
Familiárna agregácia Parkinsonovej choroby: genealogická štúdia
Familial aggregation of Parkinson’s disease: a genealogical study
Parkinson's disease (PD) is the second most common neurodegenerative disease after Alzheimer's disease. It is characterised by a high variability for the age of the onset of symptoms. The question is whether the genes predisposing to the development of PD are also responsible for the symptom onset age [1]. The onset of PD symptoms at a younger age is related to familial incidence of the disease, as the age of onset in some families with PD was lower than 50 years. In some Parkinsonism families, the onset of symptoms was reported below 50 years of age. Based on this observation, the onset of PD symptoms at a younger age is supposed to be related to familial incidence of Parkinson disease. The familial forms stand for between 10 and 15 % of all cases [2]. There are few prospective studies concerning rare incidence of large Parkinsonism families, low hereditability and large heterogeneity of PD, which endorse this hypothesis. The objective of this study is to compare the link between the early onset and familial incidence of PD, i.e. to find out whether or not genetic factors contribute to early onset of PD. 148 patients regularly monitored by the supraregional Centre for Diagnosis and Treatment of Neurodegenerative Diseases filled in a structured questionnaire. Questions concerned the sex and age, the first symptoms of the disease and the age of their onset in the monitored patients, as well as in their relatives. 100 filled questionnaires were returned for evaluation. Familiar incidence associated with low age of PD onset was confirmed in 18 cases (9 men and 9 women) out of 100 patients enrolled in the epidemiological study. Paternal heredity was discovered in 4 cases in men, and maternal heredity was discovered in 7 cases in women.
Key words:
Parkinson’s disease – familiar incidence – type of heredity – age of onset
Authors:
K. Kollárová 1; P. Kaňovský 1; A. Šantavá 2; R. Obereignerů 1; I. Nestrašil 1; P. Ressner 1
Authors‘ workplace:
Neurologická klinika LF UP a FN Olomouc
1; Ústav lékařské genetiky a fetální medicíny LF UP a FN Olomouc
2
Published in:
Cesk Slov Neurol N 2008; 71/104(2): 197-200
Category:
Short Communication
Overview
Parkinsonova choroba (PD) je po Alzheimerovej chorobe druhé najčastejšie neurodegeneratívne ochorenie. Vyznačuje sa širokou variabilitou pre vek nástupu príznakov. Je otázne, či gény podmieňujúce dispozíciu k vzniku PD sa podieľajú aj na veku nástupu príznakov [1]. Začiatok príznakov PD v mladšom veku je spojený s rodinným výskytom, pretože v niekoľkých rodinách s výskytom PD bol vek nástupu nižší ako 50 rokov. V niekoľkých parkinsonských rodinách sa začiatok príznakov popisuje v nižšom veku ako 50 rokov. Na základe tohto pozorovania sa predpokladá, že začiatok príznakov PD v mladšom veku by mohol súvisieť s rodinným výskytom Parkinsonovej choroby. Familiárne formy tvoria 10–15 % všetkých prípadov [2]. Pre raritný výskyt veľkých parkinsonských rodín, nízku heritabilitu a veľkú heterogenitu PD existuje len málo prospektívnych štúdií podporujúcich túto hypotézu. Cieľom tejto štúdie je porovnať vzťah medzi skorým nástupom a rodinným výskytom PD, a teda či genetické faktory prispievajú k skorému začiatku PD. 148 pacientov pravidelne sledovaných v nadregionálnom Centre pre diagnostiku a liečbu neurodegeneratívnych ochorení vyplnilo štruktúrovaný dotazník. Zaujímali sme sa o pohlavie, vek pacienta, prvé príznaky ochorenia a vek nástupu u sledovaných pacientov ako aj u ich príbuzných. K vyhodnoteniu bolo vrátených 100 vyplnených dotazníkov. Rodinný výskyt v súvislosti s nízkym vekom nástupu PD sa potvrdil v 18 prípadoch (9 mužov a 9 žien) zo 100 pacientov zaradených do epidemiologickej štúdie. U mužov bola v 4 prípadoch zistená paternálna heredita, u žien bola v 7 prípadoch zistená maternálna heredita.
