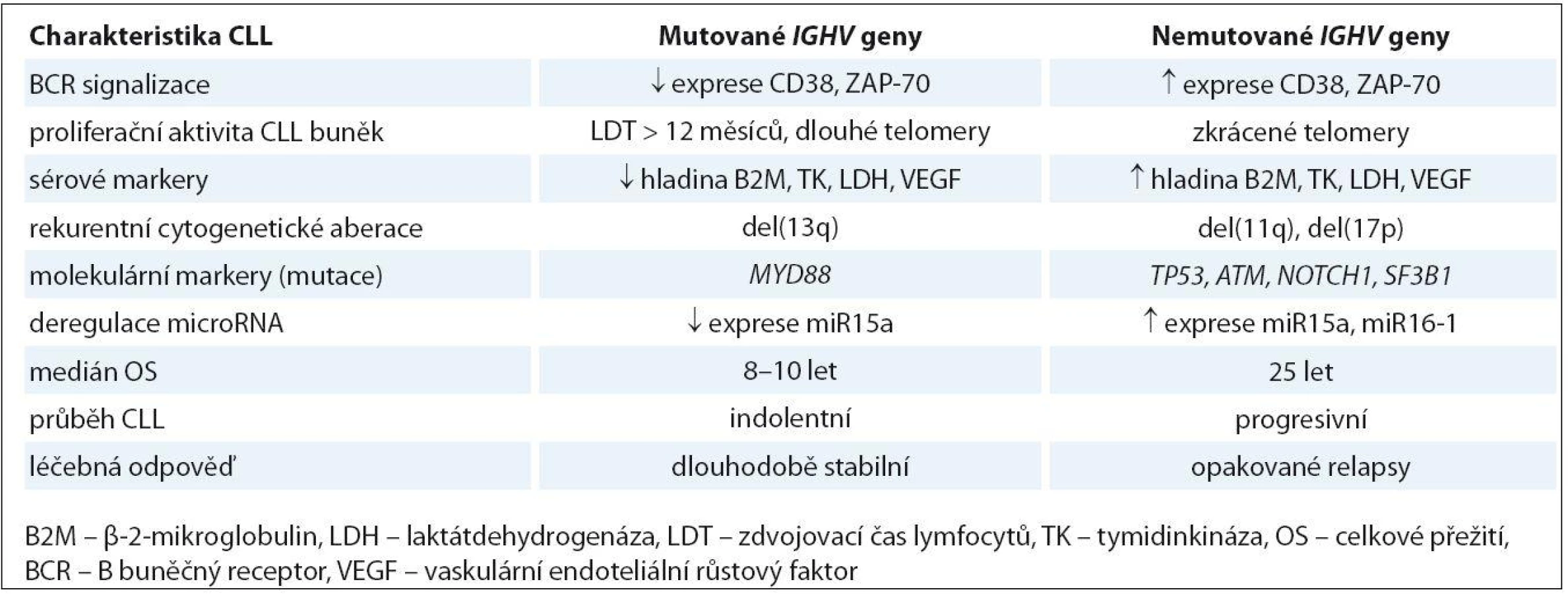
Chronická lymfocytární leukemie (CLL) patří mezi nízce maligní lymfoproliferativní onemocnění, které je zpravidla charakterizováno akumulací zralých monoklonálních B lymfocytů s typickým imunofenotypem (CD5, CD19, CD20 a CD23) v periferní krvi, kostní dřeni a lymfatických orgánech [1]. Díky velmi variabilnímu klinickému průběhu choroby a absenci kauzální genové aberace není prozatím u CLL dostupná univerzální účinná léčba. Nicméně byla popsána celá řada klinicko‑biologických faktorů, které přispívají k objasnění patogeneze CLL a stanovení prognózy pacientů (tab. 1). Zdá se, že mezi hlavní molekulární mechanizmy patří vliv mikroprostředí podílející se na antigenní stimulaci B buněčného receptoru (B-cell receptor – BCR) a vnitrobuněčná akumulace genetických defektů (obr. 1) [2,3]. Významná role antigenu ve vývoji CLL je podpořena existencí pozměněného repertoáru genů pro těžký řetězec imunoglobulinů (IGHV) při srovnání s fyziologickými B lymfocyty a rovněž také faktem, že někteří pacienti využívají sekvenčně identický, tzv. stereotypní BCR [4 – 6]. Objev cytogenetických a molekulárně‑genetických markerů výrazně napomohl objasnění heterogenity biologického pozadí CLL. Dle klasického Döhnerova modelu [7] lze podle přítomnosti cytogenetických abnormalit určit prognózu, která je nejhorší v případě delece 17p (del(17p)), následované delecí 11q (del(11q)). Nedávno publikované výsledky celogenomového a exomového sekvenování ukázaly, že kromě známých nádorových supresorů TP53 a ATM jsou u CLL rekurentně mutovány i další geny, jako je SF3B1, NOTCH1 a BIRC3, které mohou přispívat k horší prognóze pacientů [8 – 11]. Přítomnost určitých molekulárních lézí odráží klinickou fázi a rovněž klonální evoluci choroby, kdy např. premaligní léze známá jako monoklonální B lymfocytóza (MBL) je charakterizována nízkým počtem abnormalit, zatímco agresivní CLL nebo tzv. Richterův syndrom (RS) jsou spojovány s akumulací závažných genetických defektů [12 – 14]. V následujícím textu bychom rádi shrnuli poznatky o molekulární podstatě patogeneze a klonální evoluci CLL s důrazem na význam klasických i nových molekulárních prognostických markerů pro klinickou praxi.
![Klonální evoluce CLL a přítomnost molekulárních lézí v jednotlivých stadiích onemocnění. Hlavní mechanizmy patogeneze CLL jsou vypsány v šedém rámečku (podle Gaidano et al, 2012) [117].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/19581c205f06d5d5e8116c3e4097b056.jpg)
Prognostické faktory asociované s deregulací BCR signalizace
BCR receptor na povrchu lymfocytů hraje velice významnou fyziologickou úlohu v rozpoznávání antigenu a spouštění signální kaskády, která je nezbytná pro přežití a proliferaci jak normálních, tak i maligních B lymfocytů. Základní komponentou tohoto receptoru je membránový imunoglobulin, jehož struktura zahrnující preferenční využívání určitých typů subgenů pro těžký řetězec (např. VH3- 21, VH1- 69) hraje významnou roli v patogenezi CLL. Přítomnost somatických mutací v genech pro IGHV je jedním ze základních prognostických markerů, který umožňuje rozlišení dvou skupin pacientů s jasně odlišnými klinicko‑biologickými rysy. V případě mutovaného IGHV (M ‑ CLL) dochází k vývoji CLL až po reakci B lymfocytu s antigenem, který prošel germinálním centrem lymfatické uzliny. Naopak leukemické buňky s nemutovaným IGHV (U ‑ CLL) se s antigenem sice setkaly, ale nebyly po této reakci podrobeny procesu somatické hypermutace (obr. 1). Hranice pro U ‑ CLL byla stanovena arbitrárně a odpovídá více než 98% sekvenční homologii s genem zárodečné linie [15]. Tito pacienti mají vyšší frekvenci nepříznivých genových abnormalit a celkově výrazně horší prognózu než pacienti s mutovaným IGHV, kteří vykazují významně delší celkové přežití (overall survival – OS) (tab. 1) [4,5]. Zvláštní případ pak reprezentuje genový segment VH3 - 21, jehož přítomnost v leukemickém klonu představuje nepříznivou prognózu bez ohledu na jeho mutační stav [16]. V posledních letech se objevil další fakt podporující význam antigenní stimulace při vývoji a progresi CLL. Bylo zjištěno, že asi 30 % CLL pacientů využívá určitý typ tzv. stereotypního BCR receptoru, který je charakteristický identickou sekvencí VH CDR3 oblasti (variable heavy complementarity ‑ determining region 3) imunoglobulinu (tzv. subset) pro danou skupinu nepříbuzných pacientů [6,17]. Tito pacienti vykazují výrazně rozdílné genetické pozadí a také celkový průběh choroby. Například pacienti ze subsetu #2 využívající stereotypní VH3 - 21 mají nepříznivou prognózu bez ohledu na mutační stav genů IGHV a byla u nich prokázána vyšší frekvence výskytu SF3B1 mutací a poškození p53 dráhy [18,19]. Celkově tedy tato nedávná pozorování naznačují, že vývoj CLL je pravděpodobně řízen vysoce specifickou antigenní selekcí B lymfocytů spojenou se získáváním určitých genetických defektů.
