
Vzácný nález hereditární zátěže u mladé pacientky s hyperkalcemickým malobuněčným karcinomem hrdla dělohy
Rare Hereditary Burden associated with a Hypercalcemic Small-Cell Carcinoma of Cervix in a Young Female Patient
Background: Oncological diseases have, in most cases, a multifactorial etiology, composed of a combination of external and internal environmental factors. Hereditary tumorous syndromes are mostly autosomal dominant diseases with incomplete but very high penetrance.
Observation: The patient, an 18-year-old virgin female, consulted a gynecologist in June 2018 because of metrorrhagia. Magnetic resonance imaging revealed a cervical tumor with the dimensions 80 × 90 × 80 mm. Histological analysis confirmed the presence of a very rare hypercalcemic type of small-cell carcinoma of the cervix. Further investigation of the germinal exom of the patient showed pathological variations in genes PALB2 and BRCA2, presented with recommendation of detailed examination by medical genetics.
Conclusion: Clinical experience with this type of tumor is very limited, but it still comes with some useful outcome. Small cell carcinomas of the gynecologic tract are very rare, aggressive diseases, with very poor prognosis, affecting mainly young women. Their origin is most often the ovaries, based on most clinical data, but these tumor also localize to the endometrium, cervix, vagina and vulva. It is an extremely rare type of cancer, for which clinical data is scant due to the extremely low number of reported cases. In this patient, the carcinoma had an unusual genetical mutation burden, which she inherited from her parents. In the light of these findings, we recommend that patients suspected of having a small-cell of the gynecologic tract provide a detailed family history, and that genetic testing be considered in similar cases.
This work was supported by MH CR grant 16-33209A and research program of Charles University Progress Q40/06.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted: 10. 6. 2019
Accepted: 9. 9. 2019
Keywords:
hereditary neoplastic syndromes – PALB2, BRCA2 mutation – whole exome sequencing – small-cell carcinoma, hypercalcemic type – rare cervical cancer
:
L. Hruška 1; I. Sirák 1; J. Laco 2; P. Fridrichová 3; H. Nosková 4; O. Slabý 4; K. Pál 4; V. Bočkayová 1; M. Hodek 1; J. Petera 1
:
Klinika onkologie a radioterapie FN Hradec Králové
1; Fingerlandův ústav patologie, FN Hradec Králové
2; Oddělení lékařské genetiky, FN Hradec Králové
3; CEITEC – Středoevropský technologický institut, Masarykova univerzita, Brno
4
:
Klin Onkol 2019; 32(6): 456-462
:
Case Report
prolekare.web.journal.doi_sk:
https://doi.org/10.14735/amko2019456
Východiska: Nádorová onemocnění jsou ve většině případů multifaktoriální etiologie, na jejich vzniku se podílejí jak faktory vnějšího, tak vnitřního prostředí. Hereditární nádorové syndromy jsou převážně autozomálně dominantně dědičná onemocnění, s neúplnou, nicméně velmi vysokou penetrancí.
Případ: Osmnáctiletá pacientka, virgo, přichází v červnu 2018 k ošetřujícímu gynekologovi pro metroragii. Magnetická rezonance pánve odhalí tumor velikosti 80 × 90 × 80 mm vycházející z oblasti děložního hrdla. Histologicky je potvrzen velmi vzácný, tzv. hyperkalcemický typ malobuněčného karcinomu děložního hrdla. Došetření germinálního exomu odhalilo patogenní variantu genů PALB2 a BRCA2 s doporučením podrobného vyšetření cestou lékařské genetiky.
Závěr: Klinické zkušenosti s tímto typem nádoru jsou velmi omezené, ale nacházejí průnik v několika aspektech – gynekologické malobuněčné karcinomy jsou velice vzácná agresivní onemocnění s velmi špatnou prognózou, postihující převážně mladé ženy. Nejčastěji vycházejí z vaječníků, na tato onemocnění je relativně nejvíce dat, ale jsou popsány i případy lokalizace na cervixu, endometriu, v pochvě i vulvě. Jedná se o extrémně vzácný případ nádorového onemocnění, což dokumentuje i nedostatek klinických dat a popsaných kazuistik z celého světa, v našem případě spojeného s velmi málo pravděpodobnou genetickou mutační náloží, kterou pacientka získala od svých rodičů. Vzhledem k této zkušenosti doporučujeme při podobném nálezu pečlivě odebrat rodinou anamnézu a zvážit genetické testování.
Klíčová slova:
hereditární nádorové syndromy – PALB2, BRCA2 mutace – celoexomové sekvenování – malobuněčný karcinom, hyperkalcemický typ – vzácný tumor děložního hrdla
Úvod
Nádorová onemocnění jsou ve většině případů multifaktoriální etiologie a na jejich vzniku se podílejí jak faktory vnějšího, tak vnitřního prostředí. Velká většina nádorů vzniká jako náhodný proces, jen asi 5–10 % vzniká na základě vrozené dědičné dispozice [1,2].