Kľúčové slová:
Parkinsonova choroba – rodinný výskyt – typ dedičnosti – vek nástupu
Úvod
Parkinsonova choroba je chronické neurodegeneratívne ochorenie, kde hlavnou poruchou je deficit dopamínu v oblasti bazálnych ganglií. Deficit je spôsobený zníženým transportom dopamínu do oblasti striata cestou nigrostriatálnych projekcií. Jeho prvotnou príčinou je apoptotický zánik melanínových buniek v oblasti substantia nigra v mezencefale.
Okrem sporadickej formy ochorenia, ktorá je najčastejšia, je už pomerne dlho známa familiárna forma. Prvé sporadické zmienky o rodinnom výskyte Parkinsonovej choroby sa datujú už z roku 1880 [3], hoci prvou genetickou analýzou potvrdený dedičný výskyt ochorenia bol publikovaný až v roku 1997 [4]. Od tej doby sa genetická analýza v rodinách s výskytom Parkinsonovej choroby stala „hot topic“ neurogenetiky. Do súčasnej doby sa popísalo niekoľko lokusov, ktoré sú zodpovedné za vznik familiárnej formy. Do súčasnej doby bolo popísaných už niekoľko génov, ktoré sa spolupodieľajú na vzniku familiárnej formy (tab. 1) [13]. Ide o veľmi rozdielne formy dedičnosti, ktoré boli popísané jednak v multietnických rodokmeňových štúdiách rozsiahlych pedigrees [4], ako aj v malých, geneticky i geograficky izolovaných komunitách [6]. Popisuje sa prenos jednak autozomálne dominantný, recesívny, ďalej i väzba na X chromozóm, event. prenos výlučne maternálny [7], paternálny či zmiešaný [8].
![Lokusy a gény s väzbou na familiárny výskyt PD [13].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/19bf9a4af56a3613f3c92f9f008a486e.png)
Z vyššie uvedených faktov vyplýva, že súčasný počet génových mutácií, ktoré kódujú vznik familiárnej formy ochorenia, nie je konečný. Existuje pravdepodobne celá rada ďalších mutácií, ktoré zatiaľ neboli objavené. Genetická analýza je však u pacientov s familiárnym výskytom Parkinsonovej choroby veľmi zložitá a k jej realizácii v praxi sú potrebné solídne pracovné dáta. Tento základ tvoria v klinickej genetike genealogické štúdie, ktoré slúžia ako podklad k ďaľšiemu, už cielenému genetickému testovaniu. Prezentovaná štúdia vznikla v snahe zmapovať familiárny výskyt a jeho bližšie charakteristiky v populácii pacientov postihnutých Parkinsonovou chorobou, ktorí sú sledovaní v terciárnom špecializovanom diagnostickom a liečebnom centre.
Súbor a metodika
V rámci genealogickej štúdie sme oslovili 148 pacientov. Parkinsonova choroba bola u všetkých stanovená podľa diagnostických kritérií United Kingdom Parkinson's Disease Brain Bank (UK-PDBB) [9]. Pacienti podstúpili podrobné neurologické vyšetrenie, u všetkých sa realizovalo MR vyšetřenie mozgu, miechy a komplexné vyšetrenie biochemické, imunologické i likvorologické, vrátane stanovenia jednak prítomnosti a množstva všeobecných markerov zápalu, ako aj špeciálnych markerov neurodegenerácie. Pomocou neuropsychologického vyšetrenia sme zisťovali mieru deteriorácie intelektových, exekutívnych a mnestických funkcií. U všetkých pacientov bola v rámci klinického vyšetrenia hodnotená aktuálna hodnota UPDRS skóre (Unified Parkinson's Disease Rating Scale) vo fáze „ON“, ako aj hodnota skóre podľa Hoehn-Yahrovej stupnice.
Genealogická štúdia prebiehala už overenou a publikovanou dotazníkovou metódou [10]. Pacienti boli oslovení počas kontrolnej návštevy a obdržali štruktúrovaný genetický dotazník. Požiadali sme ich, aby zmienený dotazník spolu s rodinnými príslušníkmi vyplnili a zaslali späť do určitého termínu na adresu ošetrujúceho lekára. Po uplynutí tohto termínu (+ 4 týždne) boli dotazníky spracované zaslepenými hodnotiteľmi a jednotlivé dáta boli zaradené do databázy.