Další prognostické markery zapojené do BCR signalizace jsou molekuly ZAP ‑ 70 a CD38, jejichž vyšší úroveň exprese je asociována s U ‑ CLL a tedy agresivnějším průběhem onemocnění [5,20]. ZAP ‑ 70 (zeta asociovaný protein) je tyrozinkináza, která se fyziologicky vyskytuje v T lymfocytech a také v různých vývojových stadiích normálních i maligních B lymfocytů [21]. Při studiu CLL bylo prokázáno, že vysoká hladina ZAP ‑ 70 moduluje BCR signalizaci a přispívá k nutnosti dříve zahájit terapii [22]. CD38 je transmembránový glykoprotein kooperující se ZAP ‑ 70 během BCR signalizace, který po aktivaci spouští typickou signální kaskádu pro proliferaci CLL buněk. Prognóza pacientů CD38+/ ZAP ‑ 70+ je horší než u nemocných se zvýšenou expresí pouze znaku CD38 [23]. Oba tyto markery dříve sloužily jako alternativa k poněkud obtížnému stanovení mutačního stavu genů IGHV. Nicméně exprese ZAP ‑ 70 a CD38 se mění spolu s vývojem onemocnění a navíc prognosticky významná míra exprese je obtížně stanovitelná díky odlišným metodám používaným v jednotlivých laboratořích. Proto dnes slouží už jen jako doprovodné prognostické faktory ke standardizovanému vyšetření genů IGHV.
Cytoplazmatická BCR signalizace je zprostředkována řadou kináz aktivujících nezbytné pro‑proliferační a anti‑apoptické procesy v CLL buňkách [24]. Fosforylační kaskáda zahrnuje především kinázy Lyn, Syk a Btk. Zajímavé je, že Lyn není pro BCR signalizaci nezbytná tak jako ostatní zmíněné kinázy, ale naopak hraje klíčovou roli v ukončení BCR aktivace. Fosforylací jsou dále aktivovány proteiny PI3K a PLCγ2 odpovědné za tvorbu tzv. druhých poslů, které zprostředkovávají aktivaci příslušných drah (např. RAS, NFκB, AKT, MAPK, PKC atd.) [25]. Pochopení mechanizmů BCR signalizace vedlo k navržení nových terapeutických postupů založených na inhibici jednotlivých komponent této dráhy. Tyto tzv. BCR inhibitory mají pro nádorovou buňku fatální následky; vedou nejenom k inhibici signálů pro přežití, ale ovlivňují také migraci maligních buněk [26]. Mezi klinicky testovaná léčiva patří např. ibrutinib jako inhibitor Btk dosahující velmi dobrých klinických výsledků (schválen v USA v roce 2014 pro léčbu relabující/ refrakterní CLL v druhé léčebné linii), fostamatinib pro inhibici Syk a nebo PI3K inhibitor idelalisib [27].
Cytogenetické prognostické faktory
Získané genetické změny se vyskytují v době diagnózy CLL u více než 80 % nemocných a hrají významnou roli v patogenezi onemocnění. Jejich identifikace má význam pro prognózu, volbu léčebné strategie, a tedy OS nemocných. Na jejich určení se už od 70. let minulého století podílí řada cytogenetických a molekulárně‑cytogenetických metod, jako je klasická cytogenetika, fluorescenční in situ hybridizace (FISH), čipové technologie a v poslední době i nejmodernější technologie sekvenování nové generace (next-generation sequencing – NGS). Přes výskyt velkého spektra genetických změn byly určeny rekurentní změny prognostického významu.
Milníkem ve vývoji cytogenetiky CLL a jejího klinického významu byla práce z roku 2000, kdy Döhner et al určili na základě cytogenetické (FISH) analýzy 325 CLL nemocných prognostický význam nejčastějších chromozomových změn v hierarchickém uspořádání [7]. Tato „Döhnerova prognostická klasifikace“ ukázala, že nemocní s delecí 13q (del(13q)) jako jedinou změnou v karyotypu mají dobrou prognózu, zatímco pacienti s del(17p ) zahrnující gen TP53 a del(11q ) zahrnující gen ATM, mají nepříznivou prognózu. Nález trizomie chromozomu 12 (tris(12)) je spojován se střední prognózou společně s nálezem normálního karyotypu. Právě tato cytogenetická prognostická stratifikace doplnila a upřesnila již existující hodnocení klinického stadia podle Bineta a Raie [28,29].
Kromě uvedených chromozomových změn, které jsou součástí rutinního CLL panelu FISH vyšetření, existují i další chromozomové aberace, jež byly určeny klasickou cytogenetickou nebo čipovou technologií. K takovým změnám patří zmnožení krátkých ramen chromozomu 2, které bylo pozorováno u 8 – 28 % CLL nemocných [30,31]. Zmnožení zahrnuje oblast s lokalizací významných onkogenů jako MYCN, REL a nebo MSH2 a častěji se vyskytuje u nemocných v pokročilejším stadiu onemocnění (Binet B a C) a také jako sekundární změna spojená s kratším OS a transformací do RS [31,32].
Změny chromozomu 13q
Del(13q) se vyskytuje až u 50 % CLL nemocných v době diagnózy a je nejčastější změnou detekovanou metodou FISH. Během posledních několika let byla tato oblast obzvlášť intenzivně studována a byla navržena řada kandidátních genů (obr. 2), které by mohly být odpovědné za patogenezi stejně jako za prognostickou heterogenitu nemocných s del(13q) . V deletované oblasti byly určeny geny kódující microRNA (miRNA), miR 15a a miR16- 1 [33,34], ale jasná korelace s expresí a počtem alel nebyla přesně stanovena. Kromě těchto miRNA bylo studováno několik dalších genů lokalizovaných v deletované oblasti 13q, jako gen DLEU7 nebo DLEU2, které by mohly hrát významnou roli v aktivitě nádorových supresorů [35]. Jednoznačný závěr však zatím nebyl proveden. Analýzy deletované oblasti dále ukázaly, že velká delece zahrnující gen RB1 (typ I) je spojena s kratším časem do první léčby (time to first treatment – TTFT) a OS, ve srovnání s nemocnými, kteří mají kratší delece zahrnující jen geny miR15a and miR16 - 1 (typ II) [35 – 38]. Pacienti mohou mít také bialelickou del(13q), která je pozorována až ve 30 % případů [39]. Bialelická delece je často spojena s malou delecí nezahrnující RB1 gen. Klinický význam bialelické delece je stále kontroverzní, avšak častěji je pozorována u nemocných s agresivnějším průběhem onemocnění [40,41]. Prognostický význam bialelické delece je obtížné stanovit také proto, že může být ovlivněn řadou dalších faktorů, ke kterým řadíme zejména velikost klonu s touto delecí. Orlandi et al ukázali, že klon, který tvoří 65 – 90 % buněčné populace, může znamenat prognostické zhoršení u nemocných s izolovanou del(13q) [41] .
![Rekurentní cytogenetické změny u CLL s vyznačením nejvýznamnějších zasažených genů na jednotlivých chromozomech (podle Puiggros et al, 2014) [116].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/cb1e7b1ffa7b52a323dfc52a49cba076.jpg)
Trizomie chromozomu 12
Tris(12) je druhá nejčastější cytogenetická změna u nemocných s CLL s frekvencí 20 – 30 %, z čehož až v 60 % případů se vyskytuje jako jediná změna karyotypu. Pokud se objevuje spolu s dalšími cytogenetickými aberacemi, pak často s trizomií chromozomů 18 a 19, ale také s del(11q), del(13q), del(14q), del(17p) anebo s translokacemi zahrnujícími gen IGH [7]. Tris(12) je považována za marker střední prognózy, i když některé práce potvrdily výskyt u pacientů s agresivnějším průběhem onemocnění [42]. Tato aberace je považovaná za změnu časného stadia onemocnění, která podporuje akumulaci dalších změn, např. mutací genů NOTCH1 nebo TP53 [43]. Tris(12) vede dále ke zvýšené regulaci řady genů lokalizovaných na chromozomu 12 (obr. 2), přičemž za hlavní patogenetický mechanizmus této změny je považována dávka genů [44].
Delece chromozomu 11
Del(11q) je pozorována u 5 – 20 % CLL nemocných [2,7]. Tato delece je značně heterogenní a je klasifikována buď jako „velká delece“ vyskytující se častěji, nebo jako „malá delece“, která je mnohdy spojena s mutacemi na druhé párové alele. Delece se často vyskytuje společně s dalšími defekty a je součástí komplexních změn, což potvrzuje nestabilitu nádorového genomu [45,46]. Minimálně deletovaná oblast téměř vždy zahrnuje gen ATM. Homozygotní delece nebyla zatím u CLL nemocných pozorována, ale 20 – 30 % nemocných s heterozygotní del(11q) má mutaci párové ATM alely [46]. V blízkosti genu ATM, v oblasti 11q22, je lokalizován také gen BIRC3 (obr. 2), jehož role v patogenezi a prognóze CLL je v současnosti intenzivně studována. Bylo pozorováno, že nemocní s delecí nebo mutací genu BIRC3 jsou často rezistentní na léčbu fludarabinem [47].
