
Lewisův- Sumnerův syndrom – kazuistika
Lewis - Sumner Syndrome – a Case Report
Lewis-Sumner syndrome, also known as Multifocal Acquired Demyelinating Sensory And Motor neuropathy (MADSAM), is considered to be a focal asymmetric variant of the chronic inflammatory demyelinating polyneuropathy. The aim of this case report is to discuss this rare disease, its differential diagnosis and available treatment modalities.
Case report:
50 years old man experienced subacute onset of impaired sensation and weakness in his left hand. EMG examination revealed focal chronic sensory and motor lesion of median, ulnar and radial nerves of the left hand (primary demyelination with partial motor block), with signs of chronic axonopathy, located between axilla and the Erb’s point. MRI of the left brachial plexus showed spinal root enlargement. Sural nerve biopsy showed signs of demyelination. CSF was normal, except for a marked elevation of protein. Serum anti-GM1 antibody titer was below cut-off. Differential diagnosis included MMN (Multifocal Motor Neuropathy), hereditary neuropathy with liability to pressure palsy and neurofibroma of brachial plexus. The patient did not respond to corticosteroid treatment. Treatment with intravenous immunoglobulins led to clinical and electrophysiological improvement.
Key words:
Lewis-Sumner syndrome – multifocal motor neuropathy – chronic inflammatory demyelinating neuropathy
Authors:
I. Okáčová 1; B. Mičánkováihash2 1,2 1,2
Authors‘ workplace:
Neurologická klinika LF MU a FN Brno
1; CEITEC – Středoevropský technologický institut, MU, Brno
2; Ústav patologie a patologické anatomie LF MU a FN Brno
3; Radiologická klinika LF MU a FN Brno
4
Published in:
Cesk Slov Neurol N 2012; 75/108(4): 498-502
Category:
Case Report
Overview
Lewisův - Sumnerův syndrom neboli „Multifocal Acquired Demyelinating Sensory And Motor neuropathy“ (MADSAM) je považován za asymetrickou variantu chronické zánětlivé demyelinizační polyneuropatie (CIDP). Cílem prezentované kazuistiky je upozornit na toto vzácné onemocnění, poukázat na diferenciální diagnostiku a možnost terapie.
Kazuistika:
Padesátiletý muž byl vyšetřován pro poruchu citlivosti a slabost levé horní končetiny s rozvojem od 45 let věku a s postupnou progresí. EMG prokazovalo fokální chronickou senzitivně‑motorickou lézi n. medianus, n. ulnaris a n. radialis vlevo, a to primárně demyelinizační (přítomen parciální blok vedení motorických vláken) s podílem chronické axonopatie, lokalizace postižení byla mezi axillou a Erbovým bodem. Byla provedena magnetická rezonance s nálezem rozšíření struktur brachiálního plexu vlevo, postkontrastně docházelo k lehce zvýšenému nehomogennímu vysycení. Nález neměl charakter ložiskové expanze, svědčil spíše pro zánětlivé změny. Lumbální punkce prokázala lehkou hyperproteinorachii, protilátky v séru proti gangliosidům byly negativní. Nález biopsie z n. suralis odpovídal demyelinizaci v části velkých myelinizovaných vláken. Diferenciálně diagnosticky byla zvažována zejména multifokální motorická neuropatie (MMN), hereditární neuropatie s tendencí k tlakovým parézám, neurofibrom brachiálního plexu. Intravenózní podání kortikoidů bylo bez efektu, po aplikaci intravenózního imunoglobulinu (IVIG) nastalo zlepšení, a to jak klinické, tak elektrofyziologické. Terapeuticky je v plánu opakované podávání IVIG.
Klíčová slova:
Lewisův-Sumnerův syndrom – multifokální motorická neuropatie – chronická zánětlivá demyelinizační polyneuropatie
Úvod
Lewisův-Sumnerův syndrom neboli „Multifocal Acquired Demyelinating Sensory And Motor neuropathy“ (MADSAM) je autoimunitní onemocnění periferních nervů. Patří mezi chronické získané demyelinizační polyneuropatie (Chronic Acquired Demyelinating Polyneuropathy, CADP), a to asymetrickou multifokální formu se senzitivním postižením.