Výsledky
Návratnosť dotazníkov bola 70 %, tzn. že do štúdie mohlo byť takto zaradených 100 pacientov s priemerným vekom 61,7 (SD = 10,59) rokov, pričom z celkového počtu bolo 50 mužov s priemerným vekom 59,0 (SD = 10,54) a 50 žien vo veku 64,3 (SD = 10,53) rokov. Rodinný výskyt PD bol zistený v 18 prípadoch z opýtaných (18 %), tzn. u 9 mužov s priemerným vekom nástupu príznakov 51,7 (SD = 10,83) a 9 žien s priemerným vekom nástupu príznakov 53,6 rokov (SD = 14,0). V 4 prípadoch bolo zachytené jednogeneračné postihnutie, títo pacienti neboli objektom parametrickej štatistiky. Do výpočtu boli zaradení pacienti (tab. 2), u ktorých je známy medzigeneračný výskyt PD (n = 14).
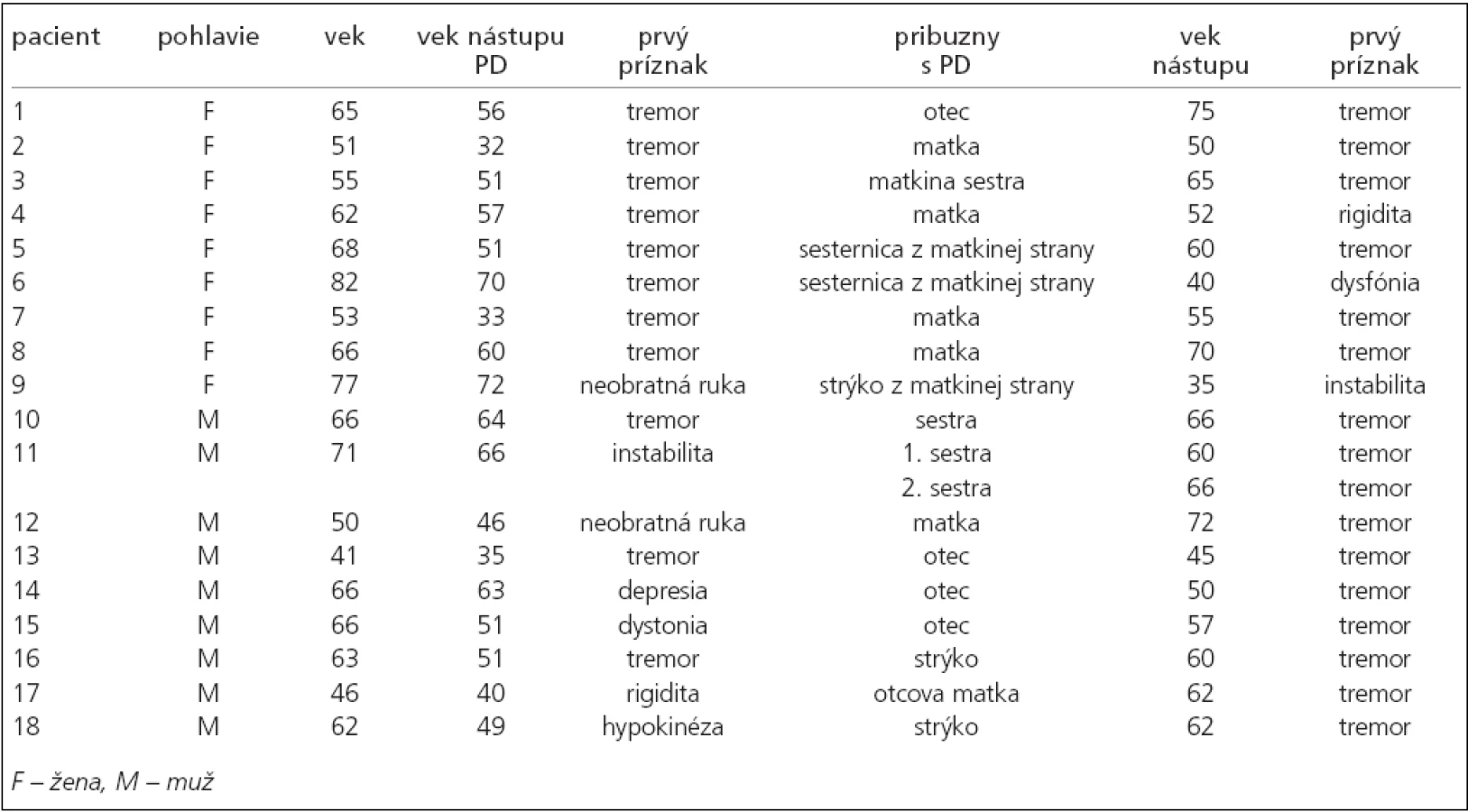
Použitím biseriálneho korelačného koeficientu sme zistili štatisticky signifikantnú vysokú koreláciu (hodnota rbis = 0,658) medzi vekom nástupu PD a jej skorším výskytom v nasledujúcej generácii, a to na hladine významnosti α = 0,01. Podrobné výsledky sú obsiahnuté v tab. 2, z ktorej vyplýva, že u 9 mužov bola zaznamenaná v nadpolovičnej väčšine prípadov paternálna a u 9 žien taktiež vo väčšine prípadov maternálna heredita.
Diskusia
Väčšina foriem PD sa nededí podľa Mendelových pravidiel. V niektorých rodinách sa nedokázala väzba na žiadny z príčinných génov. Aký je teda klinický význam doteraz identifikovaných génov? Na prvý pohľad sa zdá byť značne obmedzený. Iba v relatívne malom počte rodín sa doteraz potvrdili mutácie týchto génov a na druhej strane je ich odhalenie málo pravdepodobné u miliónov jedincov so sporadickou PD. Ide teda skôr o objav vrodených génových polymorfizmov zvyšujúcich riziko rozvoja PD. Avšak znalosť týchto génov je mimoriadne dôležitá pre ozrejmenie molekulárneho podkladu neurodegeneratívnych zmien. Ďalšia fáza liečby bude bez pochýb založená na výskume, ktorý by bez identifikácie vzácnej mutácie α-synukleínu nebolo možné uskutočniť.
V porovnaní s inými neurodegeneratívnymi ochoreniami sa zdá byt uplatnenie genetického poradenstva v praxi u PD o niečo zložitejšie. Dnes sa vo svete v rámci komplexnej diagnostiky využívajú testy k stanoveniu niekoľkých PD „podskupín“. Ide o identifikáciu génov parkin 2, PINK1, DJ1 a autozomálne dominantne „prenosný“ LRRK2. Avšak pozitívny výsledok vyšetrenia nemusí byť nutne spojený so zvýšeným rizikom, ako aj nález negatívneho výsledku možné riziko jednoznačne nevylučuje. Klinická interpretácia ostáva stále nejednoznačná. Kým sa neobjasní presná frekvencia a penetrancia mutácie pre daný gén, presymptomatická/prenatálna diagnostika sa javí byť zatiaľ predčasná [11].
Naša genealogická dotazníková štúdia preukázala celkom jednoznačnú súvislosť medzi pozitívnou rodinnou anamnézou a výskytom PD v mladšom veku. V našej skupine pacientov možno zvažovať o uplatnení známeho fenoménu anticipácie [12], keďže potomkovia boli vo veľkej väčšine prípadov mladší ako ich starší príbuzní trpiaci na PD. Toto zistenie potvrdzuje hypotézu, že pozitívna rodinná anamnéza je významným rizikovým faktorom pre možný včasný rozvoj Parkinsonovej choroby, pričom relatívne riziko tu podľa niektorých prác značne varíruje medzi 2,6 až 14,6. Existuje tu tzv. veková stratifikácia, tzn. u rodinných príslušníkov PD pacientov s vekom nástupu nižším ako 67 rokov je relatívne riziko vyššie, kým u príbuzných pacientov s vyšším vekom nástupu riziko zvýšené nie je [5].
V našej skupine pacientov je zjavné, že v prípade mužov sa jedná vo väčšine prípadov o hereditu paternálnu, kým u žien o hereditu maternálnu. Tento fakt zatiaľ nebol reflektovaný v žiadnej z veľkých publikovaných štúdií a zasluhuje bližšie skúmanie.
Je však otázne, či sa gény podmieňujúce dispozíciu k vzniku PD rovnako podieľajú aj na veku začiatku ochorenia. Je možné si predstaviť, že mutácia takéhoto génu s vysokou penetranciou by dramaticky menila aj intenzitu neurodegeneratívneho procesu, a tým by pravdepodobne ovplyvnila aj vek nástupu príznakov [11]. K potvrdeniu tejto domnienky je nutné ďalšie sledovanie pacientov a rodín s výskytom Parkinsonovej choroby v dlhšom časovom horizonte. U všetkých pacientov bola odobraná krv a izolovaná DNA, vzorky boli uschované k neskoršej analýze, možno s odstupom až niekoľkých rokov.
Závěr
V rámci získavania dostatočne reprezentatívneho súboru pacientov, oslovujem týmto kolegov z ďaľších špecializovaných centier v Českej republike aj na Slovensku, ktoré sa venujú diagnostike a liečbe pacientov s Parkinsonovou chorobou. V prípade záujmu o bližšiu spoluprácu zašleme ďaľšie informácie s príslušnými materiálmi, týkajúcimi sa danej tematiky.
MUDr. Katarína Kollárová
Neurologická klinika LF UP a FN Olomouc
I. P. Pavlova 6
775 20 0lomouc
kollarova.kata@seznam.cz
Přijato k recenzi: 3. 5. 2007
Přijato do tisku: 6. 11. 2007
Sources
1. Eriksen JL, Wszolek Z, Petrucelli L. Molecular pathogenesis of Parkinson disease. Arch Neurol 2005; 62 : 353–357.
2. Nussbaum RL, Polymeropoulos MH. Genetics of Parkinson's disease. Hum Mol Genet 1997; 6 : 1687–1691.
3. Leroux PD. Contribution à l'étude des causes de la paralysie agitante. Paris: 1880.
4. Polymerupoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia, Dutra A et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 1997; 276 : 2045–2047.
5. Gosal D, Ross OA, Toft M. Parkinson's disease: the genetics of a heterogeneous disorder. Eur J Neurol 2006; 13 : 616–627.
6. Kis B, Schrag A, Ben-Shlomo Y, Klein C, Gasperi A, Spoegler F et al. Novel three-stage ascertainment method: prevalence of PD and parkinsonism in South Tyrol. Neurology 2002; 58 : 1820–1825.
7. Wooten GF, Currie LJ, Bennett JP, Harrison MB, Trugman JM Parker WD jr. Maternal inheritance in Parkinson's disease. Ann Neurol 1997; 41 : 265–268.
8. Pankratz N, Nichols WC, Uniacke SK, Halter C, Rudolph A, Shults C et al. Significant linkage of Parkinson's disease to chromosome 2q36-37. Am J Hum Genet 2003; 72 : 1053–1057.
9. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease. A clinical pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1993; 56 : 938–939.
10. Veselá O, Růžička E, Jech R et al. Esenciální tremor v naší populaci nemocných – základní charakteristika onemocnění a vyšetření kresbou spirály. Cesk Slov Neurol N 2002; 65/98 : 180–186.
11. Pankratz N, Foroud T. Genetics of Parkinson's disease. NeuroRx. 2004; 1 : 235–242.
12. Muñoz E, Pastor P, José Martí M, Vallderiola F, Oliva R, Tolosa E. Sporadic and familial Parkinson's disease. Med Clin (Barc) 2001; 116 : 601–604.
13. Hague SM, Klaffke S, Bandmann O. Neurodegenerative disorders: Parkinson's and Huntington's disease. J Neurol Neurosurg Psychiatry 2005; 76 : 1058–1063.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2008 Issue 2
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Memantine Eases Daily Life for Patients and Caregivers
Most read in this issue
- Roztroušená skleróza
- Hemangioblastom a jeho léčba pomocí Leksellova gama nože
- Syndrom Smithové-Magenisové: kazuistika
- Výsledky léčby gliomů nízkého stupně malignity u dětí (retrospektivní analýza dat)