
Leukodystrofie – klinické a rádiologické aspekty
Leukodystrophies – Clical and Radiological Findings
Myelin disorders of the central nervous system are also known as leukoencephalopathies. This term includes diseases, in which changes of the white matter are dominant, or even exclusive, while neither the pathophysiological mechanism nor the histopathological basis are decisive. This broad selection of inborn and acquired disorders includes a group of leukodystrophies characterized by primary dysfunction of myelin and myelin-producing cells. Typically, six separate clinical entities are included in this group: X-linked adrenoleukodystrophy, metachromatic leukodystrophy, Krabbe’s globoid cell leukodystrophy, Canavan disease, Pelizaeus-Merzbacher disease and Alexander disease. The group is constantly expanding as our knowledge develops. Clinical and laboratory findings, as well as MRI scans, are crucial for exact diagnosis. Accurate analysis of MRI images accelerates the diagnostic process considerably. A common feature of leukodystrophies is their progressive nature. Children exhibit rapid loss of motor and cognitive functions. In adolescent and adult patients, psychological and mental changes are the most prominent, while motor deficit has a later onset. The course of X-linked adrenoleukodystrophy, metachromatic leukodystrophy, and Krabbe’s leukodystrophy can be managed at the early or pre-clinical stages with bone marrow or stem cell transplantation. Gene and enzyme therapy are indicated at the earliest stages of metachromatic leukodystrophy. For the remaining leukodystrophies, only symptomatic therapy is available. The purpose of this paper is to summarize current information and knowledge as well as possible therapeutic options in this group of disorders.
Key words:
leukodystrophy – leukopathy – clinical findings – magnetic resonance imaging
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Authors:
M. Kolníková; P. Sýkora
Authors‘ workplace:
Klinika detskej neurológie LF UK a DFNsP Bratislava
Published in:
Cesk Slov Neurol N 2014; 77/110(5): 534-552
Category:
Minimonography
doi:
https://doi.org/10.14735/amcsnn2014534
Poďakovanie patrí primárke KDN, MUDr. Jaroslave Payerovej za podporu pri písaní a primárovi MUDr. Dušanovi Haviarovi a kolegom z Rádiologického oddelenia DFNsP v Bratislave za poskytnutie rádiologickej dokumentácie, profesorke M. V. Knaap z univerzitného centra VU Amsterdam za možnosť konzultácie MR nálezov a genetické vyšetrenie pacienta s leukoencefalopatiou – Vanishing White Matter disease (VWM).
Overview
Ochorenia myelínu CNS sa súhrnne označujú ako leukoencefalopatie. Termín zahŕňa poruchy, pri ktorých zmeny v bielej hmote prevažujú alebo sú výlučné, pričom patofyziologický mechanizmus ani histopatologická báza nie sú rozhodujúce. Z tejto širokej skupiny vrodených a získaných porúch sa vyčleňuje skupina leukodystrofií, ktorá označuje primárnu poruchu myelínu a buniek tvoriacich myelín. Klasicky sa do tejto skupiny radí šesť klinických jednotiek: X‑adrenoleukodystrofia, metachromatická leukodystrofia, Krabbeho globoidná leukodystrofia, Canavanovej choroba, Pelizaeus ‑ Merzbacherova choroba a Alexandrova choroba. Pribúdajúcimi poznatkami sa skupina rozširuje o stále nové primárne poruchy. Pre presnú diagnostiku je dôležitý klinický nález, významné sú zmeny na MR mozgu a laboratórne vyšetrenia. Precíznou analýzou MR obrazu je možné diagnostický proces urýchliť. Jedným zo spoločných znakov leukodystrofií je progresívny priebeh. U detí nastáva zväčša rýchla strata motorických a kognitívnych funkcií. V prípade adolescentov a adultných pacientov je ťažisko zmien v psychickej a mentálnej oblasti, pričom motorický deficit nastupuje neskôr. Priebeh X‑adrenoleukodystrofie, metachromatickej leukodystrofie, Krabbeho globoidnej leukodystrofie sa dá vo včasnom štádiu alebo predklinickom období ovplyvniť transplantáciou kostnej drene alebo kmeňových buniek. V prípade metachromatickej leukodystrofie je v skorých štádiách indikovaná génová a enzýmová terapia. Pre ostatné leukodystrofie je dostupná len symptomatická liečba. Cieľom práce je zhrnutie najdôležitejších poznatkov a liečebných možností.