Onemocnění vešlo ve známost poprvé v roce 1982, kdy Lewis et al popsali mezi 40 pacienty s chronickou zánětlivou demyelinizační polyneuropatií (CIDP) pět pacientů s chronickou asymetrickou senzitivně‑motorickou neuropatií postihující především horní končetiny s multifokálním postižením periferních nervů [1]. Lewisův - Sumnerův syndrom (LSS) je považován za asymetrickou variantu CIDP [2]. Onemocnění se vyskytuje 5krát méně často než CIDP [2]. Prevalence CIDP je udávána 2 až 7/ 100 000 obyvatel. Stejně jako u CIDP je výskyt častější u mužů (sex ratio M/ F 2,83) [3 – 6] a průměr - ný věk v začátku onemocnění činí 40 – 50 let [3 – 6].
V objektivním nálezu nejčastěji dominuje senzitivně‑motorické postižení distální části horní končetiny. Iniciálními projevy onemocnění mohou být čistě senzitivní příznaky, jako parestezie nebo hypestezie [1,6]. Zhruba u poloviny pacientů je onemocnění spojeno s distální svalovou atrofií [1]. Bolest je málo častý příznak onemocnění, vyskytuje se zhruba u 22 % pacientů [5].
U pacientů s LSS bývá také popisováno postižení kraniálních nervů (n. facialis, n. oculomotorius, n. trigeminus), které se vyskytuje zhruba v 26 % případů [5,6].
V lumbální punkci nacházíme lehce zvýšenou celkovou bílkovinu, a to do 0,7 g/ l [3,6]. Protilátky proti gangliosidům (anti-GM1) bývají negativní. V elektromyografickém nálezu jsou charakteristické četné motorické kondukční bloky spolu s postižením senzitivního neurogramu. Nervová biopsie n. suralis ukazuje stejně jako u CIDP známky primární demyelinizace. Na magnetické rezonanci bývá u pacientů s Lewisovým - Sumnerovým syndromem popisována hypertrofie periferních nervů [7].
Průběh onemocnění je nejčastěji chronicky progresivní [4], méně bývá popisován relaps remitentní průběh [1]. Asi u poloviny pacientů dochází k progresi s multifokálními známkami postižení, u druhé poloviny pacientů zůstává onemocnění lokalizováno v místě prvních příznaků [1]. Asi 2/ 3 pacientů reagují na terapii kortikoidy a 50 – 70 % profituje z terapie IVIG [8]. Terapeutický efekt kortikoidů nebo IVIG je zhruba stejný jako u CIDP [2]. V literatuře je popsán jeden případ pacienta s LSS, který nereagoval ani na terapii kortikoidy, ani na terapii IVIG. Jedinou účinnou léčbou u něj byla plazmaferéza [9].
Cílem naší práce je upozornit na toto vzácné onemocnění, poukázat na diferenciální diagnostiku a možnost terapie.
Kazuistika
Pacient byl padesátiletý dosud zdravý muž, u kterého se od 45 let rozvíjely parestezie a akrální svalová slabost levé horní končetiny. Pacient neměl žádné bolesti. V objektivním nálezu byla přítomna hypestezie v distribuci nervus ulnaris, medialis a radialis vlevo. Svalová síla byla zhodnocena pomocí MRC scale (Medical Research Council scale): musculus abductor pollicis brevis MRC scale 3, extenzory prstů MRC scale 3, flexory prstů MRC scale 3 a extenzory zápěstí MRC scale 4 (vše vlevo). Současně byla u pacienta lehká atrofie dlouhých flexorů prstů vlevo, bez fascikulací.
