
Proměnlivá tvář parkinsonské neurodegenerace
The Changing Face of Parkinsonian Neurodegeneration
Parkinson’s disease continues to be described as a relatively homogenous nosological entity, characterised by the presence of four cardinal signs – bradykinesia, rigidity, tremor and postural instability – with a variable presence of other motor and non-motor symptoms (cognitive, behavioral, vegetative etc.); its pathological basis being the continuous progression of brain alpha-synucleinopathy. It seemed that this clinical entity might be relatively easily diagnosed using the United Kingdom Parkinson’s Disease Brain Bank (UK--PDBB) clinical diagnostic criteria that are based on the results of a retrospective clinical and pathological study. However, substantial opinion shift occurred as a result of the recent intensive field research. A distinction has arbitrarily been made between Dementia with Lewy bodies and Parkinson’s disease dementia. Several next mutations (PARK10–PARK13, PARK16–PARK18) encoding the manifestation of “sporadic“, late-onset and typical Parkinson’s disease have been added to the existing spectrum of familiar Parkinson’s disease. Therefore, the term “Parkinson’s disease” should be used for the “sporadic” form of the disease only. However, for the “definite” or “highly probable” level of diagnostic accuracy, the UK-PDBB criteria are less useful. The term “neurodegenerative Parkinsonism” would probably better reflect current level of diagnostic certainty referring to Parkinson’s disease.
Key words:
Parkinson’s disease – hereditary parkinsonism – neurodegenerative parkinsonism
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Authors:
K. Menšíková 1; P. Kaňovský 1; M. Kaiserová 1; I. Nestrašil 2; M. Bareš 3
Authors‘ workplace:
Neurologická klinika LF UP a FN Olomouc
1; Department of Neurology and Brain Sciences Center, University of Minnesota, MN, USA
2; I. neurologická klinika LF MU a FN u sv. Anny v Brně
3
Published in:
Cesk Slov Neurol N 2013; 76/109(1): 26-34
Category:
Review Article
Overview
Parkinsonova nemoc je stále popisována jako relativně homogenní nozologická jednotka, charakterizovaná základní tetrádou příznaků – bradykinezí, rigiditou, tremorem a posturální nestabilitou – s měnlivou přítomností a intenzitou dalších symptomů motorických a non--motorických (kognitivních, behaviorálních, vegetativních apod.), vznikajících na podkladě rozvoje mozkové tzv. alfa-synukleinopatie. Ještě nedávno se zdálo, že jde o klinickou entitu kde lze diagnózu poměrně snadno stanovit na základě univerzálně používaných tzv. United Kingdom Parkinson’s Disease Brain Bank (UK-PDBB) kritérií, vytvořených před 20 lety na základě retrospektivní klinicko-patologické studie. V posledních 10 letech ale došlo (díky intenzivnímu výzkumu) k významnému posunu v chápání parkinsonizmu a Parkinsonovy nemoci. Arbitrárně byla diferencována demence s Lewyho tělísky a Parkinsonova nemoc s demencí. K dosud existujícím známým familiárním formám Parkinsonovy nemoci se začátkem v mladém věku bylo přidáno několik dalších geneticky vázaných forem Parkinsonovy nemoci (PARK10–PARK13, PARK16–PARK18), které se svým fenotypem, začátkem a klinickým průběhem od tzv. klasické „sporadické“ Parkinsonovy nemoci prakticky neliší. Označení „Parkinsonova nemoc“ by tedy mělo být užíváno vlastně jen pro tzv. sporadickou Parkinsovou nemoc, přičemž k (relativně) jisté či spolehlivé diagnóze této formy nemoci in vivo nejsou kritéria UK-PDBB ideální; termín „neurodegenerativní parkinsonizmus“ by patrně současnou úroveň diagnostické jistoty reflektoval lépe.
Klíčová slova:
Parkinsonova nemoc – dědičný parkinsonizmus – neurodegenerativní parkinsonizmus
Úvod
Parkinsonova nemoc, podle MKSN-10 kód G20, je stále ještě vnímána prizmatem učebnic vzniklých v předminulé dekádě jako klinická nozologická entita, jejíž základní charakteristikou je tetráda dominantních příznaků – bradykineze, rigidita, tremor a posturální instabilita – variantně doplňovaná řadou dalších příznaků motorických i non-motorických.
Tato nozologická charakteristika má patologický korelát, kterým je lokalizovaná tzv. Lewyho patologie, tj. neuronální úbytek a neuronální degenerace charakterizovaná přítomností Lewyho tělísek a lokalizovaná v oblasti pars compacta substantia nigra. Všechny ostatní permutace klinicko-patologických korelátů s Lewyho tělísky bychom tedy neměli považovat za Parkinsonovu nemoc. Za co tedy ale je máme pokládat?
Určitý posun v klasifikaci se udál v posledních 10 letech. Nejdříve došlo k diferenciaci tzv. demence s Lewyho tělísky (DLB) a k tvorbě klinických diagnostických kritérií této nemoci [1], o něco později byla pojmenována Parkinsonova nemoc s demencí (PDD) a byla taktéž vytvořena a publikována klinická diagnostická kritéria této nemoci [2]. České verze obou kritérií obsahují tab. 1. a 2. Zároveň bylo arbitrárně stanoveno diferenciálnědiagnostické kritérium rychlosti rozvoje kognitivní poruchy, patrně aby bylo vůbec možno (alespoň s určitou mírou spolehlivosti) rozlišit tyto dvě nemoci. Tím vlastně byli od (dosud jednotně chápané) diagnózy Parkinsonovy nemoci odděleni všichni pacienti, kteří jeví výraznější než mírný kognitivní deficit a u kterých se tedy na základě existujících kritérií „musí“ jednat o Parkinsonovu nemoc s demencí nebo demenci s Lewyho tělísky. Ostatní pacienti by tudíž měli být považováni za homogenní skupinu jedinců, u kterých se skutečně neurodegenerativní proces odehrává lokalizovaně, převážně v substantia nigra, a u nichž lze předpokládat vysokou míru homogenity jak klinické symptomatologie a progrese nemoci, tak i patologického korelátu.