Delece chromozomu 17
Del(17p) je pozorována u 3 – 10 % nemocných v době diagnózy CLL [7,47,48], avšak až u 30 % léčených a refrakterních pacientů [48]. Delece může být přítomna de novo v době diagnózy nebo se může objevit až v průběhu onemocnění jako výsledek klonálního vývoje [49]. V současnosti převládá názor, že přítomnost více než 25 % pozitivních jader určených metodou FISH předpovídá horší průběh onemocnění [48,50]. Více než 75 % nemocných s delecí genu TP53 má mutaci v párové alele (obr. 2). Tito nemocní s bialelickým defektem mají horší průběh onemocnění, kratší dobu přežití bez progrese (progression-free survival – PFS) i OS [51]. Nicméně negativní vliv na prognózu má ale také samotná delece nebo mutace genu TP53, což potvrzuje, že každá tato změna je dostatečná pro rezistenci a klonální vývoj. Pomocí sekvenování nové generace s vysokým pokrytím (tzv. ultradeep NGS) se ukázalo, že výskyt TP53 mutací je prognosticky nepříznivý i v případě, že jsou přítomny v malých subklonech [52]. Del(17p) je často součástí komplexních karyotypů, vyskytující se společně s delecí chromozomu 4p, 18p, 20p a změnami chromozomu 8 [53,54].
Komplexní karyotyp a chromotripse
Komplexní karyotyp (CK) je obecně definován jako nález tří a více změn v karyotypu. U nemocných s CLL se vyskytuje přibližně u 16 % nemocných [55,56]. Nález CK je velmi často asociován s nemutovaným stavem IGHV, expresí CD38 a krátkým OS. Provedená analýza počtu změn v komplexním karyotypu ukázala, že prognostická hodnota CK je vyšší u nemocných s nálezem pěti a více změn, a dále byla potvrzena vysoká asociace CK s del(11q) a del(17p) [57]. Baliakas et al analyzovali celkem 1 001 neléčených CLL nemocných a CK pozorovali u 157 (15,6 %) nemocných [56]. Tento nález byl asociován s nemutovaným stavem IGHV a změnami 17p. Nález CK se ukázal jako nezávislý prognostický faktor pro přežívání bez události (event-free survival – EFS) u neléčených CLL pacientů. Cytogenetická analýza 72 nemocných s relabující/ refrakterní CLL, kteří byli léčeni kombinací s ibrutinibem, odhalila CK u 26 (36 %) nemocných, přičemž u 22 z nich byla součástí del(17p). Tato studie dále ukázala, že CK je nezávislý faktor spojený s nepříznivým EFS a OS u těchto nemocných. Toto nebylo potvrzeno u nemocných s del(17p) bez CK, což vedlo k závěru, že s nepříznivým průběhem relabující/ refrakterní CLL je spojen spíše CK než del(17p) [58].
Chromotripse je pojem odvozený z řeckých slov „chromos“ pro chromozom a „thripsis“ pro rozpad chromozomů na kousky. Tento proces úzce souvisí se vznikem komplexního karyotypu a označuje jednu katastrofickou událost, během níž se chromozom nebo více chromozomů rozpadne na mnoho kousků, které se pak spojují zpět pomocí DNA opravných procesů v náhodném pořadí. Takto mohou být vysvětleny komplexní chromozomové přestavby s řadou zlomů představující tzv. marker chromozomy. V nádorových buňkách vede chromotripse ke ztrátě nádorových supresorů a může být příčinou amplifikace onkogenů. Chromotripse se může vyskytovat u 2 – 3 % nádorů a byla pozorována až u 25 % nádorů kostí [59]. V případě CLL byl tento jev objeven nedávno pomocí metody NGS, kdy se jednalo o chromotripsi chromozomu 4 [59]. Kombinací celogenomového sekvenování, komparativní genomové hybridizace (arrayCGH) a SNPs array se povedlo analyzovat a určit chromotripsi i u řady dalších nádorů [60,61].
Poškození ATM/ p53 apoptické dráhy
Narušení apoptotické dráhy je další velice důležitý mechanizmus patogeneze CLL, které umožňuje přežívání B lymfocytů v periferní krvi. Molekulární defekty bránící správnému průběhu apoptózy zahrnují zvýšenou expresi antiapoptických proteinů rodiny Bcl ‑ 2 a aberace (delece a/ nebo mutace) nádorových supresorů TP53 a ATM, které představují významné negativní prognostické faktory CLL. ATM/ p53 signální dráha spouští v odpovědi na buněčný stres mechanizmy zabezpečující potlačení klonální expanze buněk a udržení integrity genomu [62]. Samotný prognostický význam hladiny proteinu Bcl ‑ 2 nebyl prokázán a některé studie ukazují spíše na významný poměr hladin molekul Bcl ‑ 2/ Bax při regulaci apoptózy [63].
Aberace genu TP53
Protein p53 je jaderný transkripční faktor, který se v závislosti na rozsahu a typu buněčného poškození podílí zejména na reparaci DNA, blokaci buněčného cyklu a iniciaci apoptózy. Somatické poškození genu TP53 bylo popsáno u více než poloviny onkologických malignit a v případě leukemií a lymfomů má jeho stanovení silný prognostický a prediktivní význam [64,65]. Výskyt zárodečných aberací genu TP53 je navíc spjat s autozomálně dominantními Li ‑ Fraumeni a Li ‑ Fraumeni‑like syndromy, vyznačujícími se rodinnou predispozicí k vývoji četných nádorových onemocnění v mladém věku (zpravidla v < 45 letech) [66]. Nejčastějšími způsoby deregulace proteinu p53 je narušení vazebné a/ nebo transaktivační schopnosti tohoto nádorového supresoru; popsáno je ovšem také získání onkogenní aktivity p53 u CLL mutovaných buněk [67]. U nově diagnostikované CLL jsou aberace genu TP53 detekovány u 5 – 10 % pacientů a zpravidla asociují se zdaleka nejhorší prognózou onemocnění [7,68]. Nejkratší OS přitom vykazují nemocní s bialelickým poškozením genu TP53 (medián ~19 měsíců) [68,69], kdy je del(17p) nejčastěji kombinována s mutacemi lokalizovanými v oblasti DNA ‑ vazebné domény [70]. Výskyt samostatné mutace genu TP53 je u neléčené CLL pozorován vcelku ojediněle (3 – 5 % pacientů), ovšem stejně jako bialelický defekt přispívá k rezistenci nádorových klonů na standardní chemoimunoterapii a celkovou léčebnou odpověď tak dosáhne pouze zhruba 30 – 60 % pacientů [68,71]. Při relapsu onemocnění jsou navíc často leukemické buňky nesoucí TP53 aberace v rámci klonálního vývoje selektovány [13,72]. Tyto aberace je pak možné zaznamenat u 30 – 40 % chemorefrakterní CLL a až u poloviny případů RS [14,73]. Detekce minoritních TP53 mutací je ovšem v diagnostických vzorcích často z důvodu nedostačující senzitivity konvenčně používaných metod velmi problematická a v těchto případech je doporučováno využití přístupů NGS [52,72,74]. Nezbytná součást mutační analýzy je také stanovení funkčního důsledku každé detekované TP53 varianty, ať už pomocí specifických testů (zejména FASAY) nebo dohledáním v dostupných databázích (http:/ / p53.iarc.fr/ ,http:/ / p53.free.fr/ ). Současné poznatky týkající se analýzy mutací genu TP53 včetně charakterizace nejčastěji používaných detekčních metodik, které lze využít také u ostatních nádorových onemocnění, jsou přehledně shrnuty v mezinárodně platných doporučeních Evropské iniciativy pro výzkum CLL [74]. Nemocným s jakoukoliv prokázanou patogenní TP53 aberací je standardně doporučována účast v klinických studiích testujících léčiva nezávislá na funkci p53 signální dráhy, popřípadě jsou indikováni k alogenní transplantaci krvetvorných buněk [1,75]. V minulém roce bylo v USA organizací FDA pro léčbu těchto pacientů a nemocných s relabovanou nebo refrakterní CLL schváleno použití nového tyrozinkinázového inhibitoru ibrutinibu na základě výsledků studie RESONATE [76].