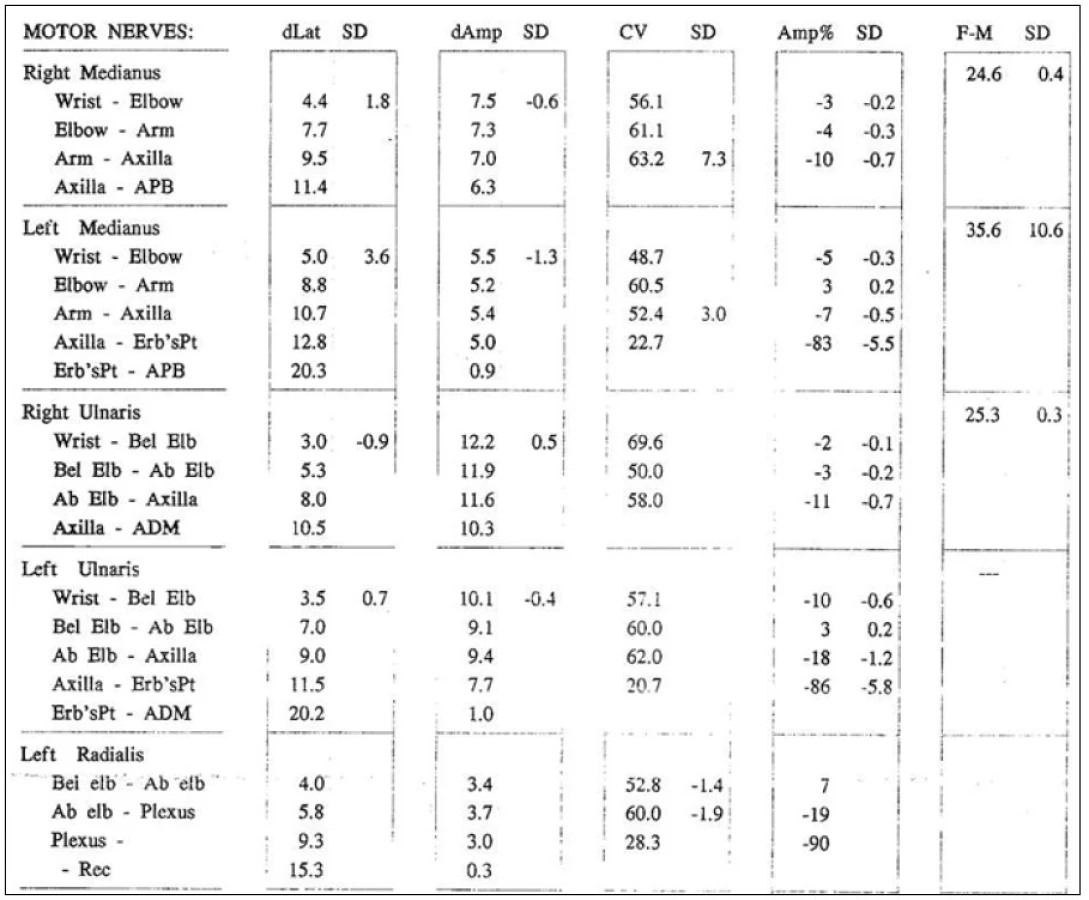
Bylo provedeno elektrofyziologické vyšetření s nálezem fokální chronické senzitivně‑motorické léze n. medianus, n. ulnaris a n. radialis vlevo, a to primárně demyelinizační (přítomen parciální blok vedení motorických vláken) s podílem chronické axonopatie, lokalizace postižení byla mezi axillou a Erbovým bodem, postižení bylo středně těžkého stupně (obr. 1). Jako vedlejší nález byl zjištěn syndrom karpálního tunelu lehkého stupně vpravo. Elektrofyziologické vyšetření na dolních končetinách bylo zcela v normě.
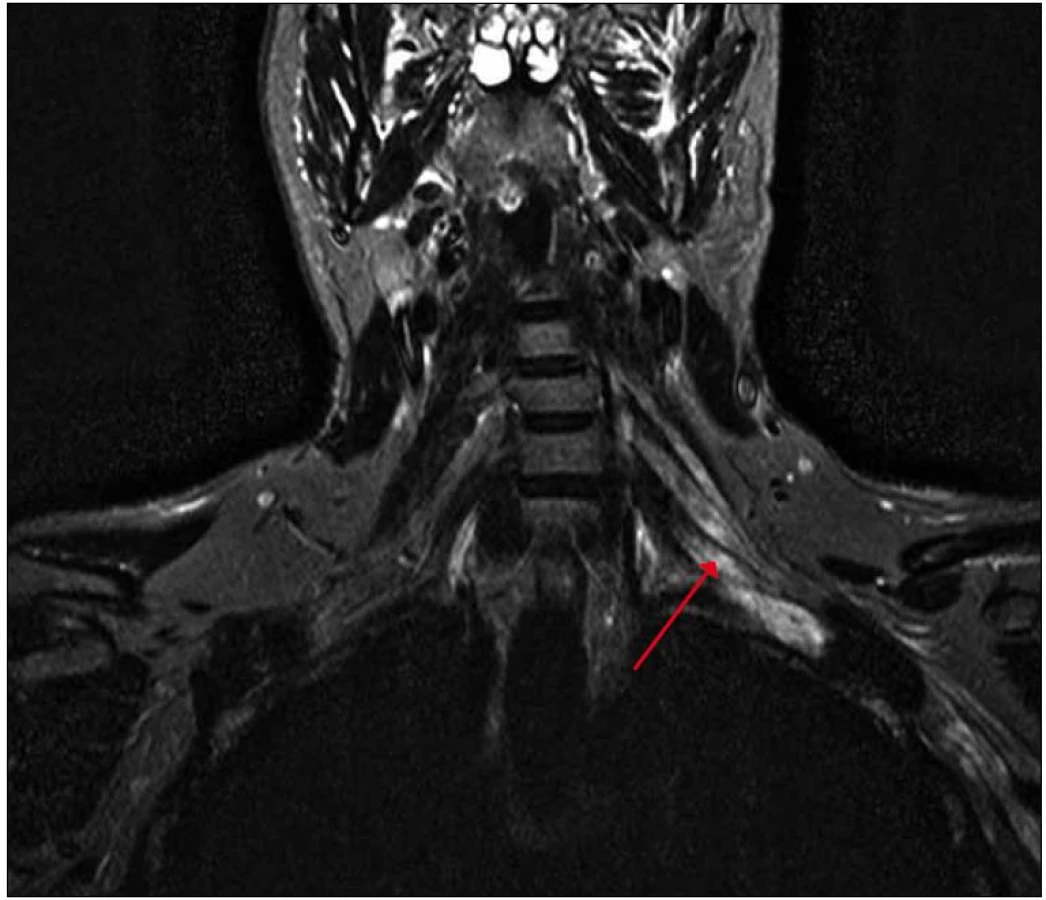
Na magnetické rezonanci krční páteře bylo popsáno rozšíření nervových pochev kořenů C6 – C8 vlevo. S odstupem tří měsíců jsme provedli magnetickou rezonanci zaměřenou cíleně na levý brachiální plexus s nálezem patologického zesílení a zvýšení signálu struktur brachiálního plexu vlevo, postkontrastně bylo prokázáno lehce zvýšené nehomogenní sycení, které svědčilo pro zánětlivé změny (obr. 2, 3).

Byla provedena biopsie nervus suralis, v histologickém nálezu byly známky demyelinizace především velkých myelinizovaných vláken, přičemž v části velkých myelinizovaných vláken byly pochvy zcela rozpadlé, jinde regresivně změněné. Pochvy malých myelinizovaných vláken zůstaly relativně ušetřeny. Axony vykazovaly známky mírných regresivních změn, normální struktura byla popsána často i tam, kde byl myelin deficitní (obr. 4, 5).


Byla provedena lumbální punkce s nálezem lehké hyperproteinorachie. Celková bílkovina v likvoru činila 0,58 g/ l (norma 0,15 – 0,45 g/ l), byly přítomny čtyři monocyty/ 1 µl (norma 0 – 3/ 1 µl).
Vyšetřili jsme protilátky IgM proti myelin asociovanému glykoproteinu (anti‑MAG), dále IgG a IgM protilátky proti gangliosidům (GM1, GM2, GM3, GD1a, GD1b, GT1b a GQ1b), všechny byly negativní. Pacient měl lehce zvýšenou glykemii nalačno, proběhlo došetření na diabetologii a uzavřeno jako porucha glukózové tolerance.
Terapie byla zahájena bolusem kortikoidů 3 × 500 mg. Efekt kortikoidů nebyl pozorován, naopak si pacient po kortikoterapii stěžoval na lehké zhoršení stavu, na výraznější oslabení svalové síly levé horní končetiny. Vzhledem k udávanému zhoršení stavu jsme u pacienta nepokračovali v kortikoterapii, ale s odstupem tří měsíců jsme podali plnou dávku IVIG (5 × 30 g), poté nastalo klinické i elektrofyziologické zlepšení. Svalová síla na LHK se zlepšila: musculus abductor pollicis brevis MRC scale 4, extenzory prstů MRC scale 5, flexory prstů MRC scale 4 a extenzory zápěstí MRC scale 5. Elektrofyziologicky došlo k regresi bloku vedení motorických vláken, ztráta motorických jednotek byla méně výrazná. S odstupem jednoho měsíce od podání plné dávky IVIG došlo k postupnému návratu obtíží. Pacientovi byla aplikována další dávka IVIG, a to 30 g. Poté opět došlo ke zlepšení, které trvalo zhruba jeden měsíc. Po měsíci zlepšování ustalo a pacient udával lehké subjektivní zhoršení. U pacienta se počítá s dlouhodobou terapií IVIG, podávanou v odstupu 4 – 6 týdnů.
Diskuze
V této práci prezentujeme kazuistiku pacienta s Lewisovým-Sumnerovým syndromem, dle našich vědomostí se jedná o první prezentovanou kazuistiku tohoto vzácného onemocnění v české literatuře. Diferenciálně diagnosticky jsme u pacienta uvažovali především o chronické zánětlivé polyneuropatii (CIDP), multifokální motorické neuropatii (MMN), tomakulózní neuropatii, neurofibromu a neurolymfomatóze.
LSS spolu s CIDP mají řadu společných znaků. Obě onemocnění jsou demyelinizační senzitivně‑motorické neuropatie s možným postižením kraniálních nervů a progresivním nebo relaps remitentním průběhem. Odpověď na terapii IVIG a kortikoidy je u obou onemocnění stejná. V biopsii z nervus suralis bývá u LSS stejně jako u CIDP popisována primární demyelinizace nervových vláken [1,3 – 6,10–13]. V klinickém nálezu se LSS od CIDP odlišuje tím, že LSS primárně postihuje většinou horní končetiny a projevuje se asymetrickým postižením. V lumbální punkci bývá u LSS celková bílkovina normální nebo lehce zvýšená, zatímco u CIDP bývá hodnota celkové bílkoviny vyšší, dosahuje hodnot více než 1 g/ l [13]. Pokud je postižení u LSS lokalizováno fokálně pouze na horní končetině, bývá celková bílkovina v likvoru jen lehce zvýšená [14]. Známky demyelinizace jsou u LSS převážně fokální na rozdíl od CIDP, kde je demyelinizace více difuzní a bývá popisována jak distálně, tak proximálně [15]. Někteří autoři se domnívají, že LSS je počáteční fokální variantou CIDP [16].
Fokální postižení pouze horní končetiny u LSS vykazuje signifikantně nižší pravděpodobnost, že dojde k postižení i dalších končetin [14].
V naší kazuistice svědčilo pro LSS asymetrické postižení pouze LHK (a jak to klinické, tak elektrofyziologické), celková bílkovina v likvoru byla jen mírně zvýšena.
MMN je motorická neuropatie, která se klinicky projevuje asymetrickou svalovou slabostí a atrofiemi v distribuci jednotlivých periferních nervů. V EMG nálezu bývají také popisovány četné motorické kondukční bloky. V některých případech jsou i u pacientů s MMN uváděny lehké distální parestezie [17]. V klinickém nálezu však vždy dominuje motorické postižení, čímž se MMN odlišuje od LSS [18]. Dalším podpůrným kritériem v diferenciální diagnostice mezi LSS a MMN je přítomnost protilátek IgM proti GM1 gangliosidu, které bývají u MMN pozitivní, u LSS bývají negativní [18]. Pacienti s MMN většinou velmi dobře reagují na terapii IVIG a kortikoterapie je u nich bez efektu [18]. U některých pacientů s MMN může po čase dojít k multifokálnímu senzitivně‑motorickému postižení [19], což naznačuje, že by i zde mohla být užší souvislost mezi MMN a LSS. V naší kazuistice pro LSS oproti MMN svědčilo senzitivní postižení, a to jak klinické, tak elektrofyziologické, lehce zvýšená celková bílkovina v likvoru, nepřítomnost protilátek IgM proti GM1 gangliosidu a abnormální nález v biopsii n. suralis.
Vzhledem k anamnestickým údajům (rozvoj parézy po otlaku levé horní končetiny) a nálezu v biopsii nervus suralis jsme také uvažovali o hereditární neuropatii s tendencí k tlakovým parézám (HNPP, tomakulózní neuropatie). U pacientů s HNPP bývá na magnetické rezonanci také popisováno rozšíření nervových pochev [20]. Onemocnění je způsobeno delecí 1,5 Mb v PMP 22 genu na chromozomu 17p11.2 – p12 [21]. U našeho pacienta jsme provedli genetické vyšetření na tomakulózní neuropatii, které bylo negativní, proti HNPP svědčí i pozitivní odezva na léčbu IVIG.
Dle prvního nálezu na magnetické rezonanci krční páteře jsme u pacienta zvažovali také nádorové postižení brachiálního plexu, například neurofibrom nebo neurolymfomatózu. Periferní neuropatie, které vznikají v důsledku systémové malignity, mají různé příčiny a mohou se manifestovat v různém stadiu malignity. Přímá infiltrace periferních nervů nebo spinálních kořenů maligními buňkami je nejméně častou manifestací malignity a nejčastěji bývá asociována s lymfomy nebo leukemií, méně často se solidními tumory. Periferní neuropatie u pacientů s lymfomy bývají popisovány v 5 – 8 %, přímá lymfomatózní infiltrace periferních nervů bývá popisována jen v 0,1 – 2 % případů. Termín neurolymfomatóza se užívá pro maligní infiltraci periferního nervového systému v souvislosti s lymfomy nebo leukémií. Maligní infiltrace postihuje míšní nervy, kořeny nebo kraniální nervy. Neuropatie se projevují jako mononeuropatie nebo asymetrické multifokální polyneuropatie. V jiných případech bývá popsána difuzní infiltrace jako asymetrická senzitivně‑motorická neuropatie [22]. Nález na magnetické rezonanci zaměřené na brachiální plexus u našeho pacienta nádorovému procesu neodpovídal, současně nález z biopsie nervus suralis svědčil pro demyelinizační postižení. K vyloučení paraneoplastické etiologie jsme provedli celotělovou pozitronovou emisní tomografii, výsledek byl negativní.
Závěr
Lewisův - Sumnerův syndrom je onemocnění projevující se senzitivně‑motorickou asymetrickou neuropatií postihující především horní končetiny. LSS je považováno za asymetrickou variantu CIDP. Cílem prezentované kazuistiky bylo upozornit na toto vzácné onemocnění, jeho terapii a uvést diferenciální diagnostiku tohoto onemocnění.
Tato práce vznikla díky projektu „CEITEC – Středoevropský technologický institut“ (CZ.1.05/1.1.00/02.0068) z Evropského fondu regionálního rozvoje.
MUDr. Iva Okáčová
Neurologická klinika
LF MU a FN Brno
Jihlavská 20
625 00 Brno
e-mail: iva.okacova@fnbrno.cz
Přijato k recenzi: 2. 11. 2011
Přijato do tisku: 27. 4. 2012
Sources
1. Lewis RA, Sumner AJ, Brown MJ, Asbury AK. Multifocal demyelinating neuropathy with persistent conduction block. Neurology 1982; 32(9): 958 – 964.
2. Viala K, Renié L, Maisonobe T, Béhin A, Neil J, Léger JM et al. Follow up study and response to treatment in 23 patients with Lewis-Sumner syndrome. Brain 2004; 127(Pt 9): 2010 – 2017.
3. Oh SJ, Claussen GC, Kim DS. Motor and sensory demyelinating mononeuropathy multiplex (multifocal motor and sensory demyelinating neuropathy) a separate entity or a variant of chronic inflammatory demyelinating polyneuropathy? J Peripher Nerv Syst 1997; 2(4): 362 – 369.
4. Gorson KC, Ropper AH, Weinberg DH. Upper limb predominant, multifocal chronic inflammatory demyelinating polyneuropathy. Muscle Nerve 1999; 22(6): 758 – 765.
5. Saperstein DS, Amato Wolfe GI, Katz JS, Nations SP, Jackson CE et al. Multifocal acquired demyelinating sensory and motor neuropathy: the Lewis-Sumner syndrome. Muscle Nerve 1999; 22(5): 560 – 566.
6. Van der Berg-Vos RM, Van den Berg LH, Franssen H, Vermeulen M, Witkamp TD, Jansen GH et al. Multifocal inflammatory demyelinating neuropathy: a distinct clinical entity? Neurology 2000; 54(1): 26 – 32.
7. Jean AP, Duprez T, Van den Bergh PY. Massive peripheral nerve hypertrophy in patient with multifocal upper limb demyelinating neuropathy (Lewis - Sumner syndrome). Acta Neurol Belg 2001; 101(4): 234 – 238.
8. Pouget J, Verschueren A, Azulay JP, Attarian S. Lewis Sumner syndrome. Rev Neurol 2001; 157(12): 1561 – 1564.
9. Park YE, Yook JW, Kim DS. A case of Lewis - Sumner syndrome showing dramatic improvement after plasma exchange. J Korean Med Sci 2010; 25(7): 1101 – 1104.
10. Thomas PK, Claus D, Jaspert A, Workman JM, King H et al. Focal upper limb demyelinating neuropathy. Brain 1996; 119(Pt 3): 765 – 774.
11. Kuwabara S, Nakajima M, Matsuda S, Hattori T. Magnetic resonance imaging at the demyelinative foci chronic inflammatory demyelinating polyneuropathy. Neurology 1997; 48(4): 874 – 877.
12. Bouchard C, Lacroix C, Planté V, Adams D, Chedru et al. Clinicopathological findings and prognosis of chronic inflammatory demyelinating polyneuropathy. Neurology 1999; 52(3): 498 – 503.
13. Vallat JM, Tabaraud F, Magy L, Torny F, Bernet Bernardy P et al. Diagnostic value of nerve biopsy for atypical chronic inflammatory demyelinating polyneuropathy, evaluation of eight cases. Muscle Nerve 2003; 27(4): 478 – 485.
14. Rajabally YA, Chavada G. Lewis - Sumner syndrome of pure upper limb onset: diagnostic, prognostic, and therapeutic features. Muscle Nerve 2009; 39(2): 206 – 220.
15. Maisonobe T, Chassande B, Verin M, Jouni M, Leger JM et al. Chronic dysimmune demyelinating polyneuropathy: a clinical and electrophysiological study of 93 patients. J Neurol Neurosurg Psychiatry 1996; 61(1): 36 – 42.
16. Kuwabara S, Ogawara K, Misawa S, Mori M, Hattori T. Distribution patterns of demyelination correlate with clinical profiles in chronic inflammatory demyelinating polyneuropathy. J Neurol Neurosurg Psychiatry 2002; 72(1): 37 – 42.
17. Nobile – Orazio E. Multifocal motor neuropathy. J Neuroimmunol 2001; 115(1 – 2): 4 – 18.
18. Verschueren A, Azulay JP, Attarian S, Boucraut J, Pellissier JF, Pouget J. Lewis – Sumner syndrome and multifocal motor neuropathy. Muscle Nerve 2005; 31(1): 88 – 94.
19. Parry GJ, Clarke S. Multifocal acquired demyelinating neuropathy masquerading as motor neuron disease. Muscle Nerve 1988; 11(2): 103 – 107.
20. van Es HW. MRI of brachial plexus. Eur Radiol 2001; 11(2): 325 – 336.
21. Mazanec R, Horáček O, Kobesová A, Smetana P. Hereditární neuropatie. Cesk Slov Neurol N 2009; 72/ 105(1): 5 – 17.
22. Ambler Z. Lymfomatózní neuropatie (neurolymfomatóza) – kazuistika. Cesk Slov Neurol N 2010; 73/ 106(1): 725 – 728.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2012 Issue 4
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Hope Awakens with Early Diagnosis of Parkinson's Disease Based on Skin Odor
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
Most read in this issue
- Mozkové arachnoidální cysty u dospělých – retrospektivní analýza výsledků chirurgické terapie
- Porucha spánku s rytmickými pohyby
- Izolovaná sfenoidální sinusitida – možná příčina bolestí hlavy a závažných komplikací
- Oswestry dotazník, verze 2.1a – výsledky u pacientů s lumbální spinální stenózou, srovnání se starší verzí dotazníku