![Klinická diagnostická kritéria demence s Lewyho tělísky (DLB) [1].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/604b08be107be9fc666b09785d5a3006.png)
![Klinická diagnostická kritéria Parkinsonovy nemoci s demencí (PDD) [2].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/f1cfbf825b531b2468c8e638f8904d66.png)
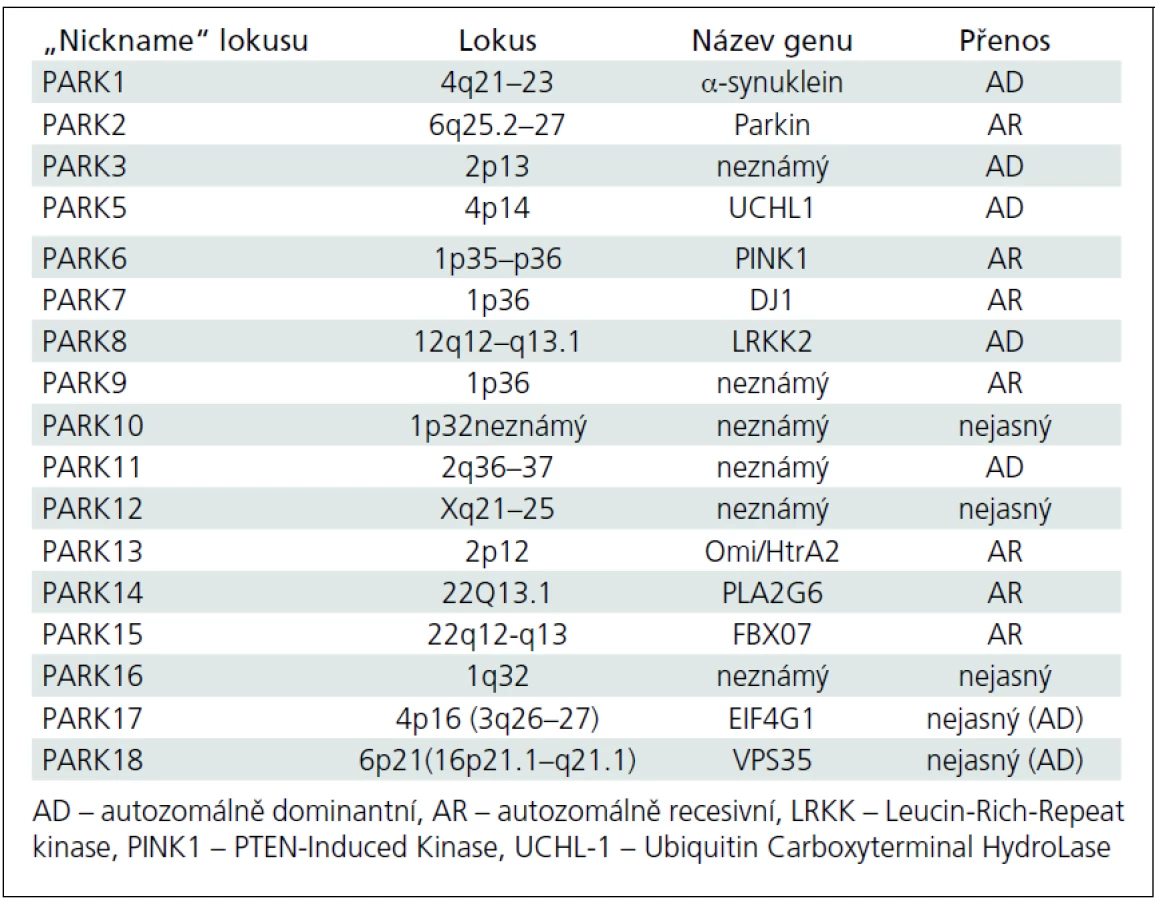
Avšak i v této přísněji definované skupině najde každý lékař (nejen ten, který se péčí o parkinsonské pacienty systematicky zabývá) vysokou variabilitu jak rychlosti rozvoje klinické symptomatologie, tak i pestrosti klinické symptomatiky; variabilní je pochopitelně i odpověď na léčbu. Tento stav implikuje otázku, zda nejsme na prahu dalšího klinického (a možná i patologického) a klasifikačního hodnocení fenotypu parkinsonské neurodegenerace. V určitém smyslu jsme možná i za tímto prahem, neboť dnes už je obvyklé označovat Parkinsonovu nemoc vzniklou v mladším věku jako tzv. juvenile onset (se začátkem do 21 let věku) nebo „young--onset“ (tzv. YOPD), se širokým začátkem po 21. roce života přibližně do 40 až 50 let věku [3–5]. Za těmito dvěma typy manifestace se zřejmě velmi často skrývají geneticky vázané formy Parkinsonovy nemoci, ale to zatím současné názvosloví neřeší. Obecně jsou také tzv. mendeliánské formy parkinsonizmu (od PARK1 až po PARK18) klinicky označovány jako Parkinsonova nemoc, přestože jsou svým klinickým charakterem, manifestací, detaily fenotypu nebo rychlostí progrese od tzv. klasické Parkinsonovy nemoci značně odlišné. V tab. 3 jsou v českém překladu srovnávacím způsobem uvedeny hlavní charakteristiky tzv. young-onset a „common“ forem Parkinsonovy nemoci [4], přehled tzv. dědičných forem Parkinsonovy nemoci obsahuje tab. 4. [6–10]. Zda je možno považovat hypotetický gen pro tzv. susceptibilitu za jedinou příčinu „common“ formy Parkinsonovy nemoci, je od r. 2001 předmětem neutuchající (leč nikoliv vášnivé) diskuze [11–13].
![Srovnání charakteristik tzv. „early-onset“ („juvenile“ a „young-onset“) a „common“ variant Parkinsonovy
nemoci [4].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/3b71f54b7e92e7b986997e9dfc034f94.png)
Problém je nejednotnost chápání nozologické terminologie. Je potřeba zde zdůraznit, že u výše zmiňovaných a dále detailněji popisovaných poruch se jedná o dědičný neurodegenerativní parkinsonizmus.Tento termín je ale jen málokdy používán. Někteří autoři použili termín „hereditary and degenerative“, tedy dědičný a degenerativní [5], jiní zase termín „parkin-related“, neboli vzniklý na základě parkinové mutace [3]. Nalezli jsme i označení „monogenetically inherited“, tedy monogeneticky dědičný; takto označené formy nemoci byly ale identické s těmi, které byly dříve nazývány jako „parkin-related“ [14]. Sami se kloníme k názoru, že nejvýstižnějšími českými termíny jsou asi dědičná Parkinsonova nemoc se začátkem a) v juvenilním, b) v mladším věku, c) v dospělém a pokročilém věku.