Inaktivace genu ATM
ATM je převážně jaderná kináza, která je aktivována po vzniku dvouřetězcových zlomů DNA a prostřednictvím fosforylace aktivuje řadu efektorových molekul zahrnutých v odpovědi na poškození DNA [77]. Kinázová kaskáda směřuje k mnoha cílovým molekulám, z nichž nejvýznamnější je protein p53 spouštějící procesy na ochranu integrity genomu [78]. Zárodečné mutace v genu ATM vedou k autozomálně recesivnímu syndromu ataxia ‑ telangiectasia, který je spojen se zvýšeným rizikem infekcí a vzniku nádorových onemocnění, zejména leukemií, lymfomů a nádorů prsu [79]. Gen ATM může být u CLL pacientů zasažen prostřednictvím del(11q) a/nebo somatické (případně germinalní) mutace. Při analýze obou typů defektů se ukázalo, že dohromady představují nejčastější abnormalitu spojenou s negativní prognózou při diagnóze CLL, kdy se vyskytují u přibližně 25 % pacientů [80]. U pacientů vyžadujících léčbu může být tato frekvence ještě vyšší [81]. Dopad ATM defektů na prognózu pacientů byl prokázán v řadě studií, které ukazují jejich negativní vliv na přežití a také jejich potenciální prediktivní charakter [80 – 84]. Nicméně analýza mutací v tomto extrémně dlouhém genu (62 kódujících exonů) je pro většinu laboratoří náročná, a proto se rutinně vyšetřuje pouze del(11q) . Přestože NGS technologie značně zjednodušily tento proces, interpretace identifikovaných variant zůstává nadále problematická. Je proto velmi vhodné využít některý z funkčních testů, který by vyčlenil skutečně patogenní mutace s jednoznačným vlivem na funkci proteinu. Tyto testy jsou založeny zejména na sledování indukce exprese p53 a jeho cílového genu p21 po poškození DNA ionizujícím zářením nebo vybranými chemoterapeutiky [85 – 88]. Právě pomocí funkčních analýz bylo zjištěno, že kompletní inaktivace ATM je u CLL pacientů zprostředkována zejména bialelickými defekty (del(11q) / mutace nebo mutace/ mutace), které vedou k trvalému narušení p53 dráhy po poškození DNA [81]. Samotná del(11q) k inaktivaci ATM nevede, protože transkripce z druhé alely je schopna zajistit dostačující syntézu funkčního proteinu, avšak může ovlivňovat průběh choroby prostřednictvím haploinsuficience dalších genů lokalizovaných v oblasti 11q22 – 23 (např. BIRC3) [80]. Klinický dopad jednotlivých ATM defektů u CLL pacientů byl podrobněji popsán teprve nedávno, protože starší studie sledovaly pouze vliv del(11q) bez analýzy možné přítomnosti ATM mutací. Efekt mutace doprovázející asi ve třetině případů deleci se projevuje ve významném zkrácení TTFT a OS ve srovnání se samotnou delecí [81]. Práce Skowronské et al prokázala signifikantní zhoršení PFS a OS pouze u pacientů s bialelickým ATM defektem [83]. Navíc bylo zjištěno, že tito pacienti jsou rezistentní na standardně používanou cytotoxickou chemoterapii, jejíž mechanizmus účinku je závislý na funkční p53 dráze [81,83]. Proto je do budoucnosti velice žádoucí hledat nové možnosti léčby s p53 nezávislým mechanizmem účinku, které by byly vhodné pro ATM ‑ defektní pacienty. Jednou z možností by mohlo být použití inhibitorů BCR signalizace [89] nebo také některého z nových terapeutických přístupů založených na konceptu syntetické letality, který k usmrcení nádorových buněk využívá dysfunkce ATM spolu s terapeutickým zvýšením genotoxického stresu [90].
Význam deregulace miRNA
MiRNA jsou krátké nekódující úseky RNA podílející se na regulaci exprese nejméně 20 – 30 % všech protein kódujících genů, zahrnutých zejména v buněčné proliferaci, diferenciaci a apoptóze. Jejich význam v iniciaci a progresi onkologických malignit podtrhuje fakt, že zhruba polovina známých lidských miRNA je lokalizována v genomových oblastech asociovaných s karcinogenezí [33]. Nádorově supresorová, popřípadě onkogenní funkce miRNA byla popsána také u CLL, kde je možné na základě specifického expresního profilu těchto markerů odlišit nádorové buňky od fyziologických B lymfocytů [91]. Pomocí vytipovaného panelu miRNA vyznačujících se v rámci CLL rozdílnou expresí lze do určité míry ve spojitosti s ostatními molekulárními markery rovněž predikovat průběh tohoto onemocnění [91,92]. Zdaleka nejčastěji (> 60 % případů) je u CLL pozorována deregulace klastru miR 15a a miR 16- 1 lokalizovaných v často deletované oblasti 13q14 [33], která vede k navýšení exprese antiapoptotického proteinu Bcl ‑ 2 [93]. Vztah mezi hladinou exprese miRNA, rekurentními genomovými defekty a klinickým průběhem CLL byl prokázán rovněž u genů kódujících miR 34b/ c (lokus 11q22.3) a miR 34a regulovaných proteinem p53 [94]. Význam aberantní exprese těchto a dalších miRNA (např. miR 150 a miR 155) u CLL je dáván do spojitosti také s aktivní BCR signalizací asociovanou se zvýšenou proliferací leukemických buněk [27,95,96].
Příčinou deregulované exprese miRNA mohou být také mutace v těchto markerech (detekované např. u miR 16- 1), které byly sporadicky nalezeny u pacientů s familiární CLL, a poukazují tak na význam genetické predispozice při jejím vývoji [91]. Navzdory prokázané úloze při patogenezi CLL není prozatím aberantní exprese miRNA u tohoto onemocnění z důvodu nestandardizované detekční metodiky rutinně vyšetřována.
Nové potenciální prognostické markery CLL
Rychle se vyvíjející technologie NGS umožnily pomocí celogenomového či celoexomového přístupu objevení dalších rekurentně se vyskytujících genových aberací u CLL [8,9,11]. Jsou to zejména mutace v genech SF3B1, NOTCH1 nebo BIRC3, které se vyskytují u 5 – 15 % nově diagnostikovaných pacientů a zejména pak v pozdějších fázích onemocnění (obr. 1). Jejich klinický dopad a možnost využití jako nových „biomarkerů“ je v současné době intenzivně studován [97 – 99]. V nedávné době bylo popsáno, že jsou asociovány s nepříznivými cytogenetickými aberacemi a také s celkově horším průběhem choroby, odrážejícím se v rychlejší progresi, rezistenci na léčbu a kratším PFS a OS pacientů (tab. 1) [19,47,98 – 100]. Na druhé straně ale není přesně znám funkční dopad těchto mutací a také chybí standardizované detekční metody, což prozatím znemožňuje zavedení těchto analýz mezi rutinní vyšetření CLL pacientů. Další rekurentně mutovaný gen MYD88, jehož proteinový produkt se podílí na iniciaci imunitní odpovědi spojené se zánětem, byl v nedávné době asociován s mutovaným IGHV a del(13q) a zřejmě je tedy spojen s příznivější prognózou CLL pacientů (tab. 1) [11,43].
Rossi et al integrovali tyto nově nalezené molekulární defekty do klasického cytogenetického hierarchického modelu a navrhli tak potenciálně dokonalejší prognostickou stratifikaci CLL pacientů [101]. Rozlišují čtyři skupiny: 1. vysoce rizikoví pacienti s defekty v genech TP53 a/ nebo BIRC3, 2. pacienti se středním rizikem s mutacemi v NOTCH1 a/ nebo SF3B1 a/ nebo s del(11q) , 3. pacienti s nízkým rizikem nesoucí tris(12) nebo normální karyotyp a 4. pacienti s velmi nízkým rizikem nesoucí pouze del(13q). V nedávné studii Baliakas et al podpořili klinickou významnost zejména SF3B1 a NOTCH1 mutací [19]. Nicméně tento prognostický model bude potřeba ještě ověřit na základě prospektivních klinických studií. Prediktivní potenciál těchto nových markerů s cílem využití v personalizované léčbě je stále předmětem diskuzí.
Mutace genu SF3B1
SF3B1 je jedna z proteinových komponent sestřihového aparátu buňky (splicezom), který se podílí na úpravě prekurzorové mRNA do její zralé formy [102]. Gen SF3B1 je lokalizován na 2. chromozomu a jeho mutace se vyskytují zejména v exonech 14 – 16 v tzv. HEAT doménách v několika preferenčně mutovaných kodonech. Nejčastější mutací je záměna K700E, vyskytující se asi ve 40 – 50 % případů. Frekvence mutací roste od diagnózy (5 – 10 %), přes progresi choroby (17 %) až k refrakterní formě CLL (20 %) [8,11,98,99]. Mutace v SF3B1 jsou asociovány s nemutovaným IGHV, s del(11q) a zdá se, že také s mutacemi genu ATM [11,99], čímž dále zhoršují už tak nepříznivou prognózu těchto pacientů; klinicky se projevují značně zkráceným TTFT a kratším OS pacientů [99,103]. Jejich funkční dopad není prozatím přesně objasněn, nicméně Paulsen et al ve své studii ukázali stěžejní roli sestřihového aparátu v udržování genomické stability, kdy při zablokování jednotlivých komponent splicesomu došlo k akumulaci poškození DNA [104]. Je tedy možné, že mutace SF3B1 vedou k narušení odpovědi na poškození DNA, podobně jako mutace v ATM [105].
Mutace genu NOTCH1
Gen NOTCH1, lokalizovaný v oblasti 9q34.3, kóduje heterodimerní transmembránový protein podílející se na regulaci buněčné proliferace (NF‑κB dráha), diferenciace a apoptózy. Aberace tohoto transkripčního faktoru byly asociovány s patogenezí řady nádorových onemocnění, kdy v rámci hematoonkologických malignit je výskyt prognosticky nepříznivých mutací genu NOTCH1 pozorován zejména u T ‑ akutní lymfoblastické leukemie (~50 % případů) [106]. V případě CLL jsou somatické onkogenní mutace genu NOTCH1 detekovány u 8 – 12 % nově diagnostikovaných pacientů [9,10] a zpravidla vedou ke konstitutivní aktivaci NOTCH1 signální dráhy. Zdaleka nejčastěji je přitom u CLL analyzována hot spot mutace c.7544_7545delCT (70 – 80 % NOTCH1 mutací) vedoucí ke vzniku předčasně zkrácené aktivní formy proteinu. Spolu s ostatními NOTCH1 defekty je tato mutace pozorována zejména u pacientů s nemutovaným stavem genů IGHV a také s tris(12) [9,100]. Z hlediska prognózy CLL je výskyt patogenních mutací genu NOTCH1 zpravidla spojen se zkráceným PFS a OS pacientů (medián ~55 měsíců) [98] a také zvýšeným rizikem Richterovy transformace [10,107]. Předběžné výsledky klinických studií poukazují též na prediktivní význam přítomnosti NOTCH1 mutací u CLL, kdy u těchto pacientů nebyl prokázán léčebný prospěch z přidání rituximabu k režimu fludarabin‑cyklofosfamid [97].
