
Molekulárně genetické vyšetření u akutní myeloidní leukemie
Molecular Genetic Testing for Acute Myeloid Leukemia
Background:
Acute myeloid leukemia (AML) is a clinically complex and very heterogeneous disease at the molecular level. Conventional cytogenetic analysis and FISH (fluorescence in situ hybridization) tests provide important information about the biological and clinical background of the disease and enable the classification of AML patients into three risk groups. However, up to half of patients have normal cytogenetics. Determining prognosis and treatment strategies in this group of patients is challenging. The development of molecular genetic methods, including next generation sequencing in the last decade, has led to the discovery of a number of recurrent mutations that have contributed to increasing the accuracy of prognosis of those patients with cytogenetically normal AML. Besides the prognostic value of these mutations, they may also be used to monitor minimal residual disease during and after treatment of AML and additionally constitute potential targets for the development of new therapeutic agents. The importance of molecular genetic testing of all patients with AML is highlighted by the WHO classification of 2008 in which subgroups of AML are purely defined by molecular genetics markers.
Aim:
In this article, we provide an overview of the most significant mutations in patients with cytogenetically normal AML. We describe their significance for prognosis, their importance in monitoring minimal residual disease, and their potential for the development of new targeted therapies. Further, we briefly draw attention to the significance of gene mutation accumulation in clonal disease development and how it affects the time of AML relapse.
Keywords:
acute myeloid leukemia – genetics – mutation – prognosis – minimal residual disease – clonal evolution
This work was supported by the program project of the Czech Ministry of Health reg. No. 15-25809A and by the project of Masaryk University, Brno MUNI/A/1028/2016.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted:
8. 9. 2016
Accepted:
30. 9. 2016
Authors:
V. Janečková; L. Semerád; I. Ježíšková; D. Dvořáková; M. Čulen; Z. Šustková; J. Mayer; Z. Ráčil
Authors‘ workplace:
Interní hematologická a onkologická klinika LF MU a FN Brno
Published in:
Klin Onkol 2016; 29(6): 411-418
Category:
Review
doi:
https://doi.org/10.14735/amko2016411
Overview
Východiska:
Akutní myeloidní leukemie (AML) je klinicky i molekulárně značně heterogenní onemocnění. Standardně používané konvenční cytogenetické vyšetření a vyšetření FISH (fluorescenční in situ hybridizace) přinášejí významnou informaci o biologické i klinické povaze onemocnění a umožňují rozdělení AML pacientů do tří rizikových skupin. Stanovení prognózy však stále zůstává složité u cca 50 % nemocných s AML, kteří mají normální cytogenetický nález. V poslední dekádě byla u nemocných s cytogeneticky normální AML objevena celá řada rekurentních mutací genů, u kterých byl následně prokázán jejich vliv na prognózu. Objev těchto mutací byl umožněn rychlým vývojem molekulárně biologických metod, zejména metod sekvenování nové generace. Kromě prognostického významu je možné v některých případech využít genové mutace při monitoraci minimální reziduální nemoci v průběhu a po ukončení léčby AML a přestavují také potenciál pro vývoj nové cílené léčby. Důležitost vyšetření genových mutací u nemocných s cytogeneticky normální AML potvrzuje fakt, že WHO klasifikace AML zařadila od roku 2008 přítomnost genových mutací u cytogeneticky normální AML do definice několika samostatných jednotek.
Cíl:
V této práci přinášíme přehled nejvýznamnějších mutací genů, které prokazujeme u nemocných s cytogeneticky normální AML, popisujeme jejich význam pro prognózu nemocných, jejich význam při sledování minimální reziduální nemoci a jejich potenciál pro vývoj nové cílené léčby. Dále je stručně popsán význam akumulace genových mutací při klonálním vývoji onemocnění a zásadní vliv tohoto fenoménu při relapsu AML.
Klíčová slova:
akutní myeloidní leukemie – genetika – mutace – prognóza – minimální reziduální nemoc – klonální vývoj
Úvod
Akutní myeloidní leukemie (AML) je klonální onemocnění krvetvorby charakterizované přítomností proliferujících, abnormálně diferencovaných buněk hematopoetického systému v kostní dřeni, periferní krvi a případně extramedulárních tkáních.
Standardní diagnostické postupy pro klasifikaci AML v současné době zahrnují nejen morfologické hodnocení nátěru periferní krve i kostní dřeně a analýzu povrchových nebo cytoplazmatických markerů pomocí průtokové cytometrie, ale také identifikaci chromozomálních změn pomocí metod konvenční cytogenetiky nebo metody FISH (fluorescenční in situ hybridizace) a vyšetření mutačního stavu vybraných molekulárních markerů.
Již od 90. let minulého století je známo, že cytogenetické aberace jsou spojeny s různým průběhem onemocnění. Na základě výsledků cytogenetického vyšetření byla v roce 1998 vytvořena první kategorizace AML do rizikových skupin dle Grimwade (tab. 1) [1]. Zařazení AML do rizikové skupiny má důležitý význam pro stanovení prognózy onemocnění a výběr vhodné léčebné strategie. Stanovení individuálního rizika onemocnění umožňuje vyčlenit skupinu nemocných s vysokým rizikem relapsu (RR) onemocnění, kteří benefitují z provedení alogenní transplantace v rámci 1. léčebné linie, a naopak skupinu nemocných s nízkým RR, u kterých alogenní transplantaci krvetvorby v rámci 1. léčebné linie standardně neprovádíme z důvodu vysoké morbidity a mortality spojené s touto vysoce intenzivní léčebnou metodou. Z cytogenetických aberací hrají základní roli v leukemogenezi rekurentní chromozomální strukturní variace, např. t (15; 17), t (8; 21), inv (16), t (16; 16), t (9; 21), t (9; 11), del5, del7, které jsou již zavedenými diagnostickými a prognostickými markery.
![Cytogenetické riziko dle Grimwade et al (1998) [1].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/50b42c7864191a875ac245457edeb061.jpg)
Problém pro stanovení prognózy přestavuje značně heterogenní skupina pacientů s cytogeneticky normální AML (CN-AML). Do této skupiny spadá 40–50 % pacientů. Je proto vyvíjena snaha o hledání dalších faktorů, které umožní lepší stratifikaci CN-AML a definují skupinu nemocných, kteří budou profitovat z intenzifikace terapie a provedení alogenní transplantace krvetvorných buněk v 1. léčebné linii. Kromě klinických prognostických faktorů, jako je přítomnost hyperleukocytózy či nedostatečná blastoredukce při časném hodnocení v průběhu první indukční terapie, hrají důležitou roli v prognostické stratifikaci molekulárně genetické markery.