Kľúčové slová:
leukodystrofie – leukopatie – klinický obraz – magnetická rezonancia
Zoznam použitých skratiek
ACTH adrenokortikotropný hormón
ADC Apparent Diffusion Coefficient
ADEM akútna diseminovaná encefalomyelitída
ADHD Attention Deficit Hyperactivity Disorder, porucha pozornosti s hyperaktivitou
AAV ‑ 2 Adeno‑Associated Virus 2
AMN adrenomyeloneuropatia
ARSA A arylsulfatáza A
BH biela hmota
BG bazálne gangliá
BMT transplantácia krvotvorných buniek
BAEP Brainstem Auditory Evoked Potentials
Cho cholín
Cr kreatín
CMV cytomegalovírusová infekcia
DWI Diffusion Weighted Image, difúzne vážený obraz
FLAIR FLuid ‑ Attenuated Inversion Recovery
GALC galaktocerebrozidáza
GFAP Glial Fibrillary Acidic Protein
MBP myelín bázický proteín
MELAS myopatia, encefalopatia, laktátová acidóza a stroke‑like epizódy
mIns myoinositol
MLD metachromatická leukodystrofia
MRS magnetická rezonančná spektroskopia
MSUD Maple Syrup Urine Disease, choroba javorového sirupu
NAA N ‑ acetylaspartát
PKAN Pantothenate Kinase ‑ Associated Neurodegeneration, neurodegenerácia spojená s pantoteín kinázou
PMD Pelizaeus ‑ Merzbacher Disease
PLP proteolipidový proteín
PVL periventrikulárna leukomalácia
tr. tractus
VLCFA Very Long Chain Fatty Acids
X ‑ ALD X‑adrenoleukodystrofia
X ‑ SPG2 X ‑ spastická paraplégia typ 2
Úvod
Myelín tvorí hlavnú súčasť bielej hmoty (BH) centrálneho nervového systému (CNS) a periférneho nervového systému (PNS). BH je tvorená veľkým počtom axónov a obsahuje množstvo neurogliových buniek, hlavne astrocytov a oligodendrocytov, ktoré myelín tvoria a udržujú. Jedna bunka oligodendroglie obaľuje svojimi výbežkami viac axónov [1 – 4]. Oligodendrocyt je kľúčovou bunkou myelinizácie CNS. Je schopný tvoriť množstvo špecifických membránových zložiek, potrebných pre koordináciu syntézy lipidov a proteínov, ale aj ich degradáciu. Astrocyt, cez sekréciu rozličných rastových faktorov, hrá významnú úlohu pri stimulácii formovania myelínu a v podpore adhézie oligodendrocytu k axónu. Skladba myelínovej dvojvrstvy je unikátna obsahom nasýtených mastných kyselín s veľmi dlhým reťazcom (VLCFA), ktoré umožňujú tesné uloženie a veľkú stabilitu membránovej štruktúry [5]. Cerebrálny myelín obsahuje veľké množstvo cerebrozidu, ktorý je typickým myelínovým lipidom mozgu. Ďalšou jeho súčasťou sú proteíny ako proteolipidový proteín (PLP) a myelín bázický proteín, ktoré tvoria podstatnú časť celkových proteínov myelínu (60 – 80 %) [5,6]. Vývoj myelínu pokračuje aj v prvých rokoch života. Neskôr neustále podlieha mnohým vonkajším a vnútorným vplyvom, dôsledkom čoho môže byť vznik rozličných porúch myelínovej štruktúry a funkcie [5].
Pokroky v klinickom a histopatologickom poznaní demyelinizujúcich porúch CNS sa datujú od druhej polovice 19. a začiatku 20. storočia. Objavy krok za krokom definovali jednotlivé ochorenia, ktoré sa týkali najskôr sklerózy multiplex. V období od roku 1897 do 1910 najprv Heubner a Pelizaeus a neskôr Merzbacher, Schilder a Krabbe popísali zriedkavé progresívne ochorenia u detí a nazvali ich difúznou sklerózou, keďže zmeny mali charakter difúznej demyelinizácie. Ďalším významným míľnikom boli pokroky v biochémii, začala sa vynárať koncepcia dedičných metabolických porúch a schopnosť vyšetriť hladinu enzýmov. Na tomto poli sú významné mená Suzuki a Suzuki (1970), ktorí dali do súvisu deficit galaktocerebrozidázy a Krabbeho chorobu. V roku 1964 Austin a kolektív dokázali deficit arylsulfatázy A ako príčinu metachromatickej leukodystrofie a nasledovali ďalšie objavy. S poznatkami sa vyvíjala aj klasifikácia demyelinizačných porúch. Prvý sa o ňu pokúsil v roku 1940 Hallervorden a rozlišoval exogénne a endogénne príčiny difúznej demyelinizácie [5].
Terminológia a delenie
Ochorenia myelínu CNS sa súhrne označujú termínom leukoencefalopatie. Zahŕňajú poruchy, pri ktorých zmeny v BH prevažujú alebo sú výlučné, pričom patofyziologický mechanizmus ani histopatologická báza nie sú rozhodujúce [4,5,7,8]. Van der Knaap a Valk vo svojej monografii navrhujú precízne rozdelenie leukoencefalopatií. Choroby BH delia na hereditárne a získané a leukodystrofie uvádzajú spoločne v časti hereditárnych porúch (členenie v tab. 1) [5]. Z klinického hľadiska považujeme za dôležité bližšie popísať a vyčleniť skupinu leukodystrofií. Pojem rozpracoval v roku 2004 Powers a leukodystrofiu definoval ako chorobu so známou alebo predpokladanou genetickou príčinou, progresívnym priebehom, prevažne splývavým postihnutím BH CNS a primárnym poškodením myelínu alebo buniek podieľajúcich sa na tvorbe myelínu [9]. Podmienky definície spĺňali dovtedy známe „klasické“ leukodystrofie: X‑adrenolekodystrofia, metachromatická leukodystrofia, globoidná leukodystrofia (Krabbeho choroba), Pelizaeus‑ Merzbacherova choroba, Canavanovej choroba a Alexandrova choroba a niekoľko nových primárnych porúch BH: leukoencefalopatia „vanishing white matter“, leukoencefalopatia s postihnutím mozgového kmeňa a vysokým laktátom, cystická leukoencefalopatia bez megalencefálie, hereditárna leukodystrofia so začiatkom v dospelosti. Zoznam sa postupne rozširoval o nové klinické jednotky [4,5,10 – 12].