Defekty genu BIRC3
BIRC3 je negativní regulátor kinázy MAPK, která aktivuje nekanonickou NF‑κB dráhu stimulující proliferaci buněk [108]. U CLL pacientů může být BIRC3 inaktivován pomocí del(11q) a/nebo mutace, podobně jak je tomu v případě ATM [19,47]. Gen BIRC3 je lokalizován do oblasti 11q22.2, ale nemusí vždy v případě del(11q) chybět. Studie Rose‑Zerilliho et al detekovala deleci BIRC3 v 83 % případů delece genu ATM a ukázala, že spíše ATM mutace než BIRC3 delece a/ nebo mutace ovlivňují přežití pacientů s del(11q) [109]. Inaktivace proteinu BIRC3 je spojena především s posunovými mutacemi nebo mutacemi vedoucímu ke vzniku stop kodonu, které způsobují ztrátu E3 ubikvitin ligázové aktivity směrem ke kináze MAPK a následně vedou ke konstitutivní aktivaci nekanonické NF‑κB dráhy. Výskyt mutací při diagnóze je nízký (5 %), zatímco u relabujících a fludarabin‑refrakterních pacientů je poměrně vysoký (25 %) [47].
Klonální evoluce molekulárních aberací u CLL
CLL je klonální onemocnění, které prochází několika klinickými stadii od bezpříznakové fáze, přes symptomatickou CLL, která může přejít až ve vysoce agresivní formu známou jako RS. V průběhu CLL je možné detekovat různé molekulární léze, které mohou být klonálně zvýhodňovány a přispívat tak k progresi onemocnění (obr. 1). Biologická variabilita CLL je tedy dána různou kombinací genetických abnormalit a přítomností subklonálních defektů [110]. Ačkoliv se prozatím u CLL nepodařilo přímo určit kauzální (tzv. founder) aberaci podílející se na prvotní iniciaci onemocnění, byly odhaleny genomové změny spojené s vývojem a progresí CLL. Tyto tzv. driver aberace se mohou vyskytovat klonálně nebo subklonálně v časnějších, resp. pozdějších fázích evoluce CLL [9,43,111].
Mezi nejčastěji deregulované buněčné mechanizmy u CLL patří zejména narušení DNA reparace a kontroly buněčného cyklu (aberace TP53, ATM, BIRC3), ovlivnění RNA zpracování a sestřihu (mutace SF3B1, XPO1), navýšení intenzity proliferace (mutace NOTCH1, FBXW7, KRAS, NRAS, MED12), modulace imunitních mechanizmů (mutace MYD88) a změna modifikace chromatinu (CHD2) [11,43]. Řadu těchto patogenetických klonálních defektů je přitom možné zaznamenat už na úrovni hematopoetických prekurzorů a také u myeloidních buněk pacientů s CLL [112,113] a mohou se uplatnit rovněž při vývoji MBL. Tento premaligní leukemický stav nevyžadující hematologickou léčbu je možné diagnostikovat až u ~13 % příbuzných pacientů s CLL, z nichž 1 – 2 % případů ročně progreduje do klinicky vyjádřené CLL [114].
V časných stadiích neléčené CLL jsou pomocí konvenčně dostupných metod ve většině nádorových buněk nalézány spíše klinicky příznivé genomové defekty (nejčastěji del(13q) a mutace genu MYD88), které jsou predominantně klonálního původu [43]. Až ve 20 % případů ovšem v průběhu CLL dochází také k nárůstu subklonálních populací nesoucích prognosticky nepříznivé genové aberace (např. TP53, ATM a SF3B1), které přispívají k agresivnímu fenotypu onemocnění včetně rezistence na standardně podávanou chemoimunoterapii (obr. 1). Riziko vývoje genomových aberací se přitom značně zvyšuje s přítomností nemutovaného IGHV a podáním terapie, která poskytuje významný selekční tlak [13,72,101]. Narušení reparačních a apoptotických mechanizmů u leukemických buněk ve spojení s navýšením intenzity jejich proliferace napomáhá akumulaci dalších patogenetických změn [43,110]. Důsledkem může být progrese CLL do malignit vyššího stupně, nejčastěji difúzního velkobuněčného B lymfomu, popřípadě prolymfocytární leukemie, které lze detekovat u 5 – 15 % pacientů a jsou spojeny s infaustní prognózou onemocnění [115].
Dosud ne zcela objasněnou otázkou klonálního vývoje CLL zůstává, zda jsou později detekované mutace následkem spontánní mutageneze nádorové buňky nebo zda jsou do DNA vnášeny agresivní genotoxickou terapií. Nejnovější studie ukazují, že minoritně mutované subklony jsou přítomny v CLL buňkách již před terapií, která následně přispívá k jejich expanzi, a to až ve chvíli, kdy je eliminována většina nádorové populace senzitivní na léčbu [43,72,111].
Závěr
Klinická heterogenita CLL se zdá být v přímé souvislosti s biologickou podstatou tohoto dosud neléčitelného onemocnění. Nejsilnějšími klinicky významnými prognostickými markery CLL nadále zůstávají analýza mutačního stavu genů IGHV ve spojení s vyšetřením stávajících rekurentních chromozomálních a genových aberací. Při predikci léčebné odpovědi je kromě del(17p) a del(11q) doporučováno také stanovení mutačního profilu kódujících oblastí genů TP53 a ATM, které společně pomáhají určit pacienty schopné profitovat z alternativních léčebných modalit. V souvislosti s novými poznatky o patogenezi CLL a vývojem sofistikovanějších detekčních přístupů lze do budoucna očekávat rozšíření portfolia vyšetřovaných molekulárních markerů. Důkladné rozdělení pacientů dle přítomnosti rizikových genomových abnormalit by mohlo napomoci při výběru efektivní cílené terapie určené pro danou skupinu pa
Práce byla realizována za podpory grantů IGA MZ ČR NT13493-4/2012, NT13576-4/2012, NT13576, AZV MZ ČR č. 15-30015A-4/2015 a 15-31834A-4/2015 a TAČR TE02000058.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
prof. RNDr. Šárka Pospíšilová, Ph.D.
Interní hematologická a onkologická klinika
LF MU a FN Brno
Jihlavská 20
625 00 Brno
e-mail: sarka.pospisilova@fnbrno.cz
Obdrženo: 30. 7. 2015
Přijato: 4. 8. 2015
Sources
1. Hallek M, Cheson BD, Catovsky D et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute ‑ Working Group 1996 guidelines. Blood 2008; 111(12): 5446 – 5456. doi: 10.1182/ blood ‑ 2007 ‑ 06 ‑ 093906.
2. Zenz T, Mertens D, Küppers R et al. From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat Rev Cancer 2010; 10(1): 37 – 50. doi: 10.1038/ nrc2764.
3. Chiorazzi N, Ferrarini M. Cellular origin(s) of chronic lymphocytic leukemia: cautionary notes and additional considerations and possibilities. Blood 2011; 117(6): 1781 – 1791. doi: 10.1182/ blood ‑ 2010 ‑ 07 ‑ 155663.
4. Hamblin TJ, Davis Z, Gardiner A et al. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999; 94(6): 1848 – 1854.
5. Damle RN, Wasil T, Fais F et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999; 94(6): 1840 – 1847.
6. Agathangelidis A, Darzentas N, Hadzidimitriou A et al. Stereotyped B ‑ cell receptors in one ‑ third of chronic lymphocytic leukemia: a molecular classification with implications for targeted therapies. Blood 2012; 119(19): 4467 – 4475. doi: 10.1182/ blood ‑ 2011 ‑ 11 ‑ 393694.
7. Döhner H, Stilgenbauer S, Benner A et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 2000; 343(26): 1910 – 1916.
8. Quesada V, Conde L, Villamor N et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet 2011; 44(1): 47 – 52. doi: 10.1038/ ng.1032.
9. Puente XS, Pinyol M, Quesada V et al. Whole ‑ genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011; 475(7354): 101 – 105. doi: 10.1038/ nature10113.
10. Fabbri G, Rasi S, Rossi D et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med 2011; 208(7): 1389 – 1401. doi: 10.1084/ jem.20110921.
11. Wang L, Lawrence MS, Wan Y et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med 2011; 365(26): 2497 – 2506. doi: 10.1056/ NEJMoa1109016.