Etiopatogenetickým podkladem pro vznik AML je kumulace mutací v genech, které hrají významnou roli v regulaci dělení a diferenciace hematopoetických buněk. Typ mutací a jejich komplexnost ovlivňuje klinický průběh onemocnění a odpověď na léčbu. Význam komplexnosti mutačního stavu hematopoetické buňky dokládají v recentní práci Wakita et al, kteří analyzovali přítomnost 28 různých mutací u 271 pacientů s AML v souvislosti s jejich prognózou [2]. Bylo prokázáno, že u mladších nemocných ve středním cytogenetickém riziku je celkové přežití (overall survival – OS) významně sníženo v případě přítomnosti tří a více mutací genů (5leté OS 18,1 vs. 45,9 %; p = 0,0006), a to i ve skupině nemocných s FLT3-ITD negativitou (5leté OS 28,3 vs. 63,2 %; p = 0,001). Předpokládá se tedy, že vyšší počet mutací je znakem vysoké genetické nestability, která souvisí s horší odpovědí choroby na léčbu.
Význam molekulárně genetického vyšetření u nemocných s AML dokládá také ta skutečnost, že mutace v genech NPM1 a CEBPA jsou od roku 2008 součástí klasifikace AML dle Světové zdravotnické organizace (WHO) [3] a spolu s mutacemi ve FLT3 jsou začleněny do prognostické klasifikace AML dle European LeukemiaNet (ELN) z roku 2010 (tab. 2) [4]. Při recentní revizi WHO klasifikace v roce 2016 byla přidána rovněž provizorní kategorie AML s mutací v genu RUNX1 [5].

Díky značnému poklesu ceny a automatizaci většiny kroků analytického postupu se široce rozšířily ve výzkumných i rutinně diagnostických laboratořích nové genomické metodické přístupy založené zejména na sekvenování nové generace. V rámci rozvoje celogenomového sekvenování byly v posledním desetiletí zjištěny další rekurentní somatické mutace, jejichž prognostický význam je intenzivně studován. Velkým přínosem je naprosto nový pohled na spektrum a frekvenci mutací, jejich kooperaci nebo vzájemnou výlučnost. Umožňují navíc sledovat skladbu buněčných subklonů a klonální evoluci v průběhu onemocnění.
Genomický pohled na AML
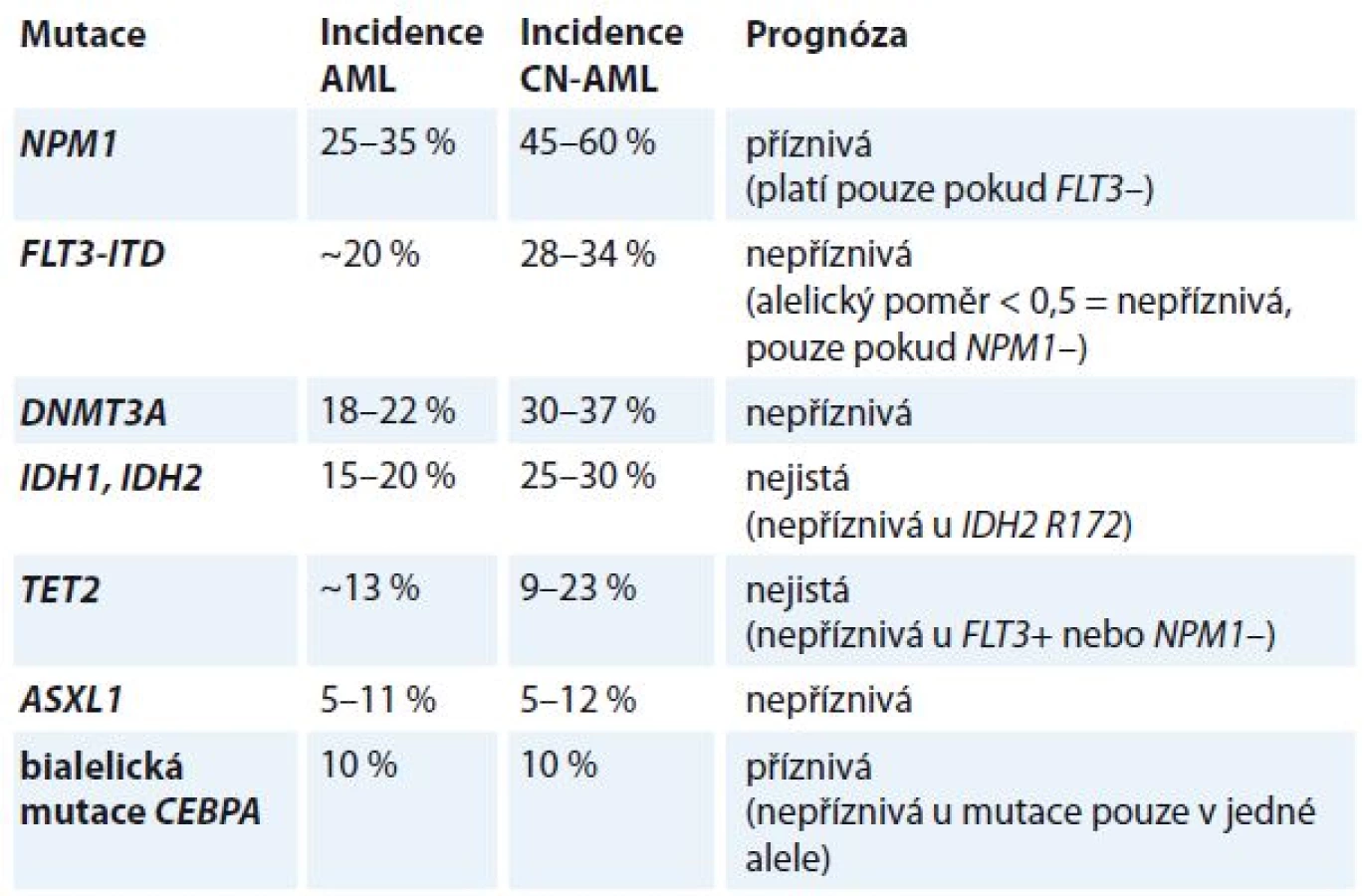
Leukemogeneze AML je vícekrokový proces, který vyžaduje spolupráci nejméně dvou tříd mutací [6,7]. Mutace třídy I aktivují dráhy signální transdukce a dávají proliferační výhodu hematopoetickým buňkám (např. mutace vedoucí k aktivaci tyrozinkinázových receptorů FLT3, c-kit a Ras), mutace třídy II ovlivňují transkripční faktory a poškozují hematopoetickou diferenciaci (např. rekurentní chromozomální aberace t (8; 21), inv (16), t (15; 17), které generují fúzní transkripty RUNX1T1, CBFB/MYH11 a PML/RARA, a rovněž mutace v genech RUNX1, CEBPA a MLL. Konsekvence mutací v dalších genech asociovaných s AML jako DNMT3A, TET2, IDH1, IDH2, NPM1, ASXL1 nejsou dosud známy. Bývají nalezeny obvykle u CN-AML (tab. 3).