Neurodegenerativní juvenilní Parkinsonova nemoc je velmi vzácná, zejména v populaci Evropy a USA, a v drtivě většině případů je její výskyt prokazatelně familární. Velmi často mívá atypické příznaky či průběh (atypické v tom smyslu, že nejsou typické pro „common“ formu PN). Naopak YOPD mívá klinický obraz odpovídající „common“ formě a není tak vzácná, její incidence se zvyšuje s věkem posuzované populace. Obě formy jsou někdy popisovány pod společným názvem „early-onset parkinsonism“; incidence takto pojaté formy onemocnění (tj. „juvenile“ a „young-onset“ dohromady) je udávána mezi 1–3 případy na 100 000 obyvatel. Celkově potom v populaci pacientů trpících Parkinsonovou nemocí tvoří tyto formy 4–5 % [14–16].
Parkinsonova nemoc ve všech svých „formách“ (můžeme-li již tento termín použít) by měla být klasickou alfa-synukleinopatií. Základní patologickou diagnostickou charakteristikou je v takovémto případě přítomnost agregátů alfa-synukleinu, které vytváří tzv. Lewyho tělíska, a ta je pro všechny tři formy („juvenile-on-set“, „young-onset“ i „common“) zatím jednotná.
Mendeliánské formy juvenilního („juvenile“) a obvyklého („common“) parkinsonizmu
Dnes je prokázáno, že 10 genových mutací může způsobit rozvoj parkinsonských symptomů v mladém věku. Jako první byla v roce 1997 popsána mutace alfa-synukleinového genu, která je dnes nazývána SNCA nebo spíše PARK1 [17]. Nicméně nejčastější je v tomto kontextu zřejmě mutace parkinového genu označovaná jako PARK2 [18], u které se rozvíjí v příslušně mladém věku obraz zcela typické Parkinsonovy nemoci, pochopitelně s pozitivní rodinnou anamnézou. Několik recentně popsaných mutací způsobuje rozvoj parkinsonizmu v pokročilejším dospělém věku, který je fenotypicky prakticky identický se „sporadickou“ formou Parkinsonovy nemoci (PARK10–PARK13). V současné době je dobře či méně dobře popisováno 18 lokusů [12] a identifikace dalších stále probíhá.
PARK1 (SNCA)
Autozomálně dominantně děděná mutace alfa-synukleinového genu na dlouhém raménku chromozomu 4q. Poprvé byla tato mutace popsána ve velkém pedigree v USA, původ mutace byl hledán v italské větvi tohoto italsko-irsko-čerokézského rodokmenu a je kladen do lázeňského městečka Contursi v Kampánii. Pacienti postižení mutací PARK1 nejčastěji manifestují prakticky identický fenotyp jako při onemocnění sporadickou Parkinsonovou nemocí, pouze začátek onemocnění bývá ve zřetelně mladším věku, nejčastěji kolem čtyřicítky. Jsou však známy i případy s predilekčním postižením kognice nebo autonomních funkcí [19]. PARK1 je typický postsynaptický parkinsonský syndrom, jak prokázaly již před lety PET studie; stejně tak patologicky se jedná o typickou alfa-synukleinopatii, jen s poněkud disperznější distribucí typických změn, především Lewyho tělísek [19,20]. V minulé dekádě bylo popsáno několik dalších možných mutací alfa-synukleinového genu, z nichž některé kódovaly manifestaci parkinsonského syndromu s fenotypem bližším demenci s Lewyho tělísky, ale tyto mutace jsou extrémně vzácné [21].
PARK2 (Parkin)
Mutace genu nazývaného parkin je patrně nejčastější monogenní příčinou vzniku Parkinsonovy nemoci v mladém věku. Gen se přenáší autozomálně recesivně a dosud bylo popsáno více než 30 jeho mutací [12]. Soudí se, že mutace PARK2 je odpovědná za rozvoj familiární Parkinsonovy nemoci v mladém věku u více než poloviny případů [22]. Vzhledem k tomu, že přenos je autozomálně recesivní¸ je zřejmě část případů tzv. sporadické Parkinsonovy nemoci způsobena mutací PARK2, odhaduje se, že se jedná o zhruba 15–20 % pacientů. Pokud tomu tak je, onemocnění začíná v mladším věku, většinou před padesátkou [23]. Parkinsonova nemoc způsobená mutací PARK2 se manifestuje velmi podobným fenotypem jako sporadická Parkinsonova nemoc, nicméně častěji bývá v začátku onemocnění přítomna dystonie, naopak méně často mívají pacienti závažnější kognitivní deficit. Poměrně často bývá přítomna autonomní dysfunkce a výraznější posturální instabilita, stejně tak častější než u sporadické Parkinsonovy nemoci je výskyt psychiatrických příznaků: poruch chování, obsesivně-kompulzivní poruchy, úzkostných poruch a i psychóz [23]. Progrese parkinsonské symptomatiky je obecně pomalejší než u sporadické choroby, nicméně poměrně brzy mohou být přítomny motorické komplikace jako poruchy chůze, freezing a dyskineze; pacienti s PARK2 mutací jsou hypersenzitivní na podávání L-DOPA, a již malá dávka může vyvolat těžký dyskinetický syndrom. Na rozdíl od sporadické nemoci je u PARK2 nemoci ušetřen čich a tento fakt může být nápomocný v iniciální diferenciální diagnostice [24]. Z patologického hlediska je zásadním rozdílem oproti sporadické nemoci absence Lewyho tělísek v místech jejich předpokládaného výskytu, tj. v substantia nigra, kde je spíše nacházena glióza doprovázející nigrální neuronální ztrátu [25].