12. D‘Arena G, Musto P. Monoclonal B ‑ cell lymphocytosis. Transl Med UniSa 2014; 8 : 75 – 79.
13. Stilgenbauer S, Sander S, Bullinger L et al. Clonal evolution in chronic lymphocytic leukemia: acquisition of high‑risk genomic aberrations associated with unmutated VH, resistance to therapy, and short survival. Haematologica 2007; 92(9): 1242 – 1245.
14. Rossi D, Spina V, Deambrogi C et al. The genetics of Richter syndrome reveals disease heterogeneity and predicts survival after transformation. Blood 2011; 117(12): 3391 – 3401. doi: 10.1182/ blood ‑ 2010 ‑ 09 ‑ 302174.
15. Ghia P, Stamatopoulos K, Belessi C et al. ERIC recommendations on IGHV gene mutational status analysis in chronic lymphocytic leukemia. Leukemia 2007; 21(1): 1 – 3.
16. Thorsélius M, Kröber A, Murray F et al. Strikingly homologous immunoglobulin gene rearrangements and poor outcome in VH3-21 - using chronic lymphocytic leukemia patients independent of geographic origin and mutational status. Blood 2006; 107(7): 2889 – 2894.
17. Stamatopoulos K, Belessi C, Moreno C et al. Over 20% of patients with chronic lymphocytic leukemia carry stereotyped receptors: Pathogenetic implications and clinical correlations. Blood 2007; 109(1): 259 – 270.
18. Strefford JC, Sutton LA, Baliakas P et al. Distinct patterns of novel gene mutations in poor ‑ prognostic stereotyped subsets of chronic lymphocytic leukemia: the case of SF3B1 and subset #2. Leukemia 2013; 27(11): 2196 – 2199. doi: 10.1038/ leu.2013.98.
19. Baliakas P, Hadzidimitriou A, Sutton LA et al. Recurrent mutations refine prognosis in chronic lymphocytic leukemia. Leukemia 2015; 29(2): 329 – 336. doi: 10.1038/ leu.2014.196.
20. Wiestner A, Rosenwald A, Barry TS et al. ZAP ‑ 70 expression identifies a chronic lymphocytic leukemia subtype with unmutated immunoglobulin genes, inferior clinical outcome, and distinct gene expression profile. Blood 2003; 101(12): 4944 – 4951.
21. Scielzo C, Camporeale A, Geuna M et al. ZAP ‑ 70 is expressed by normal and malignant human B ‑ cell subsets of different maturational stage. Leukemia 2006; 20(4): 689 – 695.
22. Rassenti LZ, Huynh L, Toy TL et al. ZAP ‑ 70 compared with immunoglobulin heavy‑chain gene mutation status as a predictor of disease progression in chronic lymphocytic leukemia. N Engl J Med 2004; 351(9): 893 – 901.
23. Deaglio S, Vaisitti T, Aydin S et al. CD38 and ZAP ‑ 70 are functionally linked and mark CLL cells with high migratory potential. Blood 2007; 110(12): 4012 – 4021.
24. Quiroga MP, Balakrishnan K, Kurtova AV et al. B ‑ cell antigen receptor signaling enhances chronic lymphocytic leukemia cell migration and survival: specific targeting with a novel spleen tyrosine kinase inhibitor, R406. Blood 2009; 114(5): 1029 – 1037. doi: 10.1182/ blood ‑ 2009‑03 ‑ 212837.
25. Dal Porto JM, Gauld SB, Merrell KT et al. B cell antigen receptor signaling 101. Mol Immunol 2004; 41(6 – 7): 599 – 613.
26. Burger JA. Inhibiting B ‑ cell receptor signaling pathways in chronic lymphocytic leukemia. Curr Hematol Malig Rep 2012; 7(1): 26 – 33. doi: 10.1007/ s11899 ‑ 011 ‑ 0104 ‑ z.
27. Mraz M, Chen L, Rassenti LZ et al. miR ‑ 150 influences B ‑ cell receptor signaling in chronic lymphocytic leukemia by regulating expression of GAB1 and FOXP1. Blood 2014; 124(1): 84 – 95. doi: 10.1182/ blood ‑ 2013 ‑ 09 ‑ 527234.
28. Binet JL, Auquier A, Dighiero G et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 1981; 48(1): 198 – 206.
29. Rai KR, Sawitsky A, Cronkite EP et al. Clinical staging of chronic lymphocytic leukemia. Blood 1975; 46(2): 219 – 234.
30. Jarosova M, Urbankova H, Plachy R et al. Gain of chromosome 2p in chronic lymphocytic leukemia: significant heterogeneity and a new recurrent dicentric rearrangement. Leuk Lymphoma 2010; 51(2): 304 – 313. doi: 10.3109/ 10428190903518311.
31. Chapiro E, Leporrier N, Radford ‑ Weiss I et al. Gain of the short arm of chromosome 2 (2p) is a frequent recurring chromosome aberration in untreated chronic lymphocytic leukemia (CLL) at advanced stages. Leuk Res 2010; 34(1): 63 – 68. doi: 10.1016/ j.leukres.2009.03.042.
32. Rinaldi A, Mian M, Kwee I et al. Genome ‑ wide DNA profiling better defines the prognosis of chronic lymphocytic leukaemia. Br J Haematol 2011; 154(5): 590 – 599. doi: 10.1111/ j.1365 ‑ 2141.2011.08789.x.
33. Calin GA, Dumitru CD, Shimizu M et al. Frequent deletions and down ‑ regulation of micro ‑ RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 2002; 99(24): 15524 – 15529.
34. Klein U, Lia M, Crespo M et al. The DLEU2/ miR ‑ 15a/ 16 - 1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 2010; 17(1): 28 – 40. doi: 10.1016/ j.ccr.2009.11.019.
35. Parker H, Rose‑Zerilli MJ, Parker A et al. 13q deletion anatomy and disease progression in patients with chronic lymphocytic leukemia. Leukemia 2011; 25(3): 489 – 497. doi: 10.1038/ leu.2010.288.
36. Ouillette P, Erba H, Kujawski L et al. Integrated genomic profiling of chronic lymphocytic leukemia identifies subtypes of deletion 13q14. Cancer Res 2008; 68(4): 1012 – 1021. doi: 10.1158/ 0008 ‑ 5472.CAN ‑ 07 ‑ 3105.
37. Ouillette P, Collins R, Shakhan S et al. The prognostic significance of various 13q14 deletions in chronic lymphocytic leukemia. Clin Cancer Res 2011; 17(21): 6778 – 6790. doi: 10.1158/ 1078 ‑ 0432.CCR ‑ 11 ‑ 0785.
38. Mosca L, Fabris S, Lionetti M et al. Integrative genomics analyses reveal molecularly distinct subgroups of B ‑ cell chronic lymphocytic leukemia patients with 13q14 deletion. Clin Cancer Res 2010; 16(23): 5641 – 5653. doi: 10.1158/ 1078 ‑ 0432.CCR ‑ 10 ‑ 0151.
39. Reddy KS. Chronic lymphocytic leukaemia profiled for prognosis using a fluorescence in situ hybridisation panel. Br J Haematol 2006; 132(6): 705 – 722.
40. Chena C, Avalos JS, Bezares RF et al. Biallelic deletion 13q14.3 in patients with chronic lymphocytic leukemia: cytogenetic, FISH and clinical studies. Eur J Haematol 2008; 81(2): 94 – 99. doi: 10.1111/ j.1600 ‑ 0609.2008.01086.x.
41. Orlandi EM, Bernasconi P, Pascutto C et al. Chronic lymphocytic leukemia with del13q14 as the sole abnormality: dynamic prognostic estimate by interphase ‑ FISH. Hematol Oncol 2013; 31(3): 136 – 142. doi: 10.1002/ hon.2032.
42. Matutes E, Oscier D, Garcia ‑ Marco J et al. Trisomy 12 defines a group of CLL with atypical morphology: correlation between cytogenetic, clinical and laboratory features in 544 patients. Br J Haematol 1996; 92(2): 382 – 388.
43. Landau DA, Carter SL, Stojanov P et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 2013; 152(4): 714 – 726. doi: 10.1016/ j.cell.2013.01.019.
44. Kienle DL, Korz C, Hosch B et al. Evidence for distinct pathomechanisms in genetic subgroups of chronic lymphocytic leukemia revealed by quantitative expression analysis of cell cycle, activation, and apoptosis‑associated genes. J Clin Oncol 2005; 23(16): 3780 – 3792.
45. Marasca R, Maffei R, Martinelli S et al. Clinical heterogeneity of de novo 11q deletion chronic lymphocytic leukaemia: prognostic relevance of extent of 11q deleted nuclei inside leukemic clone. Hematol Oncol 2013; 31(2): 88 – 95. doi: 10.1002/ hon.2028.
46. Ouillette P, Li J, Shaknovich R et al. Incidence and clinical implications of ATM aberrations in chronic lymphocytic leukemia. Genes Chromosomes Cancer 2012; 51(12): 1125 – 1132. doi: 10.1002/ gcc.21997.