![Súhrnná klasifikácia leukoencefalopatií (upravené podľa [5]).](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/906191a2989b613523873b66291e9b6c.png)
V roku 2014 Vanderver et al definíciu leukodystrofií upravili – pridali charakteristický MR nález, kde leukodystrofia musí mať na T2WI hyperintenzívny signál BH. Signál na T1WI môže byť variabilný: izo ‑ alebo hyperintenzívny, čo zodpovedá hypomyelininzujúcim leukodystrofiám, alebo hypointenzívny, ktorý majú demyelinizačné leukodystrofie.
Rozdelili poruchy BH na:
I. leukodystrofie (kompletný zoznam uvádzame v tab. 2),
![Zoznam leukodystrofií [upravené podľa 13].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/29dd3342937481a71ef826b2aec4ec48.png)

II. hereditárne ochorenia s významným postihnutím BH:
- vrodené vaskulopatie – CADASIL a iné,
- ochorenia CNS s primárnym postihnutím neurónov (kortex, ostatné sivé štruktúry) – neuronálna ceroid lipofuscinóza, gangliozidóza GM2 a iné,
- poruchy metabolizmu so zjavným sekundárnym postihnutím BH – organické acidémie poruchy aminokyselín a iné,
- choroby s postihnutím oboch, bielej aj sivej hmoty – MELAS a iné,
III. ostatné choroby BH:
- akvirované poruchy BH: infekčné, neinfekčné, toxické, traumatické [13].
V našej práci sme sa zamerali prednostne na „klasické“ leukodystrofie. Viedli nás k tomu terapeutické možnosti pre niektoré z nich. Väčšina liečebných postupov je podmienená zachytením včasného štádia ochorenia, preto sme popísali klinický a rádiologický pohľad, prípade niektoré vyšetrované zložky, ktoré sú pri danom ochorení špecifické a diagnosticky prínosné.
Nasleduje prehľad vybraných leukodystrofií.
Klinický a rádiologický obraz X‑adrenoleukodystrofie
X‑adrenoleukodystrofia (X ‑ ALD) je najčastejšia peroxizómová porucha charakterizovaná adrenálnou insuficienciou a neurologickou manifestáciou s veľmi variabilným klinickým obrazom. Je spôsobená defektom v géne ABCD1 na chromozóme Xq28, ktorý postihuje peroxizómovú beta‑oxidáciu mastných kyselín. Porucha vedie k zvýšeniu ukladania veľmi dlhých mastných kyselín (VLCFA) vo všetkých tkanivách, zvlášť v bielej hmote CNS, periférnych nervoch, v kortexe nadobličky a testes. VLCFA sú unikátne nasýtené mastné kyseliny a hrajú hlavnú úlohu v diagnóze a patogenéze ochorenia. Odhadovaná incidencia ochorenia je 1 : 17 000 [4,12,14,15]. Princípom dvoch rôznych klinických obrazov ochorenia je nezápalové axonálne poškodenie periférnych nervov a miechy verzus zápalové postihnutie BH mozgu. Je pravdepodobné, že axonálne postihnutie aké vidíme u AMN je vlastne primárne poškodenie pri X ‑ ALD. Avšak 50 % chlapcov zomiera na zápalové postihnutie mozgu ešte pred AMN manifestáciou [16,17]. Primárne postihnutie je najskôr spôsobené excesívnou akumuláciou VLCFA v tkanivách a môže poškodzovať stabilitu myelínového puzdra. VLCFA sa ukázali ako cytotoxické aj pre bunky nadobličky a zasahujú do mnohých membránových funkcií bunky. Začiatok zápalového postihnutia je s najväčšou pravdepodobnosťou ako „second ‑ hit“ prekrývajúci axonálnu patológiu a možnú myelínovú instabilitu, ktorá postupuje u všetkých chlapcov s X ‑ ALD. Cerebrálny inflamačný proces je agresívny, zápalový, demyelinizačný proces spojený s akumuláciou CD8 cytotoxických lymfocytov a stratou axónov [18] .
Klinický obraz a klasifikácia
Akumulácia VLCFA nastáva vo všetkých tkanivách organizmu, avšak postihnutá je hlavne kôra nadobličky a nervová sústava. Vznikajúce zmeny sú veľmi variabilné, čo sa týka veku i formy a môžu sa manifestovať nasledovne:
Detská cerebrálna adrenoleukodystrofia
Je najznámejšia zo všetkých foriem a dlho bola jedinou poznanou formou