PARK3
Mutace specifického genu na chromozomu 2p13 je dávána do souvislosti s manifestací familiární formy Parkinsonovy nemoci, která je fenotypicky prakticky shodná se sporadickou formou. Přenos je autozomálně dominantní s redukovanou penetrancí. Mutace byla popsána ve třech velkých rodokmenech v severním Německu a jižním Dánsku a podrobná molekulární analýza potvrdila původ mutace v této oblasti [26,27].
PARK4
Familiární Parkinsonova nemoc s autozomálně dominantním přenosem a redukovanou penetrancí, která je způsobena triplikací alfa-synukleinového genu na chromozomu 4p [28]. Původní rodokmen, ve kterém byla mutace objevena, je někdy nazýván Iowa kindred [29]. Parkinsonova nemoc začíná obvykle ve věku mezi 30 a 60 lety a fenotypicky se může podobat sporadické Parkinsonově nemoci, demenci s Lewyho tělísky nebo i esenciálnímu třesu. Patologicky se jedná o typickou alfa-synukleinopatii s poměrně difuzní přítomností Lewyho tělísek.
PARK5 (UCHL-1)
Ubikvitinová mutace popsaná původně v německé rodině. Příčinou je pravděpodobně abnormalita v tzv. ubikvitinového proteázomového systému, přičemž klíčovou je pravděpodobně porucha enzymu ubikvitin-karboxyl-terminal-hydrolázy, označovaného jako UCHL-1 [30]. Ubikvitinový proteázový systém je vysoce stabilní metabolická dráha, jejímž úkolem je eliminace vadných nebo poškozených proteinů. Kromě toho reguluje některé základní celulární procesy, jako genovou transkripci, imunitní odpověď, progresi buněčného cyklu a i neurotransmisi. Centrální složkou tohoto komplexu je peptid ubikvitin. Mutace UCHL-1 genu, který byl později nazván PARK5, byla poprvé popsána na exonu 4 v německé rodině u dvou sourozenců [31]. Mutace způsobuje manifestaci Parkinsonovy nemoci bez významnějšího kognitivního deficitu, s fenotypem velmi podobným sporadické formě. Přenos je patrně autozomálně dominantní [32].
PARK6
Typický obraz Parkinsonovy nemoci se začátkem v mladém věku je fenotyp, kterým se manifestuje mutace genu nazývaného PINK1 nebo PARK6. Gen je lokalizován na krátkém raménku chromozomu 1 a řídí expresi mitochondriálního proteinu [33]. Pravděpodobně je (hned po PARK2) druhou nejčastější autozomálně recesivní příčinou vzniku Parkinsonovy nemoci v mladém věku [34]. První příznaky se objevují mezi 30. a 40. rokem věku, ale byl popsán i vznik ve věku kolem 50 let. Podobně jako u parkinové nemoci může se u pacientů s PARK6 již v počátečních stadiích nemoci objevit dystonie a kognitivní deficit. Patologický obraz Parkinsonovy nemoci způsobené mutací PARK6 není znám.
PARK7 (DJ-1 gen)
Parkinsonova nemoc se začátkem v mladém věku a nepříliš rychlou progresí, vysokou frekvencí výskytu dystonie a psychiatrických symptomů a s velmi dobrou odpovědí na podání L-DOPA je způsobena autozomálně recesivní mutací lokusu na chromozomu 1p [35,36]. Byla popsána v italské a holandské rodině s familárním výskytem Parkinsonovy nemoci. Fenotypicky připomíná parkinovou nemoc, je však zjevně mnohem vzácnější. Patologický obraz PARK7 není znám.
PARK8 (LRRK2)
Kináza nazývaná „Leucin-Rich Repeat Kinase 2“ (LRRK2) kóduje tvorbu proteinu nazývaného dardarin. Její mutace je příčinou manifestace typické Parkinsonovy nemoci, včetně zpočátku asymetrického třesu, rigidity a bradykineze, s dobrou odpovědí na podání L-DOPA, s poněkud dřívějším začátkem než sporadická nemoc, a to nejčastěji kolem 50 let. Přenos mutace je autozomálně dominantní s variující penetrancí, četnost výskytu v jednotlivých rodinách kolísá tedy pochopitelně také [37,38]. Patologicky je LRRK2 synukleinopatií kombinovanou s tauopatií, přičemž četnost výskytu Lewyho tělísek také variuje. Jinak jsou nacházena neurofibrilární klubka nebo jen různě vyjádřená neuronální ztráta. Takto rozdílné nálezy byly referovány dokonce i u různých případů v jednom pedigree [39].
PARK9
Parkinsonova nemoc s počátkem na přelomu dětského věku a dospělosti (11–16 let) doprovázená další symptomatologií (pyramidové příznaky, kognitivní deficit, supranukleární pohledová obrna) byla popsána v jordánských a chilských rodinách s výskytem tzv. Kufor-Rakebovy nemoci [40,41]. Přenáší se autozomálně recesivně a je dopa-responzivní. Nápadnými znaky nemoci mohou být dystonie, mj. i okulární (někdy je onemocnění označováno jako tzv. dystonie – parkinsonizmus komplex), a minimyoklonus prstů. Patologický obraz nemoci není znám, podle výsledků četných MR studií se soudí, že se jedná o neurodegeneraci s akumulací železa [42].
PARK10
Parkinsonova nemoc se začátkem v pozdním věku, dopa-responzivní, s fenotypem téměř identickým se sporadickou formou nemoci, byla popsán ve velké skupině příbuzných pacientů na Islandu a v několika rodinách ve Spojených státech. Gen byl lokalizován do oblasti lokusu 1p32 a je nazýván ELAVL4 [43]. Dědičnost této formy Parkinsonovy nemoci dosud není jasná. Vzhledem k původu rodin, ve kterých byl výskyt popsán, byl zkoumán výskyt mutace ve Skandinávii a v Irsku. U irských pacientů trpících Parkinsonovou nemocí byla nalezena asociace se zkoumanou mutací, což zavdalo příčinu ke spekulacím o keltském původu této mutace [44]. Patologický obraz této Parkinsonovy nemoci není znám.