Klonální evoluce u AML
Celogenomové sekvenování u pacientů s AML ukázalo, že klonálně odvozená hematopoetická buňka akumuluje obrovský počet mutací v závislosti na věku, avšak naprostou většinu těchto mutací lze považovat za náhodné benigní události. Tyto mutace byly tedy zřejmě přítomny již v hematopoetické buňce, která byla transformována iniciující (driver) mutací [8]. Mnohé studie prokazují, že většina případů AML je charakterizována klonální heterogenitou v době diagnózy, s přítomností jak zakládajícího klonu, tak alespoň jednoho subklonu [9]. Byla publikována různá schémata klonální evoluce, která se mohou skrývat za relapsem onemocnění a být příčinou jeho rezistence [10–13]. Základní model uvádí dvě možné situace: 1. zakládající klon získává mutace a vyvíjí se v relabující klon; 2. subklony s různými mutacemi se vyskytují mezi zakládajícími klony a jeden ze subklonů z této populace expanduje v relaps [10]. U pacientů v morfologické remisi může pokračovat klonální hematopoéza těch klonálních populací, které jsou blízce příbuzné klonu zakládajícímu. Zároveň však byla pozorována expanze hematopoetické populace, která nebyla příbuzná s iniciální AML, ale nesla mutace v genech často mutovaných u AML a myelodysplastického syndromu (MDS). Tyto mutace bylo možné detekovat ve velmi nízkých frekvencích již v době diagnózy AML. Výsledky podporují hypotézu, že neleukemické hematopoetické kmenové a progenitorové buňky, které nesou určité mutace, mohou získat kompetitivní výhodu díky perzistenci těchto mutací během i po ukončení indukční chemoterapie [14]. Cytoredukční chemoterapie tak působí selekčním tlakem na leukemickou i neleukemickou hematopoetickou populaci [10,15].
V rámci Cancer Genome Atlas (TCGA) Research Network byly analyzovány genomy 200 pacientů a téměř v každém vzorku byla nalezena jedna potenciální driver mutace, která poskytuje hematopoetické progenitorové buňce růstovou výhodu vedoucí ke klonální expanzi [9]. Signifikantně mutované geny, které jsou relevantní pro patogenezi, byly organizovány do devíti funkčních kategorií – fúze transkripčních faktorů (18 % případů), NPM1 (27 %), tumor-supresorové geny (16 %), geny vztahující se k DNA metylaci (44 %), signální geny (59 %), geny modifikující chromatin (30 %), geny pro myeloidní transkripční faktory (22 %), geny pro kohezinový komplex (13 %) a geny pro komplex spliceozomu (14 %). Systém vzájemné spolupráce a výlučnosti mezi mutačními událostmi v těchto genech pak pravděpodobně přispívá k patogenezi AML u konkrétního pacienta.
Sekvenování nové generace a prognostická klasifikace CN AML
Nejvýznamnějším prognostickým parametrem byly dosud rekurentní cytogenetické aberace [16]. Předmětem intenzivního výzkumu je nyní hodnocení molekulárně genetických lézí jako prognostických a prediktivních markerů [17–19]. V současné době jsou na základě doporučení ELN v klinické praxi používány tři molekulární markery (NPM1, CEBPA a FLT3-ITD) [4]. V budoucnu je očekáváno zařazení dalších markerů (např. RUNX1, ASXL1 a TP53), které byly opakovaně asociovány s horší prognózou, přičemž prognostický význam dalších často mutovaných genů (např. DNMT3A, IDH1, IDH2, TET2) je stále nejasný.
FLT3
FLT3 je buněčný receptor s tyrozinkinázovou aktivitou patřící do stejné skupiny receptorů jako je c-kit nebo PDGF-R. FLT3 protein hraje důležitou roli v dělení a diferenciaci myeloidních buněk. Nejčastější mutací genu FLT3 je interní tandemová duplikace (FLT3-ITD), která je prokazována u 20–30 % případů AML [20,21]. Mutace vede k trvalé aktivaci kinázové domény FLT3 receptoru a následné poruše diferenciace myeloidních buněk a jejich leukemické transformaci. Druhou méně častou mutací genu FLT3 je bodová mutace v aktivační smyčce druhé tyrozinkinázové domény (FLT3-TKD). Tato mutace je prokazována u 5–8 % případů AML [6,22,23]. Obě tyto mutace v genu pro FLT3 jsou asociovány s vyšším počtem leukocytů, nižším počtem trombocytů a vyšším zastoupením blastů v kostní dřeni v době diagnózy. Relativně častěji je mutovaný gen FLT3 prokazován u akutní monocytární leukemie (AML FAB M5), a to cca ve 40 % případů. Častěji je přítomen u CN-AML ve srovnání s AML s cytogenetickou abnormalitou [22]. Mutace FLT3-ITD je také často asociována s t (6; 9) (88–90 %) a MLL-PTD. Kooperativní mutace byly nalezeny také u FLT3-TKD a CBFB/MYH1 [7].
Mutace v genu FLT3 mají jasně prokázaný negativní prognostický význam. Pravděpodobnost dosažení kompletní remise (complete remission – CR) onemocnění po indukční terapii není sice mutací FLT3 ovlivněna, četnost CR u mladších nemocných je v případě FLT-ITD negativity 66,8 % a v případě FLT3-ITD pozitivity 71,2 %. Mutace FLT3 jsou však spojeny s významně vyšším RR onemocnění. OS pacientů s CN-AML s mutací FLT3-ITD nebo TKD je tak horší oproti nemocným bez přítomnosti těchto mutací. Na OS nemocných má kromě samotné přítomnosti mutace FLT3 vliv alelický poměr FLT3 mutovaného/nemutovaného genu (FLT3 mutant/wt). Bylo prokázáno, že nemocní s mutací NPM1 a současnou mutací FLT3 s nízkým alelickým poměrem FLT3 mutant/wt mají stejně dobrou prognózu jako nízce rizikoví CN-AML nemocní s mutací NPM1 bez mutace FLT3 (5leté OS 47 ± 10 vs. 56 ± 5 %; p = nesignifikantní). Oproti tomu nemocní s mutací NPM1 a vysokým alelickým poměrem FLT3 mutant/wt mají OS významně zkrácené (5leté OS 29 ± 7 vs. 56 ± 5 %; p = 0,017). Prognóza pacientů s nízkým alelickým poměrem FLT3 mutant/wt a nemutovaným genem NPM1 je spojena s vyšší pravděpodobností relapsu onemocnění a horším OS (5leté RR 74 ± 20 vs. 48 ± 6 %; p = 0,017; 5leté OS 20 ± 12 vs. 39 ± 6 %; p = 0,014) [24].