Hereditární nádorové syndromy jsou převážně autozomálně dominantně dědičná onemocnění s neúplnou, nicméně velmi vysokou penetrancí. Podle Knudsonovy teorie germinální mutace v příslušeném genu vytvářejí první zásah, což zvýší náchylnost jedince k nádoru, jelikož nyní stačí pouze jedna somatická mutace k vyřazení obou alel příslušného genu, a buňka tak získává selekční výhodu. Charakteristický je přenos mutace bez rozdílu pohlaví, klinické projevy se mohou u jednotlivých pohlaví lišit, a ne každý nositel musí onemocnět, avšak riziko onemocnění je u takového jedince větší než u normální populace. Pro tyto nádory je typický opakovaný výskyt určitého nádorového onemocnění v rodině, nízký věk při diagnóze (často méně než 35 let) a vícenásobný či opakovaný výskyt nádorového onemocnění u téže osoby.
V různých rodinách se mohou vyskytovat a přenášet různé typy mutací. Pokud je mutace ve vysoce penetrantním genu prokázána, je v dnešní době možné nechat testovat všechny příbuzné v riziku na přítomnost, resp. nosičství stejné mutace. Díky tomuto postupu je možné u dalších členů vybrané rodiny odhalit vyšší riziko nádorového onemocnění a časně zahájit opatření, ať charakteru dispenzárního, či charakteru specifických preventivních výkonů.
Kazuistika
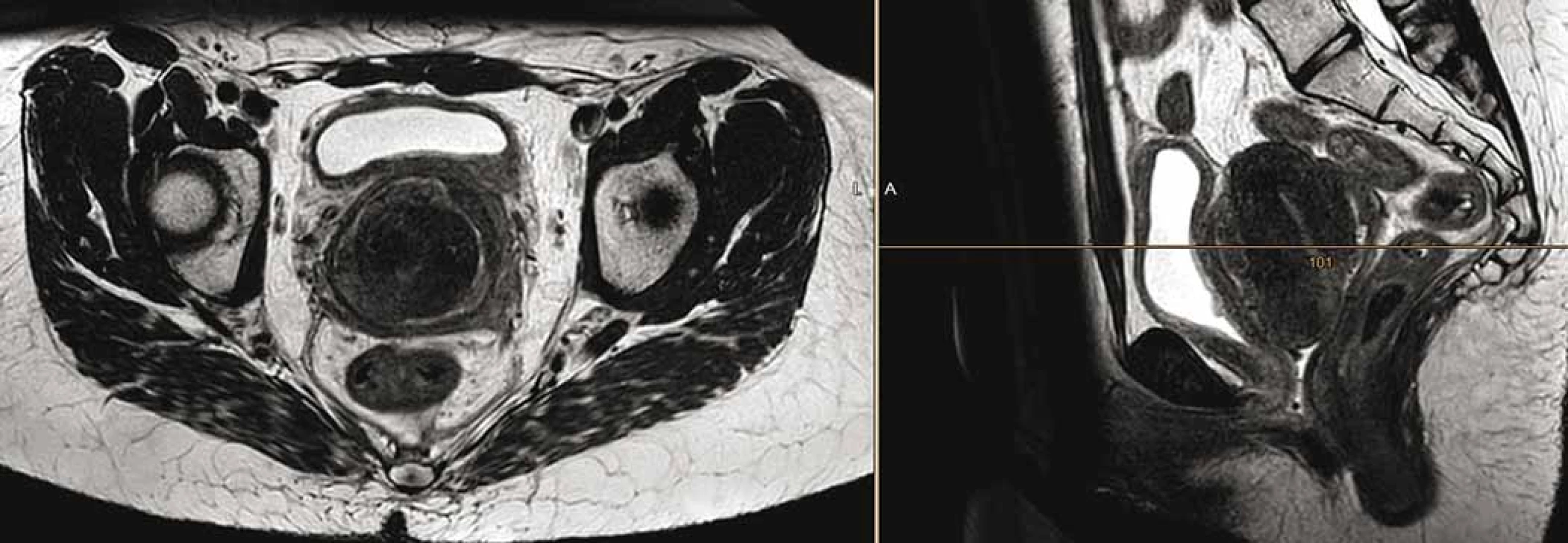
Osmnáctiletá pacientka, virgo, přichází v červnu 2018 k ošetřujícímu gynekologovi pro metroragii. Ten při vyšetření nalézá tumor v oblasti děložního hrdla, provede komplexní vyšetření vč. biopsie a posléze odesílá nemocnou na indikační seminář do vyššího pracoviště. Magnetická rezonance (magnetic resonance imaging – MRI) pánve odhalí tumor velikosti 80 × 90 × 80 mm vycházející z oblasti děložního hrdla, de facto obdávající dělohu (obr. 1), vyplňující pochvu, naléhající na močový měchýř a rektum (bez zřetelné invaze). Současně je na MRI přítomna lymfadenopatie presakrálně (30 × 15 × 15 mm) a bilaterálně parailicky (28 × 21 × 40 mm). Z druhého čtení biopsie ve Fingerlandově ústavu patologie FN Hradec Králové je potvrzen velmi vzácný, tzv. hyperkalcemický typ malobuněčného karcinomu děložního hrdla, se ztrátou exprese SMARCA4.