PARK11
Parkinsonova nemoc se začátkem v pozdním věku, fenotypem identickým se sporadickou Parkinsonovou nemocí a autozomálně dominantním přenosem s neúplnou penetrancí a velmi dobrou odpovědí na L-DOPA byla popsána v USA při podrobném zkoumání téměř dvou set pacientů, u kterých se v příbuzenstvu vyskytl sourozenec trpící rovněž Parkinsonovou nemocí. Mutace, která působí tuto Parkinsonovu nemoc, byla lokalizována na druhý chromozom do lokusu 2q36–q37 [45]. O původu této mutace není nic bližšího známo, taktéž není znám patologický obraz této Parkinsonovy nemoci.
PARK12
Parkinsonova nemoc se začátkem v pozdním věku a (pravděpodobně) s fenotypem blízkým sporadické Parkinsonově nemoci, nejasným přenosem a neznámým patologickým obrazem je dávána do souvislosti s chromozomem X a mutací genu v lokusu Xq21–25 [46].
PARK13
Parkinsonova nemoc se začátkem v pozdním věku a identickým fenotypem se sporadickou Parkinsonovou nemocí je způsobena mutací genu nazývaného Omi/HtrA2 na krátkém raménku chromozomu 2 v lokusu 2p12. Byla popsána u německých pacientů z rodin se známou familiární agregací Parkinsonovy nemoci [47].
PARK14
Parkinsonova nemoc se začátkem nejčastěji v dospělém, stále však mladém věku a fenotypem charakteru dystonie-parkinsonizmus komplex je působena mutací genu nazývaného PLA2G6, který byl lokalizován na dlouhé raménko chromozomu 22 do lokusu 22Q13.1. Přenáší se autozomálně recesivně, patologický obraz tohoto onemocnění není znám [48].
PARK15
Parkinsonova nemoc se začátkem v mladém věku, kde parkinsonský fenotyp doplňují ještě dystonie a pyramidové příznaky, je způsobena mutací genu nazývaného FBX07, který byl recentně lokalizován na dlouhé raménko chromozomu 22, do lokusu 22q12–q13. Onemocnění je dopa-responzivní, přenáší se autozomálně recesivně a jeho patologický obraz není znám [49].
PARK 16–PARK18
Recentně popsané mutace v lokusech 1q32 (PARK16), 4p16 (PARK17) a 6p21.3 (PARK18), byly označeny za další, které pravděpodobně mohou kódovat vznik Parkinsonovy nemoci s fenotypem identickým se sporadickou Parkinsonovou nemocí (tzv. susceptibility genes). Nicméně rozsáhlá genetická studie provedená v severošpanělské populaci výše uvedené asociace popřela [50] a nejnovější práce potvrzují pouze lokus 1q32 (PARK16) s tím, že uvolněná označení PARK17 a PARK18 by měla být obsazena spíše velmi recentně popsanými autozomálně dominantními mutacemi genů EIF4G1 a VPS35 v lokusem 3q26–27 a 16p21.1–q12.1 [51].
„Sporadická“ Parkinsonova nemoc
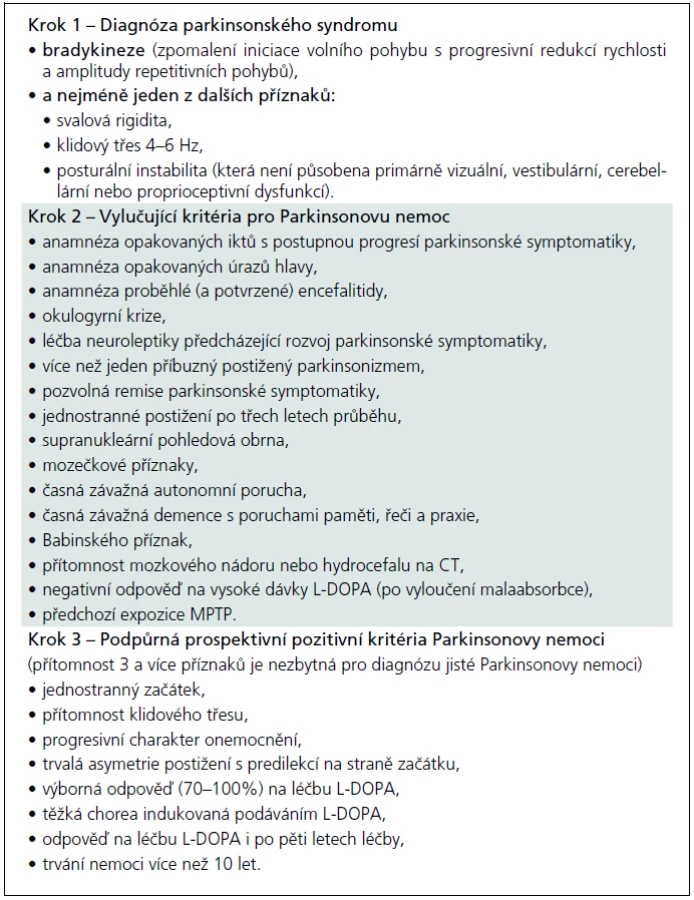
Jako „sporadickou“ Parkinsonovu nemoc lze patrně označit jakýkoliv parkinsonský syndrom manifestující se (dnes již klasickou) tetrádou „bradykineze, rigidita, tremor, posturální instabilita“, který není některým z atypických parkinsonských syndromů nebo není působen některou z výše uvedených mutací, a kde není prokázán rodinný výskyt. Jde tedy – překvapivě – o diagnózu per exclusionem. Přísně literárně by diagnóza „sporadické“ Parkinsonovy nemoci měla být stanovena na základě tzv. United Kingdom Parkinson’s disease Brain Bank Criteria, která byla postulována v sérii prací Leesovy skupiny ze začátku 90. let a jejich o deset let starších replik [52–56]. Česká verze těchto kritérií je (pro názornost) obsažena v tab. 5. Otázkou ovšem je, zda tato kritéria, vytvořená na základě post mortem a retrospektivních klinicko-patologických korelací, obstojí ve světle poznání poslední dekády. Není tím míněn jen zvyšující se počet popsaných mutací, které kódují minimálně familiární agregaci Parkinsonovy nemoci. Nejsou tím míněny také jen Braakovy práce, vnášející svým patologickým stagingem další rozměr do parkinsonské konfuze [57,58]. Jsou tím míněna hlavně četná pozorování, jež přinášejí důkazy, že určitá forma neurodegenerace se může nejenom fenotypicky, ale i patologicky manifestovat jako úplně odlišná forma neurodegenerace, která již byla komentována před více než pěti lety [59]. Dnešní stav poznání dokonce nabízí hypotézu o vzájemné potenciaci jednotlivých typů neurodegenerace, o nichž se zatím soudilo, že jsou natolik odlišné svým charakterem, že na jejich podkladě může vzniknout nová validní klasifikace neurodegenerativních onemocnění [60,61]. Od takto odvážné hypotézy je samozřejmě k přesvědčivému důkazu ještě daleko, nicméně důvodů k zamyšlení se nad UK-PDBB kritérii je více než dost. A to nejen proto, že tato kritéria stále slouží jako univerzální nástroj jak klinicko-patologického výzkumu, tak jako vstupní kritéria prakticky všech klinických studií nových molekul zkoušených k léčbě „sporadické“ Parkinsonovy nemoci. Důkladnější studium doslovného překladu autorské verze UK-PDBB kritérií odhalí, že se jedná vlastně o kritéria parkinsonského syndromu (v kroku 1) a další oddíly kritérií, vylučující i podpůrné (krok 2 a krok 3), jsou plné nespecifických, často dnes již obsolentních příznaků či anamnestických dat, zjevně derivovaných z dokumentace uchovávané v londýnské mozkové bance spolu s fixovanými mozky [52]. Americký pokus o upgrade UK-PDBB kritérií, nazývaný NINDS-PD criteria, byl jen spoře citován a prakticky zaveden nebyl, zejména pro naprostou (a na první pohled patrnou) nepoužitelnost [62].