Provedení alogenní transplantace krvetvorné tkáně (hematopoietic stem cell transplantation – HSCT) v rámci 1. léčebné linie snižuje RR onemocnění a vede k prodloužení OS u nemocných s mutací NPM1 a vysokým alelickým poměrem FLT3 mutant/wt (5leté RR 20 ± 13 vs. 80 ± 9 %; p = 0,014; 5leté OS 22 ± 10 vs. 70 ± 14 %; p = 0,03) (tab. 4). Naopak z provedení alogenní HSCT v 1. CR nebenefitují pacienti s mutací NPM1 bez mutace FLT3 (5leté RR 35 ± 7 vs. 20 ± 11 %; p = 0,49; 5leté OS 64 ± 7 vs. 79 ± 11 %; p = 0,296) a také nemocní s mutací NPM1 s nízkým alelickým poměrem FLT3 mutant/wt (5leté RR 22 ± 15 vs. 19 ± 20 %; p = 0,566; 5leté OS 67 ± 16 vs. 56 ± 17 %; p = 0,873). Pacienti s cytogeneticky normální AML bez přítomnosti mutace NPM1 obecně profitují z provedení alogenní HSCT v rámci 1. léčebné linie. Alogenní HSCT snižuje 5leté RR u pacientů s NPM1 wt a FLT3-ITD (5leté RR 42 ± 14 vs. 100 %; p = 0,00016) a také u pacientů s NPM1 wt a FLT3 wt (5leté RR 25 ± 10 vs. 45 ± 7 %; p = 0,08) [24].
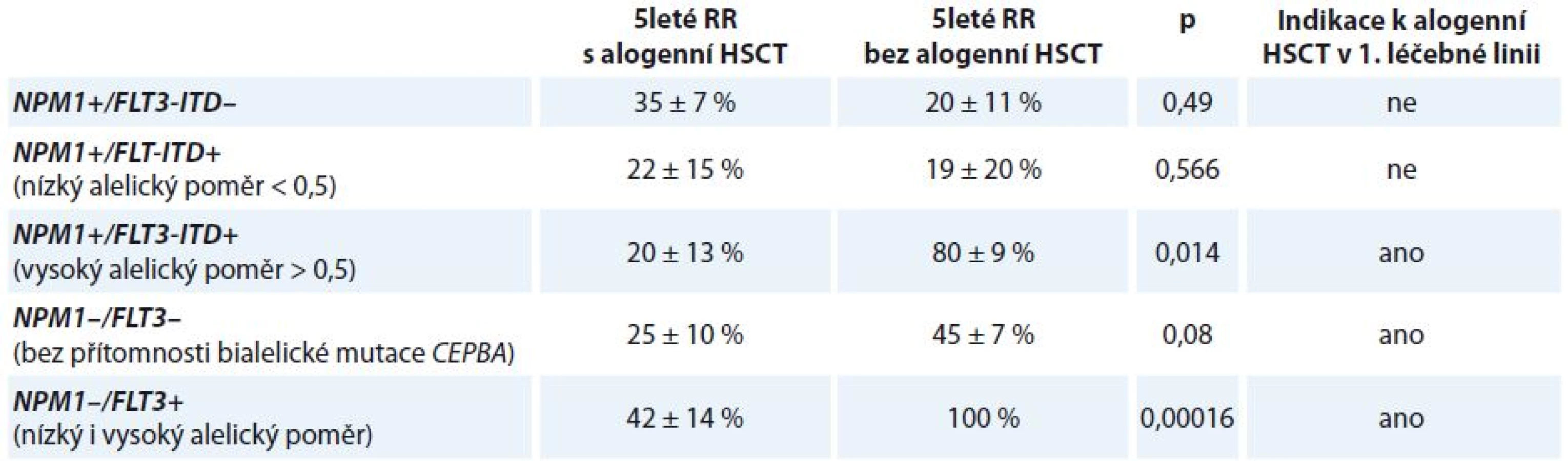
FLT3-ITD je nestabilní prognostický marker – až u 30 % pacientů, kteří nesli tuto mutaci při diagnóze choroby, byla pozorována ztráta nebo změna počtu tandemových duplikací v době relapsu [24].
NPM1
Nukleofosmin je fosfoprotein, který plní úlohu molekulárního chaperonu a transportního proteinu. NPM1 se uplatňuje v různých buněčných procesech – při biogenezi ribozomů, zdvojení centrozomu, reparaci DNA a odpovědi na buněčný stres, ovlivňuje funkci p53 a p19. NPM1 wt chrání hematopoetické buňky před apoptotickými účinky p53 v podmínkách buněčného stresu. Existuje více než 40 různých variant mutací, přičemž nejčastější je inzerce 4 bp v exonu 12 způsobující aberantní delokalizaci proteinu z jádra do cytoplazmy. Buňky jsou následně citlivější k buněčnému stresu vyvolanému chemoterapií, což znamená lepší prognózu [25].
Mutace NPM1 je nejčastější mutací u pacientů s AML (25–30 %) a je prokazována cca u 45–60 % pacientů s CN-AML mladších 60 let [26]. Nemocní s CN-AML a mutací NPM1 bez přítomnosti jiné mutace lépe odpovídají na indukční chemoterapii a mají významně nižší RR onemocnění po ukončení léčby. Nejlepší léčebná odpověď na indukční terapii byla u nemocných s mutací NPM1 a nemutovaným FLT3 (86 %), následovala skupina nemocných s nemutovaným NPM1 a mutací FLT3-ITD (76 %), ve skupině s nemutovaným NPM1 i FLT3 byla odpověď na indukci 68,5 % a nejnižší pravděpodobnost dosažení CR onemocnění po indukci byla pozorována ve skupině se současnou mutací NPM1 a FLT3-ITD (63 %) [27,28]. Na základě těchto poznatků současná prognostická klasifikace dle ELN z roku 2010 zařazuje CN-AML s mutací NPM1 bez mutace FLT3 či DNMT3A do nízce rizikové skupiny, u které není v rámci 1. léčebné linie doporučeno provedení alogenní transplantace krvetvorných buněk [4].