Při explorativní laparotomii v červenci 2018 se daří pouze odstranění zvětšených uzlin s histologicky identickým nálezem hyperkalcemického malobuněčného nádoru, radikální výkon R0 se zachováním močového měchýře není možný a nemocná je cestou multidisciplinárního týmu ihned indikována k chemoterapii. Při absenci jakýchkoliv léčebných doporučení je v 1. linii zvolen režim cisplatina/etoposid à 4 týdny se zajištěním faktor stimulující granulocytární kolonie (G-CSF) na 5 dní, s pravidelnou MRI pánve před každým dalším cyklem pro sledování léčebné odpovědi. První dva cykly zvládla pacientka zcela bez obtíží, avšak na kontrolní MRI před třetím cyklem v září 2018 byla zjištěna progrese nálezu na děloze o velikosti 98 × 80 × 75 mm s infiltrací obou parametrií, stěny močového měchýře a přední stěny rekta. Na výpočetní tomografii (computed tomography – CT) trupu vzdálená diseminace nebyla jednoznačně prokázána, až na sporné drobnoložiskové postižení obou plic do 5 mm. Pro chemorezistenci onemocnění byla pacientka konzultována onkogynekologickým týmem VFN v Praze, kde byla potvrzena definitivní inoperabilita nálezu.
Vzhledem ke gynekologickému krvácení a zhoršení mikčních potíží pacientka podstoupila paliativní radioterapii pánve a retroperitonea do 45 Gy ve 25 frakcích, s následným boostem na oblast tumoru do celkové dávky 59,4 Gy ve 33 frakcích. Radioterapii pacientka zvládla bez komplikací s klinickou úlevou od potíží. Graficky byla potvrzena parciální regrese nálezu na děloze i uzlinách (obr. 2).
Současně s radioterapií byly vzorek nádoru i krev pacientky odeslány k vyšetření somatického i germinálního exomu metodou celoexomového sekvenování (whole exome sequencing – WES) cestou výzkumného centra CEITEC Masarykovy univerzity v Brně. Bohužel pro pacientku nález vyšetření somatického exomu neodhalil žádnou známou mutaci vhodnou k efektivní cílené terapii. Současně byla prokázána nízká nádorová mutační nálož, což neumožnilo pacientku zařadit do specifického léčebného programu s anti-PD1 terapií nivolumabem (Bristol-Myers Squibb, NY, USA). Žádost o schválení nivolumabu nad rámec pojištění byla pojišťovnou zamítnuta pro nedostatek důkazů o efektivitě léčby v této indikaci. Došetření germinálního exomu odhalilo patogenní variantu genů PALB2 a BRCA2 s doporučením k podrobnému vyšetření cestou lékařské genetiky.
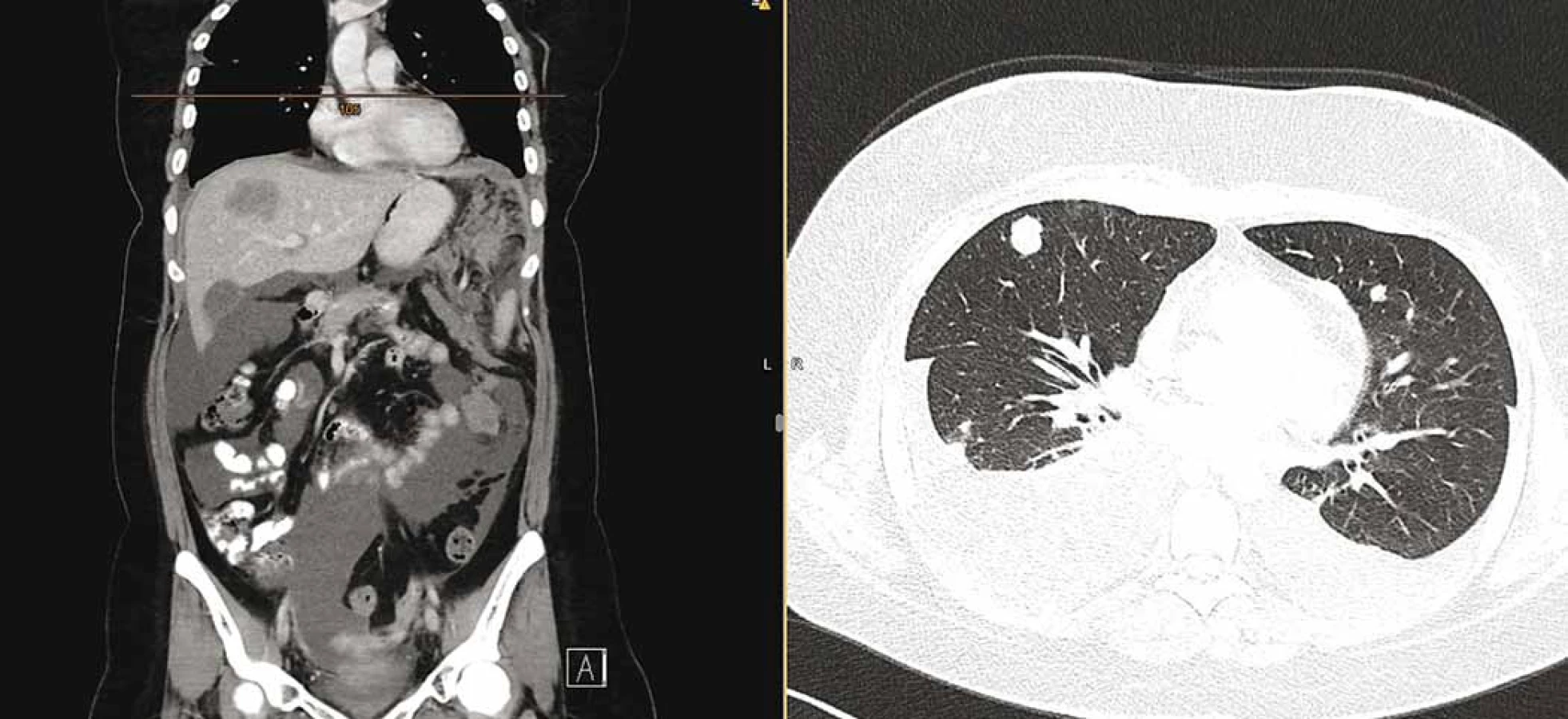
V lednu 2019 byla pacientka přijata pro celkové zhoršení stavu, dušnost, teploty, nechutenství a bolesti. Doplněné CT hlavy vyloučilo organické postižení. Na CT trupu byla sice potvrzena parciální regrese nádoru v pánvi, došlo ale k masivní systémové progresi onemocnění se zvětšenými uzlinami na krku a v mediastinu, mnohočetným ložiskovým postižením plic a pleury s oboustranným fluidothoraxem, vícečetným ložiskovým postižením jater (největší 52 × 36 mm), postižením omenta s měkkotkáňovými masami (největší 72 × 56 mm) a ascitem (obr. 3). Pacientka byla indikována k pokusu o 2. linii paliativní chemoterapie, které se již bohužel nedočkala, a umírá na rychlou progresi základního onemocnění.
Histopatologie
Malobuněčný karcinom hyperkalcemického typu je velmi vzácný a velmi agresivní typ maligního nádoru, který v rámci orgánů ženského pohlavního traktu postihuje nejčastěji ovarium. Vyskytuje se u mladých žen (průměrný věk 23 let) a u přibližně dvou třetin nemocných lze prokázat paraneoplastickou hyperkalcemii. Je důležité zmínit, že tento nádor nemá žádný vztah k malobuněčným karcinomům neuroendokrinního typu vyskytujícím se v jiných orgánech, např. v plicích.
Makroskopicky bývá nádor většinou objemný, na řezu solidní struktury, šedobílé barvy, často s ložisky nekróz a krvácením. Mikroskopicky nádor sestává ze solidně či méně často folikulárně rostoucích atypických buněk s hyperchromními jádry. Variabilní část nádorové populace bývá navíc tvořena objemnými buňkami s vezikulárními jádry s jadérky a s eozinofilní cytoplazmou, někdy až rhabdoidního vzhledu. Pokud tyto buňky převažují, hovoří někteří autoři o tzv. velkobuněčné variantě. Typická je vysoká mitotická a proliferační aktivita. Imunohistochemický profil nádoru je relativně nespecifický. Je popisována variabilní exprese cytokeratinů (CK), epiteliálního membránového antigenu, CD10, WT-1 a calretininu. Recentně byla jako důležitý diagnostický znak identifikována ztráta jaderné exprese markeru SMARCA4 (též BRG-1), kterou lze zjistit imunohistochemicky. Diferenciálně diagnosticky je z mikroskopického hlediska nutné pomocí imunohistochemie odlišit zejména juvenilní typ nádoru z buněk granulózy, který má odlišnou léčbu a prognózu.
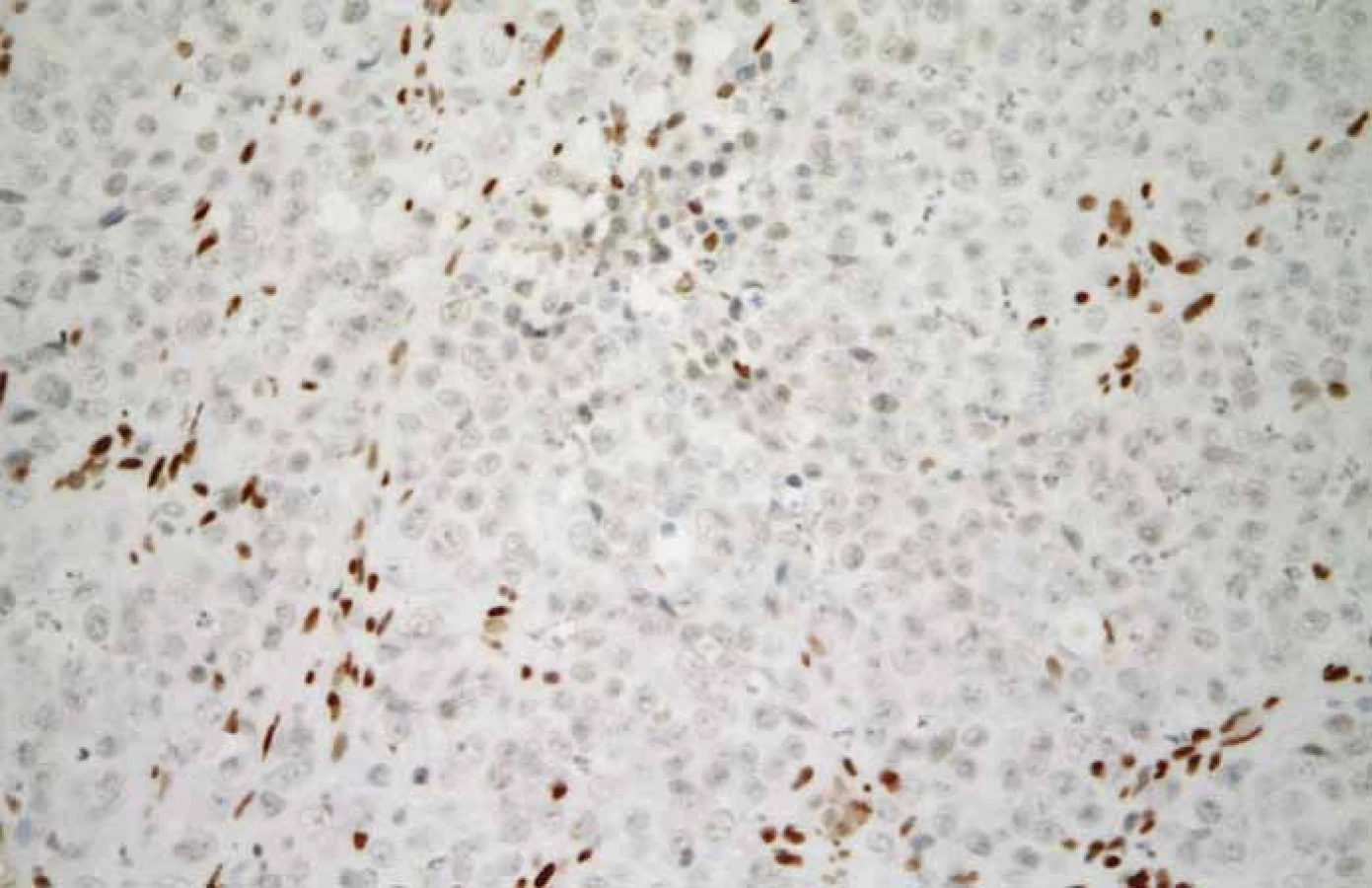
V prezentovaném případu byla diagnóza malobuněčného karcinomu hyperkalcemického typu stanovena na základě mikroskopického vzhledu nádoru v základním barvení hematoxylinem a eozinem (obr. 4) a na základě výsledku imunohistochemického vyšetření, při kterém byla zjištěna variabilní pozitivita CK, CK18, vimentinu, GATA3 a p16. Exprese SMARCB1 (též INI-1) byla zachována. Diagnosticky nejpřínosnější však byl průkaz ztráty exprese SMARCA4 (obr. 5).

Genetické vyšetření
Ve snaze odhalit mutaci pro případnou cílenou terapii byl odeslán vzorek nádoru a krve k WES. Exom je část genomu tvořená exony neboli sekvencemi, které vytvářejí mediátorovou RNA (mRNA) po vystřižení intronů [3]. Exom lidského genomu je tvořen asi 180 000 exony, což je asi 1 % celkového genomu. I když exom tvoří velice malou část celkového genomu, mutace v něm jsou dle současných odhadů zodpovědné až za 85 % nemocí podmíněných mutacemi. Sekvenování exomu je metoda pro analýzu genomu, která využívá sekvenační technologii pro selektivní detekci exomu v kódujících oblastech lidského genomu [4]. Následuje obohacení cílových fragmentů a vysokokapacitní sekvenování. Prostřednictvím této metody lze získat genetické informace pouze z exomových oblastí, což výrazně zvyšuje účinnost výzkumu protein kódujících oblastí lidského genomu a značně snižuje finanční náklady.
Celoexomové sekvenování bylo provedeno za použití soupravy TruSeq DNA Exome Kit, sekvenační kazety NextSeq 500/550 Mid Output Kit a sekvenátoru NextSeq 500 (vše Illumina, CA, USA). Vstupním materiálem pro vyšetření zárodečného exomu bylo 400 ng DNA izolované z leukocytů periferní krve a pro vyšetření nádorového exomu bylo použito 400 ng DNA izolované z parafínového bloku s vysokým obsahem nádorových buněk. Sekvenování bylo provedeno s vysokým pokrytím, kdy více než 85 % vychytávaných oblastí bylo pokryto alespoň 20krát. Vyšetření nádorového exomu odhalilo variantu ve FGFR3 c.586C>T/p.R196C (NM_000142, exon 5), která se v tumoru vyskytovala s relativně nízkou frekvencí (7 %). Varianta je popsána pouze v databázích dbSNP a COSMIC, kde byla identifikována u pacienta s maligním melanomem. Bližší popis varianty či její efekt na funkci proteinu FGFR3 není znám a je možné jej pouze predikovat za pomoci příslušných programů. Mutační nálož (počet nesynonymních mutací na 1 Mb DNA) tumoru byla nízká – 4 mutace/Mb. Vyšetřením zárodečného exomu byla u pacientky nalezena mutace v BRCA2 c.8350C>T/p.R2784W (NM_000059, exon 19) a v PALB2 c.509_510del/p.R170fs (NM_024675, exon 4). Obě tyto mutace jsou v databázích popisovány jako patogenní (PALB2) nebo pravděpodobně patogenní (BRCA2), způsobující nádory prsu a/nebo ovarií.
BRCA2
Jedná se o tumor supresorový gen, spolu s BRCA1 se produkty těchto genů účastní kontroly buněčného cyklu a oprav poškozené DNA [5]. BRCA2 je lokalizován na chromozomu 13q. Produktem je polypeptid pBRCA2. Na poškození DNA zareaguje produkt genu AT. Proteinkináza ATM spustí fosforylační kaskádu, během níž se fosforyluje protein pBRCA1. Ten následně interaguje s proteinem pRAD51, který se účastní oprav dvouřetězcových zlomů DNA procesem homologní rekombinace. Protein pBRCA2 také interaguje s tímto komplexem. Jeho úlohou je transportovat pRAD51 do místa poškození. BRCA mutace jsou typickým podkladem pro hereditární nádorové syndromy, např. HBC (hereditary breast cancer syndrome) a HBOC (hereditary breast/ovarian cancer syndrome). Riziko vzniku nádorů je u nositelek mutací celoživotně velmi vysoké, pro BRCA2 je celoživotní kumulativní riziko 45–85 % pro nádory prsu a 17–37 % pro nádory ovarií. Nosiči BRCA2 mutace mají mírně až středně zvýšené riziko vzniku nádorů prostaty, tlustého střeva, žaludku, pankreatu, žlučníku a žlučových cest, maligního melanomu.
PALB2
Produktem mutace je vadný protein PALB2, který je součástí rozsáhlého endogenního multiproteinového komplexu, který se spolu s proteiny BRCA2 a RAD51C podílí na reparačních pochodech DNA [6]. Poškození této dráhy pak vede k akumulaci alterací DNA. PALB2 mutace je asociována s vyšším rizikem nádorů, u žen zejména prsu, ale také pankreatu. U bialelické mutace vzniká autozomálně recesivní onemocnění Fanconiho anemie typ N s výskytem vrozených vad a s vysokým rizikem solidních nádorů i hematologických malignit.
Vzhledem k vysoké pravděpodobnosti familiárního původu mutací bylo pacientce i rodině pacientky doporučeno genetické vyšetření. U matky pacientky (rok narození 1971) byla prokázána mutace c.8350C>T v BRCA2 genu v heterozygotním stavu. U otce pacientky (rok narození 1968) byla prokázána mutace c.509_510delGA v PALB2 genu v heterozygotním stavu. U bratra nemocné (rok narození 1996) byla prokázána familiární heterozygotní mutace PALB2 zděděná po otci. Pacientka zdědila heterozygotní mutaci v BRCA2 genu od matky a současně heterozygotní mutaci v PALB2 genu od otce. Tato velmi málo pravděpodobná mutační nálož s familiárním výskytem nejspíše svým charakterem zapříčinila extrémně vzácný typ malignity v tak nízkém věku.
Diskuze
U pacientů s hereditárními nádorovými syndromy máme v dnešní době zavedený systém preventivních opatření [7]. Rozeberme si detailněji prevenci u hereditárních syndromů nádorů prsu a ovarií, které se nejvíce pojí s BRCA či PALB2 mutacemi. Primární prevence spočívá v preventivních chirurgických zákrocích, např. preventivní mastektomii a preventivní adnexektomii, které riziko onemocnění snižují na 1–5 %. Dále spoléháme na sekundární prevenci, kam řadíme např. samovyšetřování prsou každý měsíc po skončení menstruace od 20 let, kontroly prsou lékařem každoročně od 20 let, kontroly prsou ultrazvukem ročně od 20 let, od 25 let po půl roce, MRI prsů po roce od 25 let, od 35 let kontroly prsou mamograficky každoročně, gynekologické kontroly vč. vaginálního ultrazvuku a markeru CA125 od 20 do 30 let každoročně, později každého půl roku, mezi další vhodná vyšetření patří vyšetření okultního krvácení ve stolici od 40 let každoročně, koloskopie, gastroskopie od 45 let po 3 letech, ultrazvuk břišních orgánů, kožní kontroly, kontroly prsou u mužů a kontroly prostaty (od 45 let) každoročně, vhodné je i doplnění nádorových markerů CA125, CEA, CA15-3 kaž-doročně. Při pohledu na věkové hranice jednotlivých opatření je zřejmé, že cílová skupina je sledována nejdříve od 20 let. Z toho plyne, že i kdyby se v rodině naší pacientky předpokládal hereditární nádorový syndrom, stejně by kvůli nízkému věku nestihla genetické testování a preventivní vyšetření. Avšak díky pohotovému genetickému vyšetření může být její nejbližší rodina zařazena do preventivního programu a dále sledována.
Dalším bodem k zamyšlení je fakt, proč nebyl při WES zjištěn hypermutovaný nádorový proces, když v reparaci DNA hrají důležitou roli mutace BRCA2 i PALB2, při kterých by se dal předpokládat výskyt dalších chyb v genetickém zápisu. Což otevírá i druhou otázku, proč nádor vycházející z hereditární chyby v reparačních genech neodpovídal na cytotoxickou léčbu, která by z těchto chyb naopak měla profitovat. Na tuto klíčovou otázku bohužel odpověď neznáme a jsme ve shodě s dalšími pracemi, které tento typ nádoru také považují za velmi málo chemo-i radiosenzitivní.
Pro tento typ nádoru je typická mutace genu SMARCA4, kdy gen SMARCA4 je nádorový supresorový gen, který kóduje protein, jenž se podílí na remodelaci chromatinu [8]. Somatická nebo zárodečná mutace bývá zjištěna u 91,5 % těchto tumorů. Nosičky této mutace mají také zvýšené riziko rhabdoidních nádorů, což podporuje i fakt, že malobuněčný karcinom vaječníků hyperkalcemického typu je rhabdoidním tumorům morfologicky velmi blízký. V našem případě nebyla prokázána zárodečná mutace SMARCA4, ztráta exprese příslušného genu byla způsobena pouze somatickou mutací. Vzhledem k vysokému záchytu zárodečných mutací genu SMARCA4 je u pacientek s tímto typem nádoru doporučována genetická konzultace a testování. Ačkoli lze prediktivní testování doporučit i příbuzným, z důvodu neznámé penetrance onemocnění, zatím neexistují oficiální doporučení pro nosičky s mutací genu SMARCA4. Účinnou prevencí by jistě byla oboustranná adnexektomie, nicméně bez známé penetrance je obtížné stanovit optimální věk pro tento postup. Účinnost sledování zatím není známá.
Práce zahraničních autorů se také potýkají s myšlenkou nízké mutační nálože a možného efektu imunoterapie [9]. V 8 z 11 popisovaných případů byla vykazována exprese PD-L1 jak ve vlastním tumoru, tak v nádorovém stromatu, spojená s nádorovou infiltrací T lymfocyty. Toto zjištění může naznačovat, že se PD-L1 podílí na vzniku získané imunitní rezistence těchto tumorů. Práce naznačují, že i u tumoru s nízkou mutační náloží může díky příhodnému imunogennímu prostředí cílená blokáda PD-1/PD-L1 přinést léčebný benefit. V našem případě však byla exprese PD-1 5 %.
Klinické zkušenosti s tímto typem nádoru jsou velmi omezené, ale nacházejí průnik v několika aspektech – gynekologické malobuněčné karcinomy jsou velice vzácná, agresivní onemocnění s velmi špatnou prognózou, postihující převážně mladé ženy. Nejčastěji vycházejí z vaječníků, jichž se týká relativně nejvíce dat, ale jsou popsány i případy lokalizace na cervixu, endometriu, v pochvě i vulvě; většina dat vychází z retrospektivních studií a z dat léčby pro malobuněčný karcinom plic. Nejdůležitějším faktorem pro dlouhodobější přežití je stadium onemocnění [10].
Největší studie byla představena Youngem [11], obsahovala údaje 150 pacientů, z nichž 50 % mělo stadium choroby I, 5 % stadium II, 43 % stadium III a 1 % stadium IV; 33 % pacientů se stadiem IA stále žilo a bylo bez známek choroby i po 5 letech, ze stadií IC, II, III, IV se 5 let dožilo pouze 10 %. Pacienti, kteří podstoupili adjuvantní chemoterapii, měli lepší přežití ve stadiu IA dle Mezinárodní federace gynekologie a porodnictví (FIGO) v porovnání s těmi, kteří absolvovali pouze chirurgický výkon. Zatím chybí jasný důkaz o tom, že by adjuvantní chemoterapie ve stadiu IA mohla zlepšit celkovou prognózu.
V prvním vybraném případě byla postižena 24letá pacientka [12], s téměř 20cm tumorem vycházejícím z pravého vaječníku, bez dalších ložisek dle CT. Prvním krokem byla shodně laparotomie, při které se na rozdíl od našeho případu podařilo primární tumor resekovat bez rezidua. Pacientka podstoupila čtyři cykly adjuvantní chemoterapie etoposid/cisplatina, následované restagingovým CT, bez průkazu onemocnění. Po 5 měsících od ukončení léčby byla přijata se zácpou, anorexií a slabostí způsobenými hyperkalcemií, na CT popsány dva téměř 10cm tumory v dutině břišní, stav se komplikoval ileózním stavem, pro který byla operována, a 4. pooperační den umírá na plicní embolii cca 10 měsíců od diagnózy. V dalším případě 29letá pacientka [13] s tumorózní masou v pánvi 110 × 116 × 125 mm, s předoperačně vysokou hladinou kalcia, podstoupila R0 resekci tumoru, dále šest cyklů karboplatina/paklitaxel. Po 1,5 roce byla stále bez známek onemocnění. V třetím případě byla 35letá pacientka [14] přijata pro příznaky hyperkalcemie, se známkami jaterního selhání, s masou 135 × 180 × 120 mm s rozsevem po břišní dutině, umírá do 1 měsíce od prvních projevů.
V námi prezentovaném případě byla po celou dobu léčby hladina sérového kalcia i jeho ionizované složky v normě, což vyvolává otázku, co zde bylo jinak, když v ostatních námi zmíněných případech hyperkalcemie přítomna byla a způsobovala i příslušné komplikace. Dle dostupné literatury bývá hyperkalcemie přítomna ve dvou třetinách případů.
Popsané případy odrážejí strohá doporučení vedení léčby. Bez platných léčebných doporučení je ošetřující lékař odkázán na kazuistická sdělení, kterých je také velmi málo. Zkušeností s prezentovaným postupem – chirurgickým výkonem s R0 resekcí, adjuvantní chemoterapií doplněnou případně o zevní radioterapii – je vzhledem k raritě onemocnění velmi málo a chybějí validní klinická data.
Závěr
Jedná se o extrémně vzácný případ nádorového onemocnění, což dokumentuje i nedostatek klinických dat a popsaných kazuistik z celého světa, v našem případě spojený se vzácným nosičstvím zárodečných mutací v genech BRCA2 a PALB2, které pacientka získala od svých rodičů. Vzhledem k této zkušenosti doporučujeme při podobném nálezu pečlivě odebrat rodinou anamnézu a zvážit genetické testování. Z výše popsaného vyplývá, že i přes multimodalitní léčebné možnosti stále chybí standardní léčba tohoto typu onemocnění. V našem případě měl nádor překvapivě nízkou mutační nálož, z čehož lze předpokládat nízký efekt i benefit imunoterapie, která je dnes v mnohých bezvýchodných situacích pro pacienty jiskrou naděje.
Práce byla podpořena MZ ČR, grantem 16-33209A a výzkumným programem Univerzity Karlovy Progres Q40/06.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Libor Hruška
Klinika onkologie a radioterapie
FN Hradec Králové
Sokolská 581
500 05 Hradec Králové
e-mail: libor.hruska@fnhk.cz
Obdrženo: 10. 6. 2019
Přijato: 9. 9. 2019
Sources
1. Tomášek J, Abrahámová J, Adam Z et al. Onkologie – minimum pro praxi. Mlečice: Axonite 2015 : 44–52.
2. Genetika-biologie.cz. Šípek A. Hereditární nádorové syndromy. 2010–2014. [online]. Dostupné z: http: //www.genetika-biologie.cz/hereditarni-nadorove-syndromy.
3. Wikipedia.org. Exom. [online]. Dostupné z: https: //cs.wikipedia.org/wiki/Exom.
4. Biogen.cz. Molekulární biologie a genetika. [online]. Dostupné z: https: //biogen.cz/sekvenovani-lidskeho-exomu.
5. Wikiskripta.eu. BRCA. [online]. Dostupné z: https: //www.wikiskripta.eu/w/BRCA.
6. Wikipedia.org. PALB2. [online]. Dostupné z: https: //en.wikipedia.org/wiki/PALB2.
7. Linkos.cz. Česká onkologická společnost ČLS JEP. Hereditární syndrom nádorů prsu a/nebo ovaria. [online]. Dostupné z: https: //www.linkos.cz/lekar-a-multidisciplinarni-tym/geneticka-rizika/nadorove-syndromy/hereditarni-syndrom-nadoru-prsu-a-nebo-ovaria/.
8. Plevová P, Geržová H. Vzácné pediatrické ovariální tumory a jejich genetické příčiny. Klin Onkol 2019; 32 (Suppl 2): 2S79–2S91. doi: 10.14735/amko2019S79.
9. Jelinic P, Ricca J, van Oudenhove E et al. Immune-active microenvironment in small cell carcinoma of the ovary, hypercalcemic type: rationale for immune checkpoint blockade. J Natl Cancer Inst 2018; 110 (7): 787–790. doi: 10.1093/jnci/djx277.
10. Cohen JG, Chan JK, Kapp DS. The management of small-cell carcinomas of the gynecologic tract. Curr Opin Oncol 2012; 24 (5): 572–579. doi: 10.1097/CCO.0b0 13e3283565ed6.
11. Young RH, Oliva E, Scully RE. Small cell carcinoma of the ovary, hypercalcemic type. A clinicopathological analysis of 150 cases. Am J Surg Pathol 1994; 18 (11): 1102–1116. doi: 10.1097/00000478-199411000-00 004.
12. Kascak P, Zamecnik M, Bystricky B. Small cell carcinomama of the ovary (hypercalcemic type): malignant rhabdoid tumor. Case Rep Oncol 2016; 9 (2): 305–311. doi: 10.1159/000446694.
13. Wang JJ, Liu Q, Wu N et al. Ovarian small-cell carcinoma hypecalcemic type successfully treated: case report and literature review. Onco Targets Ther 2016; 9 : 1409–1414. doi: 10.2147/OTT.S97170.
14. Ghazi A, Ayaz A, Hamid T et al. Small cell carcinoma of the ovary hypercalcemic type (SCCOHT): a rare case after in vitro fertilization (IVF). Pak J Med Sci 2017; 33 (1): 241–244. doi: 10.12669/pjms.331.11634.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2019 Issue 6
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Metamizole vs. Tramadol in Postoperative Analgesia
- Spasmolytic Effect of Metamizole
- Metamizole in perioperative treatment in children under 14 years – results of a questionnaire survey from practice
Most read in this issue
- Extravasation (Paravasation) of Chemotherapy Drugs – Recommendations for Standard Care in the Czech Republic based on Consolations between Representatives of the Supportive Care Group of the Czech Society for Oncology, Oncology Section of the Czech Nurses Association, and the Society for Ports and Permanent Catheters
- Rare Hereditary Burden associated with a Hypercalcemic Small-Cell Carcinoma of Cervix in a Young Female Patient
- Tranzice péče o onkologické pacienty z dětského do dospělého věku
- Bortezomib and Thalidomide Treatment Results in Newly Diagnosed Transplant-Ineligible Multiple Myeloma Patients are Comparable in Long-Term Follow-Up