47. Rossi D, Fangazio M, Rasi S et al. Disruption of BIRC3 associates with fludarabine chemorefractoriness in TP53 wild‑type chronic lymphocytic leukemia. Blood 2012; 119(12): 2854 – 2862. doi: 10.1182/ blood ‑ 2011 ‑ 12 ‑ 395673.
48. Delgado J, Espinet B, Oliveira AC et al. Chronic lymphocytic leukaemia with 17p deletion: a retrospective analysis of prognostic factors and therapy results. Br J Haematol 2012; 157(1): 67 – 74. doi: 10.1111/ j.1365 ‑ 2141.2011.09000.x.
49. Tam CS, Shanafelt TD, Wierda WG et al. De novo deletion 17p13.1 chronic lymphocytic leukemia shows significant clinical heterogeneity: the M. D. Anderson and Mayo Clinic experience. Blood 2009; 114(5): 957 – 964. doi: 10.1182/ blood ‑ 2009 ‑ 03 ‑ 210591.
50. Oscier D, Wade R, Davis Z et al. Prognostic factors identified three risk groups in the LRF CLL4 trial, independent of treatment allocation. Haematologica 2010; 95(10): 1705 – 1712. doi: 10.3324/ haematol.2010.025338.
51. Zenz T, Kröber A, Scherer K et al. Monoallelic TP53 inactivation is associated with poor prognosis in chronic lymphocytic leukemia: results from a detailed genetic characterization with long‑term follow‑up. Blood 2008; 112(8): 3322 – 3329. doi: 10.1182/ blood ‑ 2008 ‑ 04 ‑ 154070.
52. Rossi D, Khiabanian H, Spina V et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood 2014; 123(14): 2139 – 2147. doi: 10.1182/ blood ‑ 2013 ‑ 11 ‑ 539726.
53. Gunnarsson R, Mansouri L, Isaksson A et al. Array‑based genomic screening at diagnosis and during follow‑up in chronic lymphocytic leukemia. Haematologica 2011; 96(8): 1161 – 1169. doi: 10.3324/ haematol.2010.039768.
54. Rudenko HC, Else M, Dearden C et al. Characterising the TP53 - deleted subgroup of chronic lymphocytic leukemia: an analysis of additional cytogenetic abnormalities detected by interphase fluorescence in situ hybridisation and array‑based comparative genomic hybridisation. Leuk Lymphoma 2008; 49(10): 1879 – 1886. doi: 10.1080/ 10428190802345902.
55. Haferlach C, Dicker F, Schnittger S et al. Comprehensive genetic characterization of CLL: a study on 506 cases analysed with chromosome banding analysis, interphase FISH, IgV(H) status and immunophenotyping. Leukemia 2007; 21(12): 2442 – 2451.
56. Baliakas P, Iskas M, Gardiner A et al. Chromosomal translocations and karyotype complexity in chronic lymphocytic leukemia: a systematic reappraisal of classic cytogenetic data. Am J Hematol 2014; 89(3): 249 – 255. doi: 10.1002/ ajh.23618.
57. Jaglowski SM, Ruppert AS, Heerema NA et al. Complex karyotype predicts for inferior outcomes following reduced ‑ intensity conditioning allogeneic transplant for chronic lymphocytic leukaemia. Br J Haematol 2012; 159(1): 82 – 87. doi: 10.1111/ j.1365 ‑ 2141.2012.09239.x.
58. Thompson PA, O’Brien SM, Wierda WG A et al. Complex karyotype, rather than del(17p), is associated with inferior outcomes in relapsed or refractory CLL patients treated with Ibrutinib‑based regimens. Blood 2014; 124(21): abstr. doi: 10.1002/ cncr.29566.
59. Stephens PJ, Greenman CD, Fu B et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011; 144(1): 27 – 40. doi: 10.1016/ j.cell.2010.11.055.
60. Rausch T, Jones DT, Zapatka M et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 2012; 148(1 – 2): 59 – 71. doi: 10.1016/ j.cell.2011.12.013.
61. Korbel JO, Campbell PJ. Criteria for inference of chromothripsis in cancer genomes. Cell 2013; 152(6): 1226 – 1236. doi: 10.1016/ j.cell.2013.02.023.
62. Lane DP. Cancer. p53, guardian of the genome. Nature 1992; 358(6381): 15 – 16.
63. Thomas A, El Rouby S, Reed JC et al. Drug‑induced apoptosis in B ‑ cell chronic lymphocytic leukemia: relationship between p53 gene mutation and bcl ‑ 2/ bax proteins in drug resistance. Oncogene 1996; 12(5): 1055 – 1062.
64. Petitjean A, Mathe E, Kato S et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat 2007; 28(6): 622 – 629.
65. Robles AI, Harris CC. Clinical outcomes and correlates of TP53 mutations and cancer. Cold Spring Harb Perspect Biol 2010; 2(3). doi: 10.1101/ cshperspect.a001016.
66. Olivier M, Goldgar DE, Sodha N et al. Li ‑ Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res 2003; 63(20): 6643 – 6650.
67. Trbusek M, Smardova J, Malcikova J et al. Missense mutations located in structural p53 DNA‑binding motifs are associated with extremely poor survival in chronic lymphocytic leukemia. J Clin Oncol 2011; 29(19): 2703 – 2708. doi: 10.1200/ JCO.2011.34.7872.
68. Zenz T, Eichhorst B, Busch R et al. TP53 mutation and survival in chronic lymphocytic leukemia. J Clin Oncol 2010; 28(29): 4473 – 4479. doi: 10.1200/ JCO.2009.27.8762.
69. Malcikova J, Smardova J, Rocnova L et al. Monoallelic and biallelic inactivation of TP53 gene in chronic lymphocytic leukemia: selection, impact on survival, and response to DNA damage. Blood 2009; 114(26): 5307 – 5314. doi: 10.1182/ blood ‑ 2009 ‑ 07 ‑ 234708.
70. Zenz T, Vollmer D, Trbusek M et al. TP53 mutation profile in chronic lymphocytic leukemia: evidence for a disease specific profile from a comprehensive analysis of 268 mutations. Leukemia 2010; 24(12): 2072 – 2079. doi: 10.1038/ leu.2010.208.
71. Gonzalez D, Martinez P, Wade R et al. Mutational status of the TP53 gene as a predictor of response and survival in patients with chronic lymphocytic leukemia: results from the LRF CLL4 trial. J Clin Oncol 2011; 29(16): 2223 – 2229. doi: 10.1200/ JCO.2010.32.0838.
72. Malcikova J, Stano ‑ Kozubik K, Tichy B et al. Detailed analysis of therapy‑driven clonal evolution of TP53 mutations in chronic lymphocytic leukemia. Leukemia 2015; 29(4): 877 – 885. doi: 10.1038/ leu.2014.297.
73. Zenz T, Häbe S, Denzel T et al. Detailed analysis of p53 pathway defects in fludarabine ‑ refractory chronic lymphocytic leukemia (CLL): dissecting the contribution of 17p deletion, TP53 mutation, p53 - p21 dysfunction, and miR34a in a prospective clinical trial. Blood 2009; 114(13): 2589 – 2597. doi: 10.1182/ blood ‑ 2009 ‑ 05 ‑ 224071.
74. Pospisilova S, Gonzalez D, Malcikova J et al. ERIC recommendations on TP53 mutation analysis in chronic lymphocytic leukemia. Leukemia 2012; 26(7): 1458 – 1461. doi: 10.1038/ leu.2012.25.
75. Dreger P, Döhner H, Ritgen M et al. Allogeneic stem cell transplantation provides durable disease control in poor ‑ risk chronic lymphocytic leukemia: long‑term clinical and MRD results of the German CLL Study Group CLL3X trial. Blood 2010; 116(14): 2438 – 2447. doi: 10.1182/ blood ‑ 2010 ‑ 03 ‑ 275420.
76. Byrd JC, Brown JR, O‘Brien S et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med 2014; 371(3): 213 – 223. doi: 10.1056/ NEJMoa1400376.
77. Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 2013; 14(4): 197 – 210.
78. Jiang L, Sheikh MS, Huang Y. Decision Making by p53: life versus death. Mol Cell Pharmacol 2010; 2(2): 69 – 77.
79. Taylor AM, Byrd PJ. Molecular pathology of ataxia telangiectasia. J Clin Pathol 2005; 58(10): 1009 – 1015.
80. Guarini A, Marinelli M, Tavolaro S et al. ATM gene alterations in chronic lymphocytic leukemia patients induce a distinct gene expression profile and predict disease progression. Haematologica 2012; 97(1): 47 – 55. doi: 10.3324/ haematol.2011.049270.
81. Austen B, Skowronska A, Baker C et al. Mutation status of the residual ATM allele is an important determinant of the cellular response to chemotherapy and survival in patients with chronic lymphocytic leukemia containing an 11q deletion. J Clin Oncol 2007; 25(34): 5448 – 5457.
82. Austen B, Powell JE, Alvi A et al. Mutations in the ATM gene lead to impaired overall and treatment‑free survival that is independent of IGVH mutation status in patients with B ‑ CLL. Blood 2005; 106(9): 3175 – 3182.