Klíčová otázka nyní je, o co se jedná v případě „sporadické“ Parkinsonovy nemoci. Je to skutečně sporadické onemocnění, které vzniká na základě jakési blíže nedefinované dispozice nebo (dosud nepoznaných) exogenních vlivů? Či se jedná o typicky polygenní onemocnění, u kterého jsme zatím popsali pouhých necelých 20 typů mutací? Odpovědi na tyto otázky jsou složité, diskuze k nim nenápadně započala již i v českém písemnictví [63–65]. Myslíme si, že v této diskuzi je především třeba se poněkud „vrátit“ k mendeliánským formám neurodegenerativního parkinsonizmu. Je nepochybné, že mutace popsané opakovaně v různých místech světa a ve velkých rodinách (PARK1, PARK2, PARK6, PARK8) jsou specifickými genetickými onemocněními (podobně jako Huntingtonova nemoc nebo Downova nemoc) s klinickým obrazem parkinsonizmu. To samé však nelze říci o mutacích, jež byly popsány v posledních letech, prakticky ve všech případech u „sporadického“ typu parkinsonizmu s patrnou familiární agregací (PARK11––PARK18). Prakticky ve všech případech byly mutace zjištěny metodou „genome-wide analysis“, tedy extenzivním pátráním po mutaci ve větší skupině pacientů trpících Parkinsonovou nemocí, a následnou asociací „podezřelé“ mutace s klinickým obrazem parkinsonizmu (do té doby u všech subjektů považovaného za „sporadický“!). Maně se nabízí úvaha, co by bylo zjištěno, pokud bychom stejné zkoumání provedli u všech pacientů se „sporadickou“ Parkinsonovou nemocí žijících např. na území bývalého rakousko-uherského impéria.
Je tedy „sporadická“ Parkinsonova nemoc skutečně sporadická v pravém významu tohoto slova? Na základě dnešních znalostí si troufáme říci, že sotva. Výraz „idiopatická“, který se ještě občas objevuje, je už zcela jistě poněkud „demodé“, přičemž je pravděpodobně lepší, než „sporadická“. Výrazem „idiopatická“ konstatujeme u daného případu pacienta postiženého neurodegenerativním parkinsonizmem naši současnou bezmoc tváří v tvář faktu, že nejsme za použití současných zobrazovacích, biochemických, genomických, proteomických a jakýchkoliv dalších metod schopni určit primární molekulární příčinu daného případu parkinsonizmu (ne parkinsonizmu obecně!). Je však více než pravděpodobné, že každý z případů „sporadické“ Parkinsonovy nemoci takovouto příčinu má. Zda se ve všech případech jedná o dosud nepopsané mutace, které by při důsledné „genome-wide“ analýze byly odhaleny, nelze říci. Jako možnost se to nepochybně nabízí, je to patrně nejvíce plauzibilní vysvětlení existence „idiopatické“ či „sporadické“ Parkinsonovy nemoci. Určitě nelze předpokládat globální výskyt dosud nepopsaného selektivního neurotoxinu, který by působil environmentální Parkinsonovu nemoc na Islandu stejně jako v Tanzanii nebo Šalomounových ostrovech.
Ve světle výše uvedených faktů i hypotéz je tedy patrně na čase začít diskutovat o tom, zda onemocnění, které se projevuje onou základní tetrádou (bradykinezí, rigiditou, tremorem a posturální instabilitou) spolu s dalšími doprovodnými symptomy jak motorickými, tak i kognitivními a vegetativními, odpovídající na léčbu L-DOPA, můžeme nadále nazývat Parkinsonovou nemocí. Zdá se, že „neurodegenerativní parkinsonizmus“ by byl lepším označením stavu, který v některých případech nelze jednoznačně nozologicky a taxonomicky klasifikovat ani post mortem [63,64].
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Práce byla podpořena grantem IGA MZ ČR č. NT12221-5.
K. Menšíková1, P. Kaňovský1, M. Kaiserová1, I. Nestrašil2, M. Bareš3
1 Neurologická klinika LF UP a FN Olomouc
2 Department of Neurology and Brain Sciences Center, University of Minnesota, MN, USA
3 I. neurologická klinika LF MU a FN u sv. Anny v Brně
MUDr. Kateřina Menšíková
Neurologická klinika
LF UP a FN Olomouc
I. P. Pavlova 6
775 20 Olomouc
e-mail: katerina.mensikova@upol.cz
Přijato k recenzi: 24. 2. 2012
Přijato do tisku: 15. 6. 2012
Sources
1. McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005; 65(12): 1863–1872.
2. Emre M, Aarsland D, Brown R, Burn DJ, Duyckaerts C, MizunoY et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov Disord 2007; 22(12): 1689–1707.
3. Uc EY, Rodnitzky RI. Juvenile parkinsonism. Semin Pediatr Neurol 2003; 10(1): 62–67.
4. Calne SM, Kumar A. Young onset Parkinson’s disease. Practical management of medical issues. Parkinsonism Relat Disord 2008; 14(2): 133–142.
5. Thomsen TR, Rodnitzky RI. Juvenile parkinsonism: epidemiology, diagnosis and treatment. CNS Drugs 2010; 24(6): 467–477.
6. Nussbaum R, Polymeropoulos M. Genetics of Parkinson’s disease. Hum Mol Genet 1997; 6(10): 1687–1691.
7. Foroud T, Uniacke SK, Liu L, Pankratz N, Rudolph A, Halter C et al. Heterozygosity for a mutation in the parkin gene leads to later onset Parkinson disease. Neurology 2003; 60(5): 796–801.
8. Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004; 364(9440): 1167–1169.
9. Mata IF, Lockhart PJ, Farrer MJ. Parkin genetics: one model for Parkinson’s disease. Hum Mol Genet 2004; 13(1): R127–R133.
10. Eriksen J, Wsolek Z, Petrucelli J. Molecular pathogenesis of Parkinson disease. Arch Neurol 2005; 62(3): 353–357.
11. Hicks AA, Pétursson H, Jónsson T, Stefánsson H, Jóhannsdóttir HS, Sainz J et al. A susceptibility gene for late-onset idiopathic Parkinson’s disease. Ann Neurol 2002; 52(5): 549–555.
12. Gasser T. Mendelian forms of Parkinson’s disease. Biochim Biophys Acta 2009; 1792(7): 587–596.
13. Corti O, Lesage S, Brice A. What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol Rev 2011; 91(4): 1161–1218.
14. Schrag A, Ben-Shlomo Y, Quinn NP. Cross sectional prevalence survey of idiopathic Parkinson’s disease and Parkinsonism in London. BMJ 2000; 321(7252): 21–22.
15. Quinn N, Critchley P, Marsden CD. Young onset Parkinson’s disease. Mov Disord 1987; 2(2): 73–91.
16. Butterfield PG, Valanis BG, Spencer PS, Lindeman CA, Nutt JG. Environmental antecedents of young-onset Parkinson’s disease. Neurology 2006; 43(6): 1150–1158.
17. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997; 276(5321): 2045–2047.
18. Healy DG, Abou-Sleiman PM, Wood NW. PINK, PANK or PARK? A clinicans guide to familial parkinsonism. Lancet Neurol 2004; 3(11): 652–662.
19. Spira PJ, Sharpe DM, Halliday G, Cavanagh J, Nicholson GA. Clinical and pathological features of a Parkinsonian syndrome in a family with an Ala53ThR alpha-synuclein mutation. Ann Neurol 2001; 49(3): 313–319.
20. Samii A, Markopoulou K, Wszolek ZK, Sossi V, Dobko T, Mak E et al. PET studies of parkinsonism associated with mutation in the alpha-synuclein gene. Neurology 1999; 53(9): 2097–2102.
21. Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 2004; 55(2): 164–173.
22. Hedrich K, Marder K, Harris J, Kann M, Lynch T, Meija-Santana H et al. Evaluation of 50 probands with early-onset Parkinson’s disease for Parkin mutations. Neurology 2002; 58(8): 1239–1246.
23. Khan NL, Graham E, Critchley P, Schrag AE, Wood NW, Lees AJ et al. Parkin disease: a phenotypic study of a large case series. Brain 2003; 126(Pt 6): 1279–1292.
24. Khan NL, Katzenschlager R, Watt H, Bhatia KP, Wood NW, Quinn N et al. Olfaction differentiates parkin disease from early-onset parkinsonism and Parkinson disease. Neurology 2004; 62(7): 1224–1226.
25. Farrer M, Chan P, Chen R, Tan L, Lincoln S, Hernandez D et al. Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol 2001; 50(3): 293–300.
26. Gasser T, Müller-Myshok B, Wszolek ZK, Oehlmann R, Calne DB, Bonifati V et al. A susceptibility locus for Parkinson’s disease maps to chromosome 2p13. Nat Genet 1998; 18(3): 262–265.
27. Klein C, Vieregge P, Hagenah J, Sieberer M, Doyle E, Jacobs H et al. Search for the PARK3 founder haplotype in a large cohort of patients with Parkinson’s disease from northern Germany. Ann Hum Genet 1999; 63(Pt 4): 285–291.
28. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J et al. Alpha-synuclein locus triplication causes Parkinson’s disease. Science 2003; 302(5646): 841.
29. Gwinn K, Devine M, Jin LW, Johnson J, Bird T, Muenter M et al. Clinical features, with video documentation, of the original familial Lewy body Parkinsonism caused by alpha-synuclein triplication (lowa kindred). Mov Disord 2011; 26(11): 2134–2136.
30. Healy DG, Abou-Sleiman PM, Wood NW. Genetic causes of Parkinson’s disease: UCHL-1. Cell Tissue Res 2004; 318(1): 189–194.
31. Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E et al. The ubiquitin pathway in Parkinson’s disease. Nature 1998; 395(6701): 451–452.
32. Maraganore DM, Lesnick TG, Elbaz A, Chartier--Harlin MC, Gasser T, Krüger R et al. UCHL1 is a Parkinson’s disease susceptibility gene. Ann Neurol 2004; 55(4): 512–521.
33. Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004; 304(5674): 1158–1160.
34. Bonifati V, Rohé CF, Breedveld GJ, Fabrizio E, De Mari M, Tassorelli C et al. Early-onset parkinsonism associated with PINK-1 mutations: frequency, genotypes and phenotypes. Neurology 2005; 65(1): 87–95.
35. Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003; 299(5604): 256–259.
36. Dekker M, Bonifati V, van Swieten J, Leenders N, Galjaard RJ, Snijders P et al. Clinical features and neuroimaging of PARK-7-linked parkinsonism. Mov Disord 2003; 18(7): 751–757.
37. Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, van der Brug M et al. Cloning of the gene containing mutations that cause PAR8-linked Parkinson’s disease. Neuron 2004; 44(4): 595–600.
38. Aasly JO, Toft M, Fernandez-Mata I, Kachergus J, Hulihan M, White LR et al. Clinical features of LRRK-2 associated Parkinson’s disease in central Norway. Ann Neurol 2005; 57(5): 762–765.
39. Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S et al. Mutations in LRRK-2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004; 44(4): 601–607.
40. Williams DR, Hadeed A, al-Din AS, Wreikat AL, Lees AJ. Kufor Rakeb disease: autosomal recessive, levodopa-responsive parkinsonism with pyramidal degeneration, supranuclear gaze palsy and dementia. Mov Disord 2005; 20(10): 1264–1271.
41. Ramirez A, Heimbach A, Gründemann J, Stiller B, Hampshire D, Cid LP et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 2006; 38(10): 1184–1191.
42. Schneider SA, Paisan-Ruiz C, Quinn NP, Lees AJ, Houlden H, Hardy J et al. ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Mov Disord 2010; 25(8): 979–984.
43. Hicks AA, Pétursson H, Jónsson T, Stefánsson H, Jóhannsdóttir HS, Sainz J et al. A susceptibility gene for late-onset idiopathic Parkinson’s disease. Ann Neurol 2002; 52(5): 549–555.
44. Haugarvoll K, Toft M, Ross OA, Stone JT, Heckman MG, White LR et al. ELAVL4, PARK10 and the Celts. Mov Disord 2007; 22(4): 585–587.
45. Pankratz N, Nichols WC, Uniacke SK, Halter C, Rudolph A, Shults C et al. Significant linkage of Parkinson disease to chromosome 2q36-q37. Am J Hum Genet 2003; 72(4): 1053–1057.
46. Pankratz N, Nichols WC, Uniacke SK, Halter C, Murrell J, Rudolph A et al. Genome-wide linkage analysis and evidence of gene-by-gene interactions in a sample of 362 multiplex Parkinson disease families. Hum Mol Genet 2003; 12(20): 2599–2608.
47. Strauss KM, Martins LH, Plun-Favreau H, Marx FP, Kautzmann S, Berg D et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum Mol Genet 2005; 14(15): 2099–2111.
48. Yoshino H, Tomiyama H, Tachibana N, Ogaki K, Li Y, Funayama M et al. Phenotypic spectrum of patients with PLA2G6 mutation and PARK14-linked parkinsonism. Neurology 2010; 75(15): 1356–1361.
49. Paisán-Ruiz C, Guevara, R, Federoff M, Hanagasi H, Sina F, Elahi E et al. Early-onset L-dopa-responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBX07 and spatacsin mutations. Mov Disord 2010; 25(12): 1791–1800.
50. Mata IF, Yearout D, Alvarez V, Coto E, de Mena L, Ribacoba R et al. Replication of MAPT and SNCA, but not PARK16–18, as susceptibility genes for Parkinson’s disease. Mov Disord 2011; 26(5): 819–823.
51. Sundal C, Fujioka S, Uitti R, Wszolek Z. Autosomal dominant Parkinson’s disease. Parkinsonism Relat Disord 2012; 18 (Suppl 1): S7–S10.
52. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuarcy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992; 55(3): 181–184.
53. Hughes AJ, Ben-Shlomo Y, Daniel SE, Lees AJ. What features improve accuracy of clinical diagnosis in Parkinson’s disease: a clinicopathologic study. Neurology 1992; 42(6): 1142–1146.
54. Hughes AJ, Daniel SE, Blankson S, Lees AJ. A clinicopathologic study of 100 cases of Parkinson’s disease. Arch Neurol 1993; 50(2): 140–148.
55. Hughes AJ, Daniel SE, Lees AJ. Improved accuracy of clinical diagnosis of Lewy body Parkinson’s disease. Neurology 2001; 57(8): 1497–1499.
56. Hughes AJ, Daniel SE, Ben-Shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialis movement disorder service. Brain 2002; 125(Pt 4): 861–870.
57. Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res 2004; 318(1): 121–134.
58. Braak H, Müller CM, Rüb U, Ackermann H, Bratzke H, de Vos RA et al. Pathology associated with sporadic Parkinson’s disease – where does it end? J Neural Transm Suppl 2006; 70 : 89–97.
59. Galpern WR, Lang AE. Interface between tauopathies and synucleinopathies: a tale of two proteins. Ann Neurol 2006; 59(3): 449–458.
60. Badiola N, de Oliveira RM, Herrera F, Guardia--Laquarta C, Concalves SA, Pera M et al. Tau enhances alpha-synuclein aggregation and toxicity in cellular models of synucleinopathy. PLoS One 2011; 6(10): e26609.
61. Jellinger K. Interactions between alpha-synuclein and other proteins in neurodegenerative disorders. Scientific World Journal 2011; 11 : 1893–1907.
62. Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson’s disease. Arch Neurol 1999; 56(1): 33–39.
63. Fiala O, Růžička E. Genetika Parkinsonovy nemoci. Cesk Slov Neurol N 2009; 72/105(5): 419–428.
64. Petrleničová D, Gmitterová K, Benetin J. Mechanizmy neurodegenerácie pri Parkinsonovej chorobe. Cesk Slov Neurol N 2010; 73/106(6): 645–649.
65. Kračunová K, Kovačovičová M, Baldovič P, Valkovič P, Kádaši Ľ, Benetin J. Výskyt mutácií v géne Leucine rich repeate kinase 2 u pacientov s Parkinsonovou chorobou na Slovensku. Cesk Slov Neurol N 2011; 74/107(4): 443–445.
66. Stern MB, Lang A, Poewe W. Toward a redefinition of Parkinson’s disease. Mov Disord 2012; 27(1): 54–60.
67. Jellinger K. Neuropathology of sporadic Parkinson’s disease: evaluation and changes of concepts. Mov Disord 2012; 27(1): 8–30.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2013 Issue 1
- Memantine Eases Daily Life for Patients and Caregivers
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Advances in the Treatment of Myasthenia Gravis on the Horizon
Most read in this issue
- Použití botulotoxinu v neurologii
- Častý výskyt lymeské neuroboreliózy u dětí v České republice
- Tetanus – staronová diagnóza? Kazuistika
- Hydrocefalus jako komplikace subarachnoidálního krvácení