CEBPA
Gen CEBPA kóduje myeloidní transkripční faktor. Je mutovaný u přibližně 10 % CN-AML a může se vyskytnout také současně s chromozomálními aberacemi, např. del (9q) a vzájemně se vylučují s balancovanými chromozomálními přestavbami [29]. V genu CEBPA se u AML vyskytují zejména dva typy mutací – 33 bp inzerce na C-konci, která zasahuje oblasti, které se podílejí na dimerizaci proteinu a jeho vazbě na DNA, a frameshift delece na N-konci s důsledkem zkrácené formy proteinu [30]. Výskyt obou těchto mutací v bialelické formě se vyskytuje u více než poloviny pacientů s prokázanou mutací CEPB a slouží jako příznivý prognostický faktor s nevýznamně delším OS a významně nižším výskytem relapsu. Pouze jedna mutace je přitom spojována s horší prognózou. Tento biologický paradox dosud nebyl zcela objasněn [31,32].
Mutace v epigenetických regulátorech
Epigenetické regulátory zodpovídají za regulaci transkripce prostřednictvím dvou hlavních mechanizmů – metylace DNA (enzymy kódované geny DNMT3A, IDH1, IDH2 a TET2) a modifikace histonů (ASXL1). Výsledky studií zabývajících se klonální evolucí podporují hypotézu, že mutace v genech, které jsou zahrnuty v epigenetické regulaci (tj. DNMT3A, ASXL1, IDH1, IDH2 a TET2), jsou přítomny již v preleukemické hematopoetické kmenové buňce a objevují se velmi časně ve vývoji AML [10,11,33]. Tyto původní preleukemické kmenové buňky jsou schopny diferenciace do více linií, dokáží přežít chemoterapii a expandovat během remise až k rozvoji potenciálního relapsu. Současné studie ukazují, že klonální hematopoéza se somatickými mutacemi, obvykle v genech DNMT3A, TET2 a ASXL1, vzrůstá spolu s věkem a je asociována se zvýšeným rizikem rozvoje hematologické malignity [34]. Prognostický význam těchto mutací však nebyl jednoznačně stanoven.
DNMT3A
DNMT3A patří mezi DNA metyltransferázy, které katalyzují adici metylové skupiny na cytozinové zbytky CpG dinukleotidů. Funkční experimenty ukazují, že ztráta DNMT3A (DNA metyltransferáza 3 alfa) poškozuje diferenciaci hematopoetických buněk a působí zvýšenou akumulaci těchto buněk v kostní dřeni [35]. Mutace v DNMT3A nejčastěji (60–64 %) postihují arginin R882 a způsobují redukovanou aktivitu enzymu způsobující globální hypometylaci [36,37]. Ostatní méně časté mutace se nacházejí po celém genu, především v exonech 13–23. Zahrnují missense, nonsense, frameshift a mutace měnící místo sestřihu a jsou obvykle heterozygotní. Různé typy mutací mohou mít různý dopad na funkci enzymu. Delece DNMT3A v myších modelech vede k inhibici diferenciace hematopoetických kmenových buněk, avšak samotný deficit tohoto genu nevede ke vzniku AML. Zdá se tedy, že pro vznik AML je nutný vznik další tzv. driving mutace v jiných genech.
Mutace v genu DNMT3A jsou prokazovány u 17–20 % nemocných s AML, ve skupině pacientů s normálním cytogenetickým nálezem je prokazována až ve 27–30 % případů [38–40]. Jedná se tak o třetí nejčastější prokazovanou mutaci u nemocných s AML. Mutace DNMT3A je spojena s vyšším věkem nemocných, vyšším počtem leukocytů a krevních destiček v periferní krvi v době diagnózy. Významně častěji je prokazována u nemocných se současnou mutací genu NPM1, IDH1/2 nebo mutací FLT3-ITD. Asociace mutace DNMT3A je silná zejména v případě mutace NPM1, až 80 % pacientů s mutací DNMT3A má současně přítomnou mutaci NPM1.
Z hlediska prognostického významu mutace DNMT3A jsou výsledky jednotlivých klinických studií diskrepantní. V původní práci Thol et al z roku 2011 bylo prokázáno signifikantně horší přežití nemocných s mutací DNMT3A a současnou mutací NPM1 a FLT3-ITD oproti těmto pacientům s nemutovaným genem DNMT3A (medián OS 12,3 vs. 41,1 měsíce; p < 0,0001) [38,39]. Ve stejné práci však nebyl prokázán vliv mutace DNMT3A na OS nízce rizikových pacientů s mutací NPM1 bez FLT3-ITD. Tyto výsledky nekorelují s dosud nejrozsáhlejší prací Gaidzik et al, ve které vliv mutace na OS a dobu do relapsu u CN-AML prokázán nebyl [41]. Gale et al vysvětlují diskrepantní výsledky tzv. Simpsonovým paradoxem, který je dán vysokou koincidencí mutace DNMT3A s prognosticky příznivou mutací NPM1 [37]. Aby mohl být tedy ukázán negativní prognostický význam mutace DNMT3A, musí být výsledky stratifikovány podle NPM1 genotypu. Pokud hodnotíme skupinu CN-AML pacientů jako celek, nebyl prokázán vliv mutace DNMT3A na OS pacientů (p = 0,7). Pokud rozdělíme nemocné na dvě skupiny podle přítomnosti či nepřítomnosti mutace NPM1, byl v obou těchto skupinách prokázán negativní prognostický vliv mutace DNMT3A na OS nemocných. Mutace DNMT3A je tedy obecně považována za negativní prognostický marker u nemocných s AML.
V rámci optimalizace dávek daunorubicinu u standardní indukční terapie 3 + 7 bylo prokázáno, že nemocní s mutací DNMT3A výrazně více profitují z podání vyšší dávky daunorubicinu oproti jiným geneticky definovaným skupinám [38,39].
IDH1, IDH2
IDH1 a IDH2 kódují NADP-dependentní izocitrát-dehygrogenázy, které katalyzují oxidativní dekarboxylaci izocitrátu na 2-oxoglutarát v citrátovém cyklu. Mutace v IDH1 a IDH2 jsou hemizygotní, způsobující v IDH1 záměnu argininu R132 a v IDH2 argininu R172 nebo R140 [42]. Nové mutace byly nalezeny pomocí masivního paralelního sekvenování zejména u CN-AML jak v IDH1 [43], tak IDH2 [44]. IDH1 a IDH2 se uplatňují především v buněčném metabolizmu a syntéze lipidů, v ochraně buňky před oxidativním stresem [45]. IDH1 a IDH2 způsobují aberantní hypermetylaci a potvrdila se vzájemná výlučnost mutací v IDH1 a IDH2 a v TET2 [46,47].
Mutace genů IDH1 a IDH2 byla prokázána u 7,6, resp. u 8,7 % nemocných s AML [42]. Je asociována s vyšším věkem nemocných (p = 0,002), s nižším počtem leukocytů (p = 0,04) a vyšším počtem trombocytů v době diagnózy (p = 0,007). Mutace IDH1 a IDH2 je častěji prokazována u nemocných s CN-AML, zejména u nemocných s mutovaným NPM1 bez FLT3-ITD (p < 0,001). Asociace s mutací NPM1 neplatí pro mutaci IDH2 v kodonu 172. Mutovaný IDH1 zároveň silně asociuje s MLL-PTD [48].
Význam mutace IDH1 a IDH2 pro určení prognózy není zcela jasně definován, výsledky jednotlivých prací se liší. Paschka et al ve své práci neprokazují vliv mutace na dosažení remise onemocnění po indukční terapii [42]. Negativní vliv mutace IDH1 a IDH2 na OS byl v jeho práci prokázán pouze ve skupině nemocných s CN-AML s mutovaným NPM1 bez mutace FLT3-ITD (5leté OS 41 vs. 65 %; p = 0,03). Pokud hodnotíme skupinu nemocných s CN-AML jako celek, nebyl vliv mutace IDH1 a IDH2 na OS nemocných prokázán. Patel et al naopak u nemocných s AML ve středním cytogenetickém riziku prokázali významně lepší 3leté OS nemocných, kteří byli nositeli mutace NPM1 a současně mutace IDH1 a IDH2, ve srovnání s nemocnými, kteří měli mutovaný pouze NPM1 bez IDH1 a IDH2 (3leté OS 89 vs. 31 %; p < 0,001) [49]. V tomto srovnání však nebyl brán v potaz negativní vliv mutace FLT3-ITD na OS nemocných. Analýza jednotlivých geneticky definovaných podskupin v práci Patel et al potvrdila negativní prognostický význam mutace IDH2 v kodonu 172 na OS nemocných ve středním riziku, což je pravděpodobně dáno malou asociací této konkrétní mutace s mutací genu NPM1.
TET2
Enzym TET2 je dioxygenáza, která dokáže konvertovat 5-metylcytozin (5-mC) na 5-hydroxymetylcytozin (5-hmC), který je meziproduktem DNA demetylace [50]. Pacienti nesoucí mutaci v TET2 jsou tedy charakterizováni hypermetylovaným fenotypem s místně specifickou metylací cytozinu cílových genů a zároveň poklesem 5-hmC. Pokles 5-hmC je tedy pravděpodobně průvodním jevem stárnutí hematopoetického systému, na němž se podílí právě mutace v TET2. Ztráta TET2 vyvolává vzrůstající sebeobnovu hematopoetických kmenových buněk a kompetitivní růstovou výhodu [51].
Mutace genu TET2 jsou prokazovány v 7–13 % případů AML. Tato mutace je asociována s vyšším věkem nemocných, vyšším počtem leukocytů a nižším počtem trombocytů v době diagnózy. Častěji je také prokazována u nemocných s mutací NPM1, CEPBA a ASXL1 [46]. Naopak mutace TET2 není téměř nikdy prokazována u nemocných s mutovaných IDH1/2.
Prognostický vliv mutace TET2 je nejasný. Chou et al prokázali negativní prognostický význam a horší OS nemocných s mutací TET2 ve středním cytogenetickém riziku (medián OS – medián OS nebyl dosažen vs. 14,7 měsíce; p = 0,021) [52]. Silná asociace mutace TET2 s horším OS byla pozorována zejména ve skupině pacientů s FLT3-ITD pozitivitou (medián OS 5,0 vs. 16,9 měsíce; p = 0,001) a ve skupině pacientů s nemutovaným NPM1 (medián OS 12,3 vs. 61,0 měsíce; p = 0,055). Tyto výsledky však nebyly potvrzeny v práci Gaidzik et al, která neprokázala žádný vliv mutace TET2 na OS nemocných s AML, a to ani ve skupině nemocných ve středním cytogenetickém riziku či s normálním cytogenetickým nálezem [53]. Do této práce však byli zahrnuti pouze mladší nemocní do 60 let, což z důvodu asociace mutace TET2 s vyšším věkem nemocných pravděpodobně ovlivnilo výsledky.
Chou et al dále dokládají nevhodnost mutace TET2 pro sledování minimální reziduální nemoci v průběhu a po ukončení léčby. Mezi 13 pacienty, kteří byli vstupně TET2 pozitivní a u kterých došlo po ukončení léčby k relapsu onemocnění, šest pacientů ztratilo při relapsu onemocnění původní mutaci TET2 [52].
ASXL1
ASXL1 (aditional sex comb-like 1) patří do rodiny Polycomb proteinů podílejících se na regulaci remodelace chromatinu. Ve spolupráci s HP1 ovlivňuje metylaci histonů prostřednictvím regulace aktivity demetylázy LSD1 [54]. Funkce ASXL1 v hematopoéze je však dosud nejasná. Mutace v genu ASXL1 byly nalezeny u chronické myelomonocytární leukemie (CMML), MDS, sekundárních AML, které se vyvinuly z CMML, i de novo AML [55,56].
Mutace ASXL1 jsou typu delecí nebo bodových mutací. Jejich výskyt u pacientů s AML je 9–18 % [57–59]. U nemocných se středně rizikovou AML je mutace ASXL1 asociována s mužským pohlavím (23,5 vs. 9,9 %; p < 0,001), s vyšším věkem nemocných (medián 71,8 vs. 61,8; p < 0,001). Mutace je významně častěji prokazována u nemocných s mutací RUNX1, naopak její přítomnost negativně koreluje se současnou přítomností mutací NPM1, FLT3-ITD, FLT3-TKD a DNMT3A. Významně častěji je mutace ASXL1 prokazována u AML s trizomií chromozomu 8 (52,7 %) [58].
Mutace ASXL1 je spojena s výrazně horší prognózou pacientů s AML. Bylo prokázáno významně zkrácené OS nemocných se středně rizikovou AML (medián OS 11,0 vs. 62,2 měsíce; p < 0,001) [57]. Zkrácení OS bylo prokázáno ve všech geneticky definovaných rizikových a věkových skupinách. Mutace ASXL1 je tedy považována za významný negativní prognostický marker u nemocných s AML a pacienti s touto mutací by měli být směřování k provedení alogenní HSCT v rámci léčby 1. linie.
Závěr
Hledání nových molekulárně genetických markerů u nemocných s AML je důležité pro stanovení individuálního léčebného přístupu, zejména u nemocných ve středním cytogenetickém riziku. Z důvodu vysoké peritransplantační morbidity a mortality je nutné vyčlenit skupinu nemocných, kteří v rámci 1. léčebné linie neprofitují z provedení alogenní transplantace. Metody celogenomového sekvenování umožnily prokázat celou řadu mutací genů, které hrají významnou roli v patogenezi AML a mají význam pro prognostickou stratifikaci. Kromě prognostického významu představují mutované geny také potenciál pro vývoj nové cílené léčby. V rámci klinických studií jsou v současné době hodnoceny inhibitory FLT3, IDH1 a IDH2. Nové terapeutické přístupy jsou z důvodu neuspokojivé prognózy nemocných s AML vysoce očekávány.
Velké klinické studie prokázaly příznivý vliv na OS nemocných pouze u mutace genu NPM1 a bialelické mutace CEBPA, naopak jasně negativní vliv na prognózu má mutace genu FLT3 a DNMT3A. Mutace NPM1, FLT3-ITD a DNMT3A se na pracovišti Interní hematologické a onkologické kliniky (IHOK) FN Brno rutinně vyšetřují u všech nemocných s nově diagnostikovanou AML, u kterých je plánováno zahájení intenzivní terapie. Z důvodu velmi nízké incidence bialelické mutace CEBPA se tato mutace standardně nevyšetřuje. Z nových molekulárně genetických markerů je negativní prognostický význam prokázán pouze u mutace ASXL1, která rovněž z důvodů nízké incidence není na IHOK vyšetřována. U ostatních markerů jsou výsledky jednotlivých prací diskrepantní a jejich prognostický význam není zcela jasně definován.
Tato práce byla podpořena programovým projektem MZ ČR s reg. č. 15-25809A a z projektu Masarykovy univerzity, Brno MUNI/A/1028/2016.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Lukáš Semerád
Interní hematologická a onkologická klinika
LF MU a FN Brno
Jihlavská 20
625 00 Brno
e-mail: lukas.semerad@fnbrno.cz
Obdrženo: 8. 9. 2016
Přijato: 30. 9. 2016
Sources
1. Grimwade D, Walker H, Oliver F et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The medical research council adult and children’s leukaemia working parties. Blood 1998; 92 (7): 2322–2333.
2. Wakita S, Yamaguchi H, Ueki T et al. Complex molecular genetic abnormalities involving three or more genetic mutations are important prognostic factors for acute myeloid leukemia. Leukemia 2016; 30 (3): 545–554. doi: 10.1038/leu.2015.288.
3. Swerdlow SH, Campo E, Harris NL et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon (France): IARC 2008.
4. Döhner H, Estey EH, Amadori S et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010; 115 (3): 453–474. doi: 10.1182/blood-2009-07-235358.
5. Arber DA, Orazi A, Hasserjian R et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016; 127 (20): 2391–2405. doi: 10.1182/blood-2016-03-643544.
6. Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002; 100 (5): 1532–1542.
7. Takahashi S. Current findings for recurring mutations in acute myeloid leukemia. J Hematol Oncol 2011; 4 : 36. doi: 10.1186/1756-8722-4-36.
8. Welch JS, Ley TJ, Link DC et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012; 150 (2): 264–278. doi: 10.1016/j.cell.2012.06.023.
9. Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013; 368 (22): 2059–2074. doi: 10.1056/NEJMoa1301689.
10. Ding L, Ley TJ, Larson DE et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012; 481 (7382): 506–510. doi: 10.1038/nature10738.
11. Shlush LI, Zandi S, Mitchell A et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014; 506 (7488): 328–333. doi: 10.1038/nature13038.
12. Xie M, Lu C, Wang J et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014; 20 (12): 1472–1478. doi: 10.1038/nm.3733.
13. Grimwade D, Ivey A, Huntly BJ. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood 2016; 127 (1): 29–41. doi: 10.1182/blood-2015-07-604496.
14. Wong TN, Miller CA, Klco JM et al. Rapid expansion of preexisting nonleukemic hematopoietic clones frequently follows induction therapy for de novo AML. Blood 2016; 127 (7): 893–897. doi: 10.1182/blood-2015-10-677021.
15. Greaves M, Maley CC. Clonal evolution in cancer. Nature 2012; 481 (7381): 306–313.
16. Grimwade D, Walker H, Harrison G et al. The predictive value of hierarchical cytogenetic classification in older adults with acute myeloid leukemia (AML): analysis of 1,065 patients entered into the United Kingdom Medical Research Council AML11 trial. Blood 2001; 98 (5): 1312–1320.
17. Meyer SC, Levine RL. Translational implications of somatic genomics in acute myeloid leukaemia. Lancet Oncol 2014; 15 (9): e382–e394. doi: 10.1016/S1470-2045 (14) 70008-7.
18. Marcucci G, Haferlach T, Döhner H. Molecular genetics of adult acute myeloid leukemia: prognostic and therapeutic implications. J Clin Oncol 2011; 29 (5): 475–486. doi: 10.1200/JCO.2010.30.2554.
19. Klco JM, Miller CA, Griffith M et al. Association between mutation clearance after induction therapy and outcomes in acute myeloid leukemia. JAMA 2015; 314 (8): 811–822. doi: 10.1001/jama.2015.9643.
20. Kottaridis PD, Gale RE, Frew ME et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood 2001; 98 (6): 1752–1759.
21. Nakao M, Yokota S, Iwai T et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia 1996; 10 (12): 1911–1918.
22. Thiede C, Steudel C, Mohr B et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood 2002; 99 (12): 4326–4335.
23. Yamamoto Y, Kiyoi H, Nakano Y et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood 2001; 97 (8): 2434–2439.
24. Cloos J, Goemans BF, Hess CJ et al. Stability and prognostic influence of FLT3 mutations in paired initial and relapsed AML samples. Leukemia 2006; 20 (7): 1217–1220.
25. Li J, Zhang X, Sejas DP et al. Negative regulation of p53 by nucleophosmin antagonizes stress-induced apoptosis in human normal and malignant hematopoietic cells. Leuk Res 2005; 29 (12): 1415–1423.
26. Falini B, Mecucci C, Tiacci E et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med 2005; 352 (3): 254–266.
27. Krönke J, Bullinger L, Teleanu V et al. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood 2013; 122 (1): 100–108. doi: 10.1182/blood-2013-01-479188.
28. Thiede C, Koch S, Creutzig E et al. Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML). Blood 2006; 107 (10): 4011–4020.
29. Pabst T, Mueller BU, Zhang P et al. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet 2001; 27 (3): 263–270.
30. Fasan A, Haferlach C, Alpermann T et al. The role of different genetic subtypes of CEBPA mutated AML. Leukemia 2014; 28 (4): 794–803. doi: 10.1038/leu.2013.273.
31. Wouters BJ, Löwenberg B, Erpelinck-Verschueren CA et al. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood 2009; 113 (13): 3088–3091. doi: 10.1182/blood-2008-09-179895.
32. Green CL, Koo KK, Hills RK et al. Prognostic significance of CEBPA mutations in a large cohort of younger adult patients with acute myeloid leukemia: impact of double CEBPA mutations and the interaction with FLT3 and NPM1 mutations. J Clin Oncol 2010; 28 (16): 2739–2747. doi: 10.1200/JCO.2009.26.2501.
33. Corces-Zimmerman MR, Hong WJ, Weissman IL et al. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci U S A 2014; 111 (7): 2548–2553. doi: 10.1073/pnas.1324297111.
34. Genovese G, Kähler AK, Handsaker RE et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014; 371 (26): 2477–2487. doi: 10.1056/NEJMoa1409405.
35. Challen GA, Sun D, Jeong M et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet 2012; 44 (1): 23–31. doi: 10.1038/ng.1009.
36. Yan XJ, Xu J, Gu ZH et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet 2011; 43 (4): 309–315. doi: 10.1038/ng.788.
37. Gale RE, Lamb K, Allen C et al. Simpson’s paradox and the impact of different dnmt3a mutations on outcome in younger adults with acute myeloid leukemia. J Clin Oncol 2015; 33 (18): 2072–2083. doi: 10.1200/JCO.2014.59. 2022.
38. Thol F, Damm F, Lüdeking A et al. Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. J Clin Oncol 2011; 29 (21): 2889–2896. doi: 10.1200/JCO.2011.35.4894.
39. Ley TJ, Ding L, Walter MJ et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med 2010; 363 (25): 2424–2433. doi: 10.1056/NEJMoa1005143.
40. Marcucci G, Metzeler KH, Schwind S et al. Age-related prognostic impact of different types of DNMT3A mutations in adults with primary cytogenetically normal acute myeloid leukemia. J Clin Oncol 2012; 30 (7): 742–750. doi: 10.1200/JCO.2011.39.2092.
41. Gaidzik VI, Schlenk RF, Paschka P et al. Clinical impact of DNMT3A mutations in younger adult patients with acute myeloid leukemia: results of the AML Study Group (AMLSG). Blood 2013; 121 (23): 4769–4777. doi: 10.1182/blood-2012-10-461624.
42. Paschka P, Schlenk RF, Gaidzik VI et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol 2010; 28 (22): 3636–3643. doi: 10.1200/JCO.2010.28.3762.
43. Mardis ER, Ding L, Dooling DJ et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009; 361 (11): 1058–1066. doi: 10.1056/NEJMoa0903840.
44. Marcucci G, Maharry K, Wu YZ et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol 2010; 28 (14): 2348–2355. doi: 10.1200/JCO.2009.27. 3730.
45. Reitman ZJ, Yan H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J Natl Cancer Inst 2010; 102 (13): 932–941. doi: 10.1093/jnci/djq187.
46. Metzeler KH, Maharry K, Radmacher MD et al. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol 2011; 29 (10): 1373–1381. doi: 10.1200/JCO.2010.32.7742.
47. Figueroa ME, Abdel-Wahab O, Lu C et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010; 18 (6): 553–567. doi: 10.1016/j.ccr.2010.11.015.
48. Schnittger S, Haferlach C, Ulke M et al. IDH1 mutations are detected in 6.6% of 1414 AML patients and are associated with intermediate risk karyotype and unfavorable prognosis in adults younger than 60 years and unmutated NPM1 status. Blood 2010; 116 (25): 5486–5496. doi: 10.1182/blood-2010-02-267955.
49. Patel JP, Gönen M, Figueroa ME et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012; 366 (12): 1079–1089. doi: 10.1056/NEJMoa1112304.
50. Ko M, Huang Y, Jankowska AM et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010; 468 (7325): 839–843. doi: 10.1038/nature09586.
51. Busque L, Patel JP, Figueroa ME et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet 2012; 44 (11): 1179–1181. doi: 10.1038/ng.2413.
52. Chou WC, Chou SC, Liu CY et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood 2011; 118 (14): 3803–3810. doi: 10.1182/blood-2011-02-339747.
53. Gaidzik VI, Paschka P, Späth D et al. TET2 mutations in acute myeloid leukemia (AML): results from a comprehensive genetic and clinical analysis of the AML study group. J Clin Oncol 2012; 30 (12): 1350–1357. doi: 10.1200/JCO.2011.39.2886.
54. Lee SW, Cho YS, Na JM et al. ASXL1 represses retinoic acid receptor-mediated transcription through associating with HP1 and LSD1. J Biol Chem 2010; 285 (1): 18–29. doi: 10.1074/jbc.M109.065862.
55. Gelsi-Boyer V, Trouplin V, Adélaïde J et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol 2009; 145 (6): 788–800. doi: 10.1111/j.1365 - 2141.2009.07697.x.
56. Abdel-Wahab O, Manshouri T, Patel J et al. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res 2010; 70 (2): 447–452. doi: 10.1158/0008-5472.CAN-09-3783.
57. Schnittger S, Eder C, Jeromin S et al. ASXL1 exon 12 mutations are frequent in AML with intermediate risk karyotype and are independently associated with an adverse outcome. Leukemia 2013; 27 (1): 82–91. doi: 10.1038/leu.2012.262.
58. Chou WC, Huang HH, Hou HA et al. Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood 2010; 116 (20): 4086–4094. doi: 10.1182/blood-2010-05-283291.
59. Rocquain J, Carbuccia N, Trouplin V et al. Combined mutations of ASXL1, CBL, FLT3, IDH1, IDH2, JAK2, KRAS, NPM1, NRAS, RUNX1, TET2 and WT1 genes in myelodysplastic syndromes and acute myeloid leukemias. BMC Cancer 2010; 10 : 401. doi: 10.1186/1471-2407-10-401.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2016 Issue 6
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- Molekulárně genetické vyšetření u akutní myeloidní leukemie
- Molekulární genetika kolorektálního karcinomu
- Pacientka s primárním intraventrikulárním gliosarkomem s dlouhodobým přežíváním – kazuistika
- Kvalita života, úzkost a deprese u pacientů s diferencovaným karcinomem štítné žlázy během krátkodobé hypotyreózy indukované vysazením levothyroxinu