83. Skowronska A, Parker A, Ahmed G et al. Biallelic ATM inactivation significantly reduces survival in patients treated on the United Kingdom Leukemia Research Fund Chronic Lymphocytic Leukemia 4 trial. J Clin Oncol 2012; 30(36): 4524 – 4532.
84. Skowronska A, Austen B, Powell JE et al. ATM germline heterozygosity does not play a role in chronic lymphocytic leukemia initiation but influences rapid disease progression through loss of the remaining ATM allele. Haematologica 2012; 97(1): 142 – 146. doi: 10.3324/ haematol.2011.048827.
85. Best OG, Gardiner AC, Majid A et al. A novel functional assay using etoposide plus nutlin‑3a detects and distinguishes between ATM and TP53 mutations in CLL. Leukemia 2008; 22(7): 1456 – 1459. doi: 10.1038/ sj.leu.2405092.
86. Navrkalova V, Sebejova L, Zemanova J et al. ATM mutations uniformly lead to ATM dysfunction in chronic lymphocytic leukemia: application of functional test using doxorubicin. Haematologica 2013; 98(7): 1124 – 1131. doi: 10.3324/ haematol.2012.081620.
87. Pettitt AR, Sherrington PD, Stewart G et al. p53 dysfunction in B ‑ cell chronic lymphocytic leukemia: inactivation of ATM as an alternative to TP53 mutation. Blood 2001; 98(3): 814 – 822.
88. Stankovic T, Stewart GS, Fegan C et al. Ataxia telangiectasia mutated ‑ deficient B ‑ cell chronic lymphocytic leukemia occurs in pregerminal center cells and results in defective damage response and unrepaired chromosome damage. Blood 2002; 99(1): 300 – 309.
89. Byrd JC, Furman RR, Coutre SE et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 2013; 369(1): 32 – 42. doi: 10.1056/ NEJMoa1215637.
90. Navrkalova V, Raskova Kafkova L, Divoky V et al. Oxidative stress as a therapeutic perspective for ATM ‑ deficient chronic lymphocytic leukemia patients. Haematologica 2015; 100(8): 994 – 996. doi:10.3324/ haematol.2015.130260.
91. Calin GA, Ferracin M, Cimmino A et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med 2005; 353(17): 1793 – 1801.
92. Zenz T, Mohr J, Eldering E et al. miR ‑ 34a as part of the resistance network in chronic lymphocytic leukemia. Blood 2009; 113(16): 3801 – 3808. doi: 10.1182/ blood ‑ 2008 ‑ 08 ‑ 172254.
93. Cimmino A, Calin GA, Fabbri M et al. miR ‑ 15 and miR ‑ 16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A 2005; 102(39): 13944 – 13949.
94. Fabbri M, Bottoni A, Shimizu M et al. Association of a microRNA/ TP53 feedback circuitry with pathogenesis and outcome of B ‑ cell chronic lymphocytic leukemia. JAMA 2011; 305(1): 59 – 67. doi: 10.1001/ jama.2010.1919.
95. Li S, Moffett HF, Lu J et al. MicroRNA expression profiling identifies activated B cell status in chronic lymphocytic leukemia cells. PLoS One 2011; 6(3). doi: 10.1371/ journal.pone.0016956.
96. Cui B, Chen L, Zhang S et al. MicroRNA ‑ 155 influences B ‑ cell receptor signaling and associates with aggressive disease in chronic lymphocytic leukemia. Blood 2014; 124(4): 546 – 554.
97. Stilgenbauer S, Schnaiter A, Paschka P et al. Gene mutations and treatment outcome in chronic lymphocytic leukemia: results from the CLL8 trial. Blood 2014; 123(21): 3247 – 3254. doi: 10.1182/ blood ‑ 2014 ‑ 01 ‑ 546150.
98. Oscier DG, Rose‑Zerilli MJ, Winkelmann N et al. The clinical significance of NOTCH1 and SF3B1 mutations in the UK LRF CLL4 trial. Blood 2013; 121(3): 468 – 475. doi: 10.1182/ blood ‑ 2012 ‑ 05 ‑ 429282.
99. Rossi D, Bruscaggin A, Spina V et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine ‑ refractoriness. Blood 2011; 118(26): 6904 – 6908. doi: 10.1182/ blood‑2011 ‑ 08 ‑ 373159.
100. Weissmann S, Roller A, Jeromin S et al. Prognostic impact and landscape of NOTCH1 mutations in chronic lymphocytic leukemia (CLL): a study on 852 patients. Leukemia 2013; 27(12): 2393 – 2396. doi: 10.1038/ leu.2013.218.
101. Rossi D, Rasi S, Spina V et al. Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood 2013; 121(8): 1403 – 1412. doi: 10.1182/ blood ‑ 2012 ‑ 09 ‑ 458265.
102. Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell 2009; 136(4): 701 – 718. doi: 10.1016/ j.cell.2009.02.009.
103. Mansouri L, Grabowski P, Degerman S et al. Short telomere length is associated with NOTCH1/ /SF3B1/ TP53 aberrations and poor outcome in newly diag-nosed chronic lymphocytic leukemia patients. Am J Hematol 2013; 88(8): 647 – 651. doi: 10.1002/ ajh.23466.
104. Paulsen RD, Soni DV, Wollman R et al. A genome ‑ wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell 2009; 35(2): 228 – 239. doi: 10.1016/ j.molcel.2009.06.021.
105. Te Raa GD, Derks IA, Navrkalova V et al. The impact of SF3B1 mutations in CLL on the DNA ‑ damage response. Leukemia 2015; 29(5): 1133 – 1142. doi: 10.1038/ leu.2014.318.
106. Weng AP, Ferrando AA, Lee W et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004; 306(5694): 269 – 271.
107. Rossi D, Rasi S, Fabbri G et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood 2012; 1192): 521 – 529. doi: 10.1182/ blood ‑ 2011 ‑ 09 ‑ 379966.
108. Conze DB, Zhao Y, Ashwell JD. Non ‑ canonical NF ‑ kB activation and abnormal B cell accumulation in mice expressing ubiquitin protein ligase ‑ inactive c ‑ IAP2. PLoS Biol 2010; 8(10). doi: 10.1371/ journal.pbio.1000518.
109. Rose‑Zerilli MJ, Forster J, Parker H et al. ATM mutation rather than BIRC3 deletion and/ or mutation predicts reduced survival in 11q ‑ deleted chronic lymphocytic leukemia: data from the UK LRF CLL4 trial. Haematologica 2014; 99(4): 736 – 742. doi: 10.3324/ haematol.2013.098574.
110. Schuh A, Becq J, Humphray S et al. Monitoring chronic lymphocytic leukemia progression by whole genome sequencing reveals heterogeneous clonal evolution patterns. Blood 2012; 120(20): 4191 – 4196. doi: 10.1182/ blood ‑ 2012 ‑ 05 ‑ 433540.
111. Ouillette P, Saiya ‑ Cork K, Seymour E et al. Clonal evolution, genomic drivers, and effects of therapy in chronic lymphocytic leukemia. Clin Cancer Res 2013; 19(11): 2893 – 2904. doi: 10.1158/ 1078 ‑ 0432.CCR ‑ 13 ‑ 0138.
112. Damm F, Mylonas E, Cosson A et al. Acquired initiating mutations in early hematopoietic cells of CLL patients. Cancer Discov 2014; 4(9): 1088 – 1101. doi: 10.1158/ 2159 ‑ 8290.CD ‑ 14 ‑ 0104.
113. Kikushige Y, Ishikawa F, Miyamoto T et al. Self ‑ renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell 2011; 20(2): 246 – 259. doi: 10.1016/ j.ccr.2011.06.029.
114. Rawstron AC, Bennett FL, O‘Connor SJ et al. Monoclonal B ‑ cell lymphocytosis and chronic lymphocytic leukemia. N Engl J Med 2008; 359(6): 575 – 583. doi: 10.1056/ NEJMoa075290.
115. Chigrinova E, Rinaldi A, Kwee I et al. Two main genetic pathways lead to the transformation of chronic lymphocytic leukemia to Richter syndrome. Blood 2013; 122(15): 2673 – 2682. doi: 10.1182/ blood ‑ 2013 ‑ 03 ‑ 489518.
116. Puiggros A, Blanco G, Espinet B. Genetic abnormalities in chronic lymphocytic leukemia: where we are and where we go. Biomed Res Int 2014; 2014 : 435983. doi: 10.1155/ 2014/ 435983.
117. Gaidano G, Foà R, Dalla ‑ Favera R. Molecular pathogenesis of chronic lymphocytic leukemia. J Clin Invest 2012; 122(10): 3432 – 3438. doi: 10.1172/JCI64101.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2015 Issue Supplementum 3
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- Difuzní velkobuněčný B lymfom – moderní způsoby diagnostiky a molekulárně cílené léčby
- Folikulární lymfom
- Lymfom z plášťových buněk – současný stav poznání a možnosti léčby
- Záchranná léčba a role transplantací u lymfomů