
Diagnóza Lynchova syndromu od patologa
Lynch Syndrome – the Pathologist’s Diagnosis
Lynch syndrome (formerly known as hereditary non-polyposis colorectal cancer) is the most common hereditary colorectal cancer syndrome. The syndrome is caused by a germline mutation of one of the mismatch repair (MMR) genes responsible for DNA replication error repair. Impaired function of the proteins encoded by these genes leads to microsatellite instability (MSI), which is associated with increased incidence of neoplasms: mainly colorectal cancer. According to recent estimates, up to 5% of all colorectal cancers are associated with Lynch syndrome. Due to this relatively high frequency, familial occurence, absence of premorbid phenotype, and development of malignant tumors at a reproductive age, a correct diagnosis is important not only from an ethical but also from an economical point of view. Unfortunately, clinical means of diagnosis, namely, the revised Bethesda guidelines designed to detect patients suitable for genetic testing for Lynch syndrome, lack sufficient sensitivity. The methods associated with modern pathology are more sensitive than the clinical criteria used to detect patients suspected of having Lynch syndrome. Pathological diagnostics are based on direct or indirect detection of MSI. Indirect methods include analysis of morphological signs associated with MSI in histological samples from colorectal carcinoma patients and immunohistochemical investigation of MMR protein expression. To rule out sporadic cases caused by epigenetic inactivation of an MMR gene, molecular genetic investigation of the BRAF gene and methylation analysis of the MLH1 promoter are performed during diagnostic workup. A suspicion of Lynch syndrome based on the results of the methods mentioned above should be proven by detection of a germline mutation in an MMR gene in peripheral blood leukocytes.
Key words:
colorectal cancer – Lynch syndrome – HNPCC – MSI – microsatellite instability
This work was supported by IGA NT14227 with contribution of SVV 260171/2015.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted:
9. 6. 2015
Accepted:
20. 3. 2016
Authors:
M. Dušek 1,2; L. Hadravský 1; Karin Černá 2
![]() ; J. Stehlík 2; M. Švajdler 1,2; B. Kokošková 1,2; M. Dubová 1; M. Michal 1; O. Daum 1,2
; J. Stehlík 2; M. Švajdler 1,2; B. Kokošková 1,2; M. Dubová 1; M. Michal 1; O. Daum 1,2
Authors‘ workplace:
Šiklův ústav patologie, LF UK a FN Plzeň
1; Bioptická laboratoř, s. r. o., Plzeň
2
Published in:
Klin Onkol 2016; 29(3): 180-186
Category:
Reviews
doi:
https://doi.org/10.14735/amko2016180
Overview
Lynchův syndrom (dříve nazývaný hereditární nepolypózní kolorektální karcinom) je nejčastější genetickou příčinou familiárního výskytu kolorektálního karcinomu. Způsobuje jej zárodečná mutace některého z genů, které jsou zodpovědné za opravy chyb ve struktuře DNA vznikající při její replikaci. V důsledku toho dochází k dysfunkci opravného komplexu způsobující rozvoj nestability mikrosatelitů (MSI), která je asociována se zvýšením rizika vzniku nádorů, zejména kolorektálního karcinomu. V současné době se odhaduje, že až 5 % kolorektálních karcinomů vzniká v souvislosti s Lynchovým syndromem. Vzhledem k této poměrně vysoké četnosti, absenci premorbidního fenotypu, familiárnímu výskytu a prezentaci maligních nádorů v produktivním věku je včasná diagnostika Lynchova syndromu důležitá nejen z etického, ale i ekonomického hlediska. Klinická kritéria představovaná zejména revidovanými Bethesda guidelines, která byla navržena pro detekci pacientů vhodných ke genetickému vyšetření možnosti Lynchova syndromu, nejsou však dostatečně senzitivní. Vyšší senzitivity lze dosáhnout aplikací metod moderní patologie. Tato diagnostika je založena na přímém nebo nepřímém průkazu MSI. Mezi metody nepřímého průkazu MSI patří jednak detekce morfologických znaků asociovaných s MSI při histologickém vyšetření vzorků kolorektálních karcinomů, jednak imunohistochemické vyšetření exprese MMR proteinů, které navíc umožní i identifikaci dysfunkčního proteinu. K vyloučení sporadických MSI-H karcinomů způsobených somatickou epigenetickou inaktivací MMR genu z dalšího testování slouží hlavně vyšetření genu BRAF a analýza metylace promotoru genu MLH1. Podezření na Lynchův syndrom vyplývající z výsledků těchto vyšetření by mělo být nakonec potvrzeno detekcí zárodečné mutace některého z MMR genů v periferní krvi pacienta.
Klíčová slova:
kolorektální karcinom – Lynchův syndrom – HNPCC – MSI – nestabilita mikrosatelitů
Lynchův syndrom – definice
Lynchův syndrom (LS) je autozomálně dominantně dědičné onemocnění vytvářející predispozici ke vzniku maligních nádorů, patří tedy mezi familiární karcinomové syndromy. Nejčastějším karcinomem vznikajícím při LS je kolorektální karcinom (colorectal cancer – CRC), je však zvýšené riziko vzniku i dalších malignit, zejména karcinomů endometria, tenkého střeva, ovaria, ledvinné pánvičky a močovodu, nádorů mozku a kůže. Na podkladě LS vzniká podle současných odhadů až 5 % CRC. Fakt, že je dosud do značné míry přehlížen, zejména v porovnání s méně častou familiární adenomatózní polypózou (FAP), která je zodpovědná pouze za 1 % CRC, je způsoben tím, že karcinomy při LS nevznikají v terénu polypózy (definované jako > 100 polypů), což však neznamená, že nemohou být přítomny žádné polypy. Absence polypózy nebo jiného premorbidního fenotypu, tedy benigních změn, které by umožňovaly diagnostikovat tento syndrom ještě před vznikem maligního tumoru (jako je tomu třeba v případě FAP nebo neurofibromatózy 1. typu) výrazně ztěžuje jeho včasnou klinickou diagnostiku. Na rozdíl od familiárních karcinomových syndromů s premorbidním fenotypem tak může být LS diagnostikován prakticky až při nálezu maligního tumoru, případně při genetickém vyšetření rodinných příslušníků již diagnostikovaného probanda. Výjimkou z tohoto pravidla je fenotypická varianta LS projevující se vznikem kožních sebaceózních nádorů, označovaná jako Muir-Torreho syndrom (MTS) [1].
Základní klinické charakteristiky syndromu karcinomové rodiny byly definovány dr. Lynchem takto [2]:
- zvýšená incidence adenokarcinomů, zejména kolorektálních a endometriálních,
- zvýšené riziko multiplicity nádorů,
- autozomálně dominantní dědičnost,
- vznik karcinomů v mladším věku.
Ačkoli byl LS dlouhou dobu znám spíše pod pojmem hereditární nepolypózní kolorektální karcinom (hereditary non-polyposis colorectal cancer – HNPCC) [3], v současné době se od tohoto označení upouští a preferuje se eponymon Lynchův syndrom, a to jednak z důvodu možnosti výskytu extrakolonických malignit, jednak kvůli příliš zakořeněné asociaci diagnózy HNPCC s Amsterdamskými kritérii, která je ve světle dnešních poznatků již neudržitelná, a konečně i jako vyjádření úcty otci Lynchova syndromu.
V současnosti je tedy diagnóza LS založena především na molekulárně genetickém vyšetření (viz následující kapitolu), přičemž pro případy splňující Amsterdamská kritéria, ale bez prokazatelného genetického poškození definujícího LS, se doporučuje termín familiární kolorektální karcinom typu X [4].
Molekulární biologie LS
Detailně je molekulárně biologický podklad LS popsán v textu určeném primárně patologům [5], pro pochopení dále uváděných diagnostických algoritmů je zde však třeba alespoň stručně vysvětlit základní pojmy.
MMR (mismatch repair)
MMR (neboli mismatch repair) proteiny jsou odpovědné za opravy v DNA vznikajících při replikaci (replication error repair – RER). Nejdůležitější z nich se spojují do funkčních heterodimerů MLH1-PMS2 a MSH2-MSH6. Naprostá většina případů LS je způsobena zárodečnou mutací genů kódujících tyto proteiny, tedy tzv. mismatch repair (MMR) genů [6,7]. Inaktivace obou alel některého z MMR genů vede k dysfunkci celého komplexu a ke vzniku tzv. MSI-H tumorů, tedy nádorů charakteristických vysokým stupněm tzv. nestability mikrosatelitů (microsatelite instability – MSI) [8,9].
Mikrosatelity
Mikrosatelity jsou v genomu hojně se vyskytující úseky DNA tvořené několikanásobným opakováním jednoho až čtyř, vzácněji i více nukleotidů. Tyto krátké repetitivní sekvence jsou snadno zranitelné při replikaci DNA, protože DNA polymeráza v oblasti repetic „sklouzává“ a v důsledku toho dochází ke vzniku delších či kratších úseků. Ve „zdravé“ buňce s funkčním MMR systémem jsou však tyto alterace ihned detekovány a opraveny.
MSI
Délky jednotlivých mikrosatelitů (tedy počty opakování těchto sekvencí) jsou u zdravých jedinců ve všech buňkách stejné (mezi jedinci se však liší). Pokud ovšem nedochází ke korekci chyb vznikajících při replikaci, potom může délka mikrosatelitů v rámci jednoho jedince kolísat, což je stav označovaný jako nestabilita mikrosatelitů (MSI). MSI není však jednoznačně daným stavem, který by přímo způsoboval vznik nádorů. Jde spíše o semikvantitativní vyjádření genetického poškození DNA uvedeným mechanizmem. Stanovení stupně MSI je tedy arbitrární a spočívá ve stanovení nestability mezinárodně kodifikovaných markerů, přičemž na základě počtu postižených markerů se rozlišují stavy (hlavně nádory) se stabilními mikrosatelity (microsatellite stable – MSS), s nízkým stupněm nestability (microsatellite instability, low – MSI-L) a s vysokým stupněm nestability mikrosatelitů (microsatellite instability, high – MSI-H) [10].
MSI-H tumory
Tumory s MSI-H vznikají dvěma různými mechanizmy, a to buď jako sporadické nádory vyvolané genetickými a/ nebo epigenetickými změnami v somatické buňce, nebo jako familiárně se vyskytující nádory v rámci LS způsobené zárodečnou mutací některého z MMR genů. Teprve tehdy, je-li u osob nesoucích jednu zárodečně mutovanou alelu MMR genu během jejich života inaktivována i alela druhá, dochází ke vzniku maligních tumorů. Příčinou této inaktivace může být somatická mutace v druhé alele genu, ztráta heterozygozity (LOH) nebo metylace promotoru, jak tomu bývá u genu MLH1 [11].
Nejčastěji postiženými geny při LS jsou MLH1 a MSH2 (dohromady více než 80 %) [12], dále následuje MSH6 (10 %), a zbylé případy představují vzácná zárodečná postižení dalších MMR genů (PMS2, PMS1, MSH3, MLH3).
Vzácně mohou dysfunkci MMR proteinů a tím pádem i LS způsobovat komplikovanější mechanizmy, jako zárodečná hypermetylace promotoru genu MLH1 vedoucí k jeho epigenetické inaktivaci [13,14] nebo zárodečné delece 3’ konce genu EPCAM (TACSTD1), které zase vedou k epigenetické inaktivaci MSH2 [15,16].
Variabilita klinických projevů LS
Klasický typ LS se prezentuje především CRC vznikajícím v tlustém střevě bez polypózy (tedy hereditárním nepolypózním CRC). Mohou však být přítomny i extrakolonické malignity, a to relativně častěji u pacientů se zárodečnou mutací v MSH2 než MLH1 [17]. Mutace v MSH6 jsou zodpovědné za poněkud atypické prezentace LS, neboť mají jednak nízkou penetranci, dále jsou 6krát častěji asociovány s karcinomy endometria než s nádory kolorekta, a navíc jsou u těchto pacientů CRC (v porovnání s LS způsobeným mutacemi jiných MMR genů) častěji levostranné. Důležité také je, že mutace v MSH6 nevedou vždy k MSI-H, pravděpodobně díky tvorbě alternativního heterodimeru MSH2-MSH3, což může negativně ovlivnit jejich diagnostiku [18,19]. V některých případech je kombinace klinických znaků natolik výrazná, že dala vznik novým klinickým syndromům, které ve skutečnosti představují pouze varianty LS.
Muir-Torreho syndrom
MTS zahrnuje kombinaci nejméně jednoho kožního nádoru se sebaceózní diferenciací a minimálně jednoho viscerálního tumoru. Již v roce 1981 dr. Henry Lynch poukázal na společnou možnou etiologii MTS a LS, když identifikoval pacienty s fenotypem MTS v rodině postižené LS [20]. MTS jako varianta LS je unikátní v tom, že jako jediná představuje premorbidní fenotyp, tedy vykazuje znaky umožňující diagnózu LS ještě před rozvojem CRC.
Kožní léze asociované s MTS jsou detailně popsány v textech určených primárně patologům [1] a gastroenterologům [21], obecně lze shrnout, že mnohočetné kožní nádory se sebaceózní diferenciací vyskytující se u jedinců před 50. rokem života nebo postihující tělní partie mimo obličej jsou silným indikátorem MTS [22].
Turcotův syndrom 1. typu
Turcotův syndrom (TS) je klasicky charakterizován společným výskytem nádorů mozku a CRC. Podtyp označovaný jako TS 1. typu je blíže specifikován vazbou mozkového nádoru (hlavně gliomu) s CRC bez polypózy, přičemž může být také způsoben zárodečnou mutací některého z MMR genů, mutace byly detekovány zejména v MLH1 a PMS2 [23]. Za těchto podmínek se TS 1. typu jeví ve většině případů jako fenotypická varianta LS, zejména u dětských a mladistvých pacientů pak ještě spíše bývá součástí syndromu bialelického mismatch repair deficitu.
Syndrom bialelického mismatch repair deficitu
Syndrom bialelického mismatch repair deficitu (biallelic mismatch repair deficiency – BMMR-D, také constitutional mismatch repair deficiency – CMMRD) je velmi vzácně se vyskytující zárodečná bialelická mutace genů MMR. Vzhledem k tomu, že jde o onemocnění dědičné autozomálně recesivně, vyskytuje se zejména jako následek incestu. Tento stav je charakteristický fenotypickým obrazem připomínajícím neurofibromatózu 1. typu, zejména skvrnami café-au-lait, vznikem CRC již v mladém věku (průměrný věk v době diagnózy 16 let), mozkovými nádory, především glioblastomy vznikajícími již v prvních dvou dekádách života, a hematologickými malignitami (hlavně T lymfomy) [24,25].
Z diagnostického hlediska je důležité, že imunohistochemicky stanovený deficit MMR proteinu má vyšší senzitivitu než vyšetření MSI [26], přičemž tento imunohistochemický test lze provést i v nenádorové tkáni, např. v kožní biopsii [27].
Možnosti diagnostiky LS
Tradiční klinická diagnostika LS
Ke klinické diagnostice HNPCC primárně sloužila Amsterdamská kritéria [28], která byla pro zvýšení senzitivity, zejména s přihlédnutím k možnosti prezentace syndromu extrakolonickou malignitou, v roce 1998 revidována na Amsterdamská kritéria II [29]. Ale protože se zlatým standardem diagnózy LS stalo molekulárně genetické vyšetření, v popředí zájmu se ocitl záchyt co největšího množství pacientů pro toto vyšetření, nikoli samotná klinická diagnóza LS.
K identifikaci pacientů s CRC, u kterých by měla být vyšetřena nestabilita mikrosatelitů (MSI), případně provedeno molekulárně genetické vyšetření, byla v roce 1996 vypracována a v roce 2002 revidována tzv. Bethesda guidelines (BG, resp. RBG), která berou v potaz nejen klinická kritéria, ale (v případě RBG) i morfologické znaky tumoru [30]. Bohužel ani tato širší kritéria nezachytí všechny případy LS [31], zejména v případě postižení MSH6 a PMS2 [32 – 36]. Podle současných odhadů až 25 % pacientů s LS není zachyceno systémem kritérií RBG. Jednou z velkých slabin jak Amsterdamských kritérií, tak (R)BG je důraz na údaje získané rodinou anamnézou, které mohou trpět značnými nedostatky, ať už kvůli nedostatečné informovanosti pacienta, nezájmu vyšetřujícího lékaře, časté nejistotě ohledně biologického otcovství nebo i nízké penetranci zárodečné mutace (zejména v případě genu MSH6).
Moderní patologická diagnostika LS
Mezi hlavní argumenty pro současné snahy o zavedení senzitivnějšího systému depistáže, byť i za cenu snížení specificity, které je v tomto případě možné familiární tendence ke vzniku maligních nádorů ospravedlnitelné jak z hlediska etického, tak ekonomického, patří tato fakta:
- falešná negativita v případě LS nemá za následek nerozpoznání tohoto syndromu pouze u vyšetřovaného pacienta, ale i u jeho případných příbuzných;
- přibližně 1 ze 660 lidí je nositelem germinální mutace některého z MMR genů [37];
- riziko vzniku CRC u LS je 60 – 80 % [38,39];
- k progresi z adenomu do karcinomu pravděpodobně dochází během 2 – 3 let, na rozdíl od 8 – 10 let u sporadických případů [40,41];
- průměrný věk v době diagnózy je 45 let, tedy asi o 20 let méně než u sporadického CRC, navíc se zvýšeným rizikem synchronního a metachronního CRC [40].
V současné době jsou k dispozici tři základní modely vyhledávání pacientů s podezřením na LS, přičemž všechny mají společné, že detekují MSI-H tumory, a to buď přímo (tedy molekulárně genetickým stanovením MSI), nebo zprostředkovaně. Do této druhé skupiny patří jednak imunohistochemická detekce MMR proteinů, jednak histologický průkaz morfologických znaků asociovaných s MSI.
V každém z modelů diagnostikujících MSI-H tumory je ale nutné před samotnou finančně nákladnou detekcí germinálních mutací MMR genů vyloučit možnost sporadických forem, protože čtyři z pěti MSI-H CRC jsou sporadické nádory způsobené zdaleka nejčastěji metylací promotoru MLH1. V současné době umožňuje rozlišení sporadických a LS asociovaných MSI-H karcinomů hlavně zapojení dvou metod molekulární patologie do managementu CRC. První z nich je analýza genu BRAF, konkrétně průkaz substituce V600E, která je přítomna až u 1/ 2 sporadických MSI-H CRC, ale (téměř) nikdy u LS. Mutovaný protein navíc může být v nádoru prokázán i monoklonální protilátkou [42]. Druhou metodu představuje průkaz hypermetylace promotoru MLH1, která je markerem sporadických MSI-H CRC, a naopak až na výjimky nebývá přítomna u LS [43]. Nověji je také k dispozici protilátka proti annexinu A10 umožňující odlišit sporadické MSI-H karcinomy od LS [44].
1. Stanovení MSI
Rozlišení tumorů na MSS, MSI-L a MSI-H na základě stanovení nestability mezinárodně kodifikovaných markerů se samozřejmě v diagnostice LS využívalo již dříve, ale cíleně, u pacientů splňujících BG (resp. RBG). Některé laboratoře zavedly plošné vyšetření všech CRC touto metodou k depistáži LS.
Mezi nevýhody tohoto systému patří výrazný nárůst zátěže laboratoří molekulární genetiky, absence informace o tom, který z MMR genů je postižen, a konečně i fakt, že (navzdory obecnému přesvědčení) ne všechny LS asociované nádory musí vykazovat MSI-H (zejména jde o pacienty s germinální mutací genu MSH6). Praktické využití plošného vyšetřování MSI naráží též na nezbytnost porovnání stavu markerů ve tkáni nádoru s nenádorovou tkání. To vyžaduje buď přítomnost nenádorové tkáně v materiálu (např. chirurgický okraj střevního resekátu), nebo odběr periferní krve pacienta (zejména v případě endoskopicky získaných vzorků).
2. Imunohistochemická detekce MMR proteinů
Na našem pracovišti používáme jako vstupní vyšetření pro zařazení pacienta do diagnostického managementu LS imunohistochemické vyšetření exprese hlavních MMR proteinů (tedy MLH1, PMS2, MSH2 a MSH6) u všech CRC a karcinomů endometria.
Senzitivita imunohistochemického vyšetření MMR proteinů a stanovení MSI je srovnatelná [45]. V současné době jsou obě metody (stanovení MSI a imunohistochemické vyšetření) vnímány jako komplementární, protože v kombinaci mají vyšší senzitivitu než při samostatném použití [46,47]. Také Jeruzalémská kritéria, podle nichž by měly být imunohistochemicky vyšetřeny všechny CRC u pacientů mladších 70 let, považují imunohistochemii za vhodnou vstupní diagnostickou metodu [48]. Argumenty pro upřednostnění imunohistochemického vyšetření jako iniciální diagnostické metody jsou tyto:
- vyšší záchyt případů s mutací MSH6, které mohou uniknout při detekci MSI pomocí PCR, protože MSH2 může také tvořit komplex s MSH3 a tím pádem nemusí nutně vést ke stavu MSI-H [45,49];
- imunohistochemická detekce MMR proteinů, na rozdíl od stanovení MSI, umožňuje určit postižený gen pro molekulárně genetické vyšetření, což výrazně sníží náklady při následné detekci případné zárodečné mutace;
- při iniciálním vyšetření není zapotřebí kontrolní nenádorová tkáň;
- imunohistochemické vyšetření odhalí i případy BMMR-D, které se často neprojeví nestabilitou mikrosatelitů.
Další kroky diagnostického managementu založeného na iniciální imunohistochemické detekci MMR proteinů demonstruje schéma 1.

3. Histologická detekce morfologických znaků asociovaných s MSI-H
Pro většinu pracovišť patologie, která vyšetřují bioptické vzorky karcinomů tlustého střeva, endometria a dalších nádorů, však nejsou výše uvedené diagnostické metody přímo dostupné. I tato pracoviště se však mohou podílet na depistáži LS, a to detekcí histologických znaků charakteristických pro CRC s vysokým stupněm nestability mikrosatelitů, tedy takzvané „MSI-H histologii“, která samotná má vyšší senzitivitu než souhrn zbývajících čtyř kritérií RBG [50].
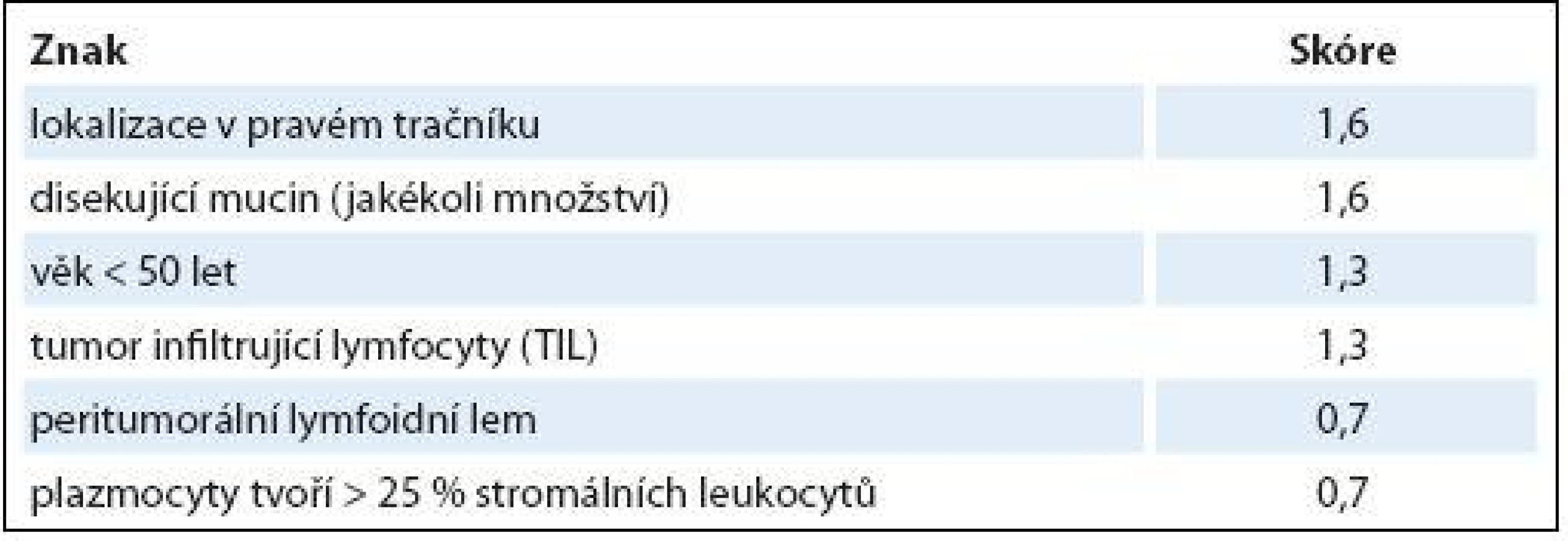
Z různých studií, jejichž cílem bylo nalézt racionální histologický algoritmus detekce MSI-H karcinomů [51], se v současné době jako nejužitečnější model jeví Model PREDICT (Pathological RolE in the Determination of Instability in Colorectal Tumors) [50], zejména ve své zjednodušené formě jako Semi PREDICT skóre (tab. 1). Histologickými znaky, na nichž je tento model založen, jsou: přítomnost mucinu disekujícího stroma v jakémkoli množství, přítomnost tumor infiltrujících lymfocytů (TIL), peritumorální lymfoidní lem a zastoupení plazmatických buněk mezi leukocyty ve stromatu převyšující 25 %. Detailněji je morfologie MSI-H CRC popsána v článku určeném patologům [5].
Mezi výhody histologického vyšetření patří i možnost levného, rychlého a jednoduchého vyloučení sporadických MSI-H karcinomů podmíněných většinou somatickou metylací promotoru genu MLH1. Zatímco tyto vznikají ze „sesilních serrated adenomů“ (do češtiny někdy překládaných do ještě horší formy „přisedlé pilovité adenomy“), prekurzorovou lézí karcinomů v terénu LS je „konvenční“ (tubulární, tubulovilózní nebo vilózní) adenom. Jsou-li tedy v periferii tumoru zbytky prekurzorového sesilního serrated adenomu, jde s největší pravděpodobností o sporadický MSI-H karcinom [52]. Nicméně tento jednoduchý diagnostický znak vylučující LS nemusí být vždy dostupný, ať již z důvodu nedostatečného samplingu, nebo destrukce původního adenomu pokročilým adenokarcinomem.
Histologické vyšetření samo o sobě samozřejmě nemůže vést k diagnóze LS,může pouze vést k suspekci, která by měla vést k odeslání vzorku nádoru (a případně i nenádorové tkáně) na pracoviště patologie vyššího typu zabývající se diagnostikou LS. Zde pak následují kroky popsané ve schématu 1.
Problémy s algoritmy – suspektní LS a Lynch-like syndrom
Tyto dva termíny se obsahově částečně překrývají, bohužel jsou dnes některými autory používány jako synonyma, ač se jejich význam liší.
Termín „suspektní Lynchův syndrom“ (SLS) se dříve používal pro případy, kdy se nepodařilo prokázat molekulární podklad LS u pacienta splňujícího Amsterdamská kritéria a/ nebo (R)BG. Zjednodušeně řečeno, většinou se jedná o familiárně se vyskytující často vícečetné karcinomy, zejména CRC, které nevznikají v terénu polypózy. Vysvětlení je samozřejmě řada, od environmentálních vlivů až po jiné familiární karcinomové syndromy, zejména tzv. MUTYH asociovanou polypózu (MAP), která se jednak nemusí prezentovat polypózou, jednak CRC vznikající při bialelické mutaci genu MUTYH může být také MSI-H [53].
Pojem „Lynch-like syndrom“ (LLS) je užší a lépe definovaný. Do této skupiny patří případy CRC, které vykazují známky dysfunkce MMR systému, tedy imunohistochemicky detekovaný deficit některého z MMR proteinů a/ nebo průkaz MSI-H, spolu s vyloučením možnosti sporadického MSI-H tumoru průkazem absence mutace genu BRAF a hypermetylace promotoru genu MLH1, ale u nichž byly molekulárně genetickým vyšetřením periferní krve vyloučeny zárodečné mutace MMR genů a 3’ konce genu EPCAM. Kromě možnosti falešně pozitivních výsledků imunohistochemického vyšetření na jedné straně a existencí mutací nedetekovaných současnými metodami může být tento fenomén vysvětlen dvěma stavy prokázanými v posledních dvou letech: somatickým mozaicizmem [54] a somatickými bialelickými mutacemi MMR genů, jejichž možnost se dříve popírala [54 – 57].
Závěr
LS je familiární karcinomový syndrom způsobený zárodečnou mutací některého z genů, jehož proteinový produkt se účastní opravy chyb v DNA vzniklých při replikaci. Vzhledem k tomu, že výskyt LS v populaci se nyní odhaduje až na 5 % a vede ke vzniku maligních nádorů již v produktivním věku, sílí v současné době tlak na zvýšení senzitivity jeho detekce.
Efektivní algoritmus diagnostiky LS by měl být vysoce senzitivní, dostatečně specifický, (relativně) levný a logisticky jednoduchý. Z hlediska senzitivity a specificity se ukázalo zcela nedostatečným spoléhat se na klinická kritéria (zejména rodinnou anamnézu). Proto se současné postupy zaměřují na detekci MSI-H tumorů, a to buď přímo, zprostředkovaně pomocí imunohistochemie, nebo na základě histologického průkazu morfologických znaků specifických pro MSI-H karcinomy.
Přestože se již podařilo i v našich podmínkách zavést účinný algoritmus pro diagnostikuS, nefunguje dosud dostatečně systém zpětné vazby s klinickými lékaři, kteří by měli organizovat další průběh vyšetření rodiny nemocného s LS. Na vině je do značné míry přávání pacienta po kolektomii mezi chirurgem, praktickým lékařem a onkologem, během kterého „zapadne“ žádost laboratoře molekulární genetiky o periferní krev, která je nezbytná k definitivnímu průkazu LS. K optimalizaci diagnostiky LS je tedy nezbytné zainteresovat do diagnostiky LS i klinické lékaře, bez jejichž aktivní účasti není možná ani kompletní diagnostika pacienta ani další vyšetření jeho rodinných příslušníků.
Práce byla realizována za podpory Interní grantové agentury MZ ČR (IGA MZ ČR) pod grantovým číslem IGA NT14227 a s přispěním SVV 260171/2015.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
doc. MUDr. Ondřej Daum, Ph.D.
Šiklův ústav patologie LF UK a FN Plzeň
Dr. E. Beneše 13
305 99 Plzeň
e-mail: daum@fnplzen.cz
Obdrženo: 9. 6. 2015
Přijato: 20. 3. 2016
Sources
1. Kacerovská D, Kazakov DV, Černá K et al. Muir-Torre syndrom – fenotypická varianta Lynchova syndromu. Cesk Patol 2010; 46(4): 86 – 94.
2. Lynch HT, Krush AJ. Cancer family „G“ revisited: 1895 – 1970. Cancer 1971; 27(6): 1505 – 1511.
3. Lynch HT, Drouhard TJ, Schuelke GS et al. Hereditary nonpolyposis colorectal cancer in a Navajo Indian family. Cancer Genet Cytogenet 1985; 15(3 – 4): 209 – 213.
4. Lindor NM, Rabe K, Petersen GM et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA 2005; 293(16): 1979 – 1985.
5. Daum O, Beneš Z, Hadravský L et al. Lynchův syndrom v rukách patologa. Cesk Patol 2014; 50(1): 18 – 24.
6. Fishel R, Lescoe MK, Rao MR et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993; 75(5): 1027 – 1038.
7. Leach FS, Nicolaides NC, Papadopoulos N et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 1993; 75(6): 1215 – 1225.
8. Peltomaki P, Lothe RA, Aaltonen LA et al. Microsatellite instability is associated with tumors that characterize the hereditary non-polyposis colorectal carcinoma syndrome. Cancer Res 1993; 53(24): 5853 – 5855.
9. Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science 1993; 260(5109): 816 – 819.
10. Boland CR, Thibodeau SN, Hamilton SR et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998; 58(22): 5248 – 5257.
11. Kruse R, Rutten A, Hosseiny-Malayeri HR et al. „Second hit“ in sebaceous tumors from Muir-Torre patients with germline mutations in MSH2: allele loss is not the preferred mode of inactivation. J Invest Dermatol 2001; 116(3): 463 – 465.
12. Nystrom-Lahti M, Wu Y, Moisio AL et al. DNA mismatch repair gene mutations in 55 kindreds with verified or putative hereditary non-polyposis colorectal cancer. Hum Mol Genet 1996; 5(6): 763 – 769.
13. Gazzoli I, Loda M, Garber J et al. A hereditary nonpolyposis colorectal carcinoma case associated with hypermethylation of the MLH1 gene in normal tissue and loss of heterozygosity of the unmethylated allele in the resulting microsatellite instability-high tumor. Cancer Res 2002; 62(14): 3925 – 3928.
14. Hitchins MP, Wong JJ, Suthers G et al. Inheritance of a cancer-associated MLH1 germ-line epimutation. N Engl J Med 2007; 356(7): 697 – 705.
15. Ligtenberg MJ, Kuiper RP, Chan TL et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3‘ exons of TACSTD1. Nat Genet 2009; 41(1): 112 – 117. doi: 10.1038/ ng.283.
16. Kovacs ME, Papp J, Szentirmay Z et al. Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to Lynch syndrome. Hum Mutat 2009; 30(2): 197 – 203. doi: 10.1002/ humu.20942.
17. Kastrinos F, Stoffel EM, Balmana J et al. Phenotype comparison of MLH1 and MSH2 mutation carriers in a cohort of 1,914 individuals undergoing clinical genetic testing in the United States. Cancer Epidemiol Biomarkers Prev 2008; 17(8): 2044 – 2051. doi: 10.1158/ 1055-9965.EPI-08-0301.
18. Hampel H, Frankel W, Panescu J et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res 2006; 66(15): 7810 – 7817.
19. Berends MJ, Wu Y, Sijmons RH et al. Molecular and clinical characteristics of MSH6 variants: an analysis of 25 index carriers of a germline variant. Am J Hum Genet 2002; 70(1): 26 – 37.
20. Lynch HT, Fusaro RM, Roberts L et al. Muir-Torre syndrome in several members of a family with a variant of the Cancer Family Syndrome. Br J Dermatol 1985; 113(3): 295 – 301.
21. Kokošková B, Daum O, Beneš Z et al. Moderní diagnostika Lynchova syndromu. Gastroent Hepatol 2014; 68(2): 157 – 165.
22. Schwartz RA, Torre DP. The Muir-Torre syndrome: a 25-year retrospect. J Am Acad Dermatol 1995; 33(1): 90 – 104.
23. De Rosa M, Fasano C, Panariello L et al. Evidence for a recessive inheritance of Turcot‘s syndrome caused by compound heterozygous mutations within the PMS2 gene. Oncogene 2000; 19(13): 1719 – 1723.
24. Gallinger S, Aronson M, Shayan K et al. Gastrointestinal cancers and neurofibromatosis type 1 features in children with a germline homozygous MLH1 mutation. Gastroenterology 2004; 126(2): 576 – 585.
25. Bandipalliam P. Syndrome of early onset colon cancers, hematologic malignancies & features of neurofibromatosis in HNPCC families with homozygous mismatch repair gene mutations. Fam Cancer 2005; 4(4): 323 – 333.
26. Bakry D, Aronson M, Durno C et al. Genetic and clinical determinants of constitutional mismatch repair deficiency syndrome: Report from the constitutional mismatch repair deficiency consortium. Eur J Cancer 2014; 50(5): 987 – 996. doi: 10.1016/ j.ejca.2013.12.005.
27. Durno CA, Sherman PM, Aronson M et al. Phenotypic and genotypic characterisation of biallelic mismatch repair deficiency (BMMR-D) syndrome. Eur J Cancer 2015; 51(8): 977 – 983. doi: 10.1016/ j.ejca.2015.02.008.
28. Vasen HF, Mecklin JP, Khan PM et al. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum 1991; 34(5): 424 – 425.
29. Vasen HF, Watson P, Mecklin JP et al. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999; 116(6): 1453 – 1456.
30. Umar A, Boland CR, Terdiman JP et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004; 96(4): 261 – 268.
31. Hampel H, Frankel WL, Martin E et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 2005; 352(18): 1851 – 1860.
32. Liu T, Yan H, Kuismanen S et al. The role of hPMS1 and hPMS2 in predisposing to colorectal cancer. Cancer Res 2001; 61(21): 7798 – 7802.
33. van der Klift H, Wijnen J, Wagner A et al. Molecular characterization of the spectrum of genomic deletions in the mismatch repair genes MSH2, MLH1, MSH6, and PMS2 responsible for hereditary nonpolyposis colorectal cancer (HNPCC). Genes Chromosomes Cancer 2005; 44(2): 123 – 138.
34. Dovrat S, Figer A, Fidder HH et al. Mutational analysis of hMSH6 in Israeli HNPCC and HNPCC-like families. Fam Cancer 2005; 4(4): 291 – 294.
35. Hegde MR, Chong B, Blazo ME et al. A homozygous mutation in MSH6 causes Turcot syndrome. Clin Cancer Res 2005; 11(13): 4689 – 4693.
36. Hendriks YM, Jagmohan-Changur S, van der Klift HM et al. Heterozygous mutations in PMS2 cause hereditary nonpolyposis colorectal carcinoma (Lynch syndrome). Gastroenterology 2006; 130(2): 312- – 322.
37. de la Chapelle A. The incidence of Lynch syndrome. Fam Cancer 2005; 4(3): 233 – 237.
38. Jenkins MA, Baglietto L, Dowty JG et al. Cancer risks for mismatch repair gene mutation carriers: a population-based early onset case-family study. Clin Gastroenterol Hepatol 2006; 4(4): 489 – 498.
39. Quehenberger F, Vasen HF, van Houwelingen HC. Risk of colorectal and endometrial cancer for carriers of mutations of the hMLH1 and hMSH2 gene: correction for ascertainment. J Med Genet 2005; 42(6): 491 – 496.
40. Lynch HT, de la Chapelle A. Genetic susceptibility to non-polyposis colorectal cancer. J Med Genet 1999; 36(11): 801 – 818.
41. Jass JR, Stewart SM. Evolution of hereditary non-polyposis colorectal cancer. Gut 1992; 33(6): 783 – 786.
42. Kuan SF, Navina S, Cressman KL et al. Immunohistochemical detection of BRAF V600E mutant protein using the VE1 antibody in colorectal carcinoma is highly concordant with molecular testing but requires rigorous antibody optimization. Hum Pathol 2014; 45(3): 464 – 472. doi: 10.1016/ j.humpath.2013.10.026.
43. Domingo E, Laiho P, Ollikainen M et al. BRAF screening as a low-cost effective strategy for simplifying HNPCC genetic testing. J Med Genet 2004; 41(9): 664 – 668.
44. Pai RK, Shadrach BL, Carver P et al. Immunohistochemistry for annexin A10 can distinguish sporadic from Lynch syndrome-associated microsatellite-unstable colorectal carcinoma. Am J Surg Pathol 2014; 38(4): 518 – 525. doi: 10.1097/ PAS.0000000000000148.
45. Shia J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry. J Mol Diagn 2008; 10(4): 293 – 300. doi: 10.2353/ jmoldx.2008.080031.
46. Funkhouser WK Jr., Lubin IM, Monzon FA et al. Relevance, pathogenesis, and testing algorithm for mismatch repair-defective colorectal carcinomas: a report of the association for molecular pathology. J Mol Diagn 2012; 14(2): 91 – 103. doi: 10.1016/ j.jmoldx.2011.11.001
47. Halvarsson B, Lindblom A, Rambech E et al. Microsatellite instability analysis and/ or immunostaining for the diagnosis of hereditary nonpolyposis colorectal cancer? Virchows Arch 2004; 444(2): 135 – 141.
48. Boland CR, Shike M. Report from the Jerusalem workshop on Lynch syndrome-hereditary nonpolyposis colorectal cancer. Gastroenterology 2010; 138(7): 2197e1 – 2197e7. doi: 10.1053/ j.gastro.2010.04.024.
49. Boland CR, Koi M, Chang DK et al. The biochemical basis of microsatellite instability and abnormal immunohistochemistry and clinical behavior in Lynch syndrome: from bench to bedside. Fam Cancer 2008; 7(1): 41 – 52.
50. Hyde A, Fontaine D, Stuckless S et al. A histology-based model for predicting microsatellite instability in colorectal cancers. Am J Surg Pathol 2010; 34(12): 1820 – 1829. doi: 10.1097/ PAS.0b013e3181f6a912.
51. Roman R, Verdu M, Calvo M et al. Microsatellite instability of the colorectal carcinoma can be predicted in the conventional pathologic examination. A prospective multicentric study and the statistical analysis of 615 cases consolidate our previously proposed logistic regression model. Virchows Archiv 2010; 456(5): 533 – 541. doi: 10.1007/ s00428-010-0896-6.
52. Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology 2007; 50(1): 113 – 130.
53. Castillejo A, Vargas G, Castillejo MI et al. Prevalence of germline MUTYH mutations among Lynch-like syndrome patients. Eur J Cancer 2014; 50(13): 2241 – 2250. doi: 10.1016/ j.ejca.2014.05.022.
54. Sourrouille I, Coulet F, Lefevre JH et al. Somatic mosaicism and double somatic hits can lead to MSI colorectal tumors. Fam Cancer 2013; 12(1): 27 – 33. doi: 10.1007/ s10689-012-9568-9.
55. Geurts-Giele WR, Leenen CH, Dubbink HJ et al. Somatic aberrations of mismatch repair genes as a cause of microsatellite-unstable cancers. J Pathol 2014; 234(4): 548 – 559. doi: 10.1002/ path.4419.
56. Mensenkamp AR, Vogelaar IP, van Zelst-Stams WA et al. Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in Lynch syndrome-like tumors. Gastroenterology 2014; 146(3): 643 – 646. doi: 10.1053/ j.gastro.2013.12.002.
57. Haraldsdottir S, Hampel H, Tomsic J et al. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology 2014; 147(6): 1308 – 1316. doi: 10.1053/ j.gastro.2014.08.041.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2016 Issue 3
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- História onkológie na Slovensku
- Diagnóza Lynchova syndromu od patologa
- Ložisková amyloidóza v dutině nosní
- Vyhodnocení výživových zvyklostí ve studii karcinomu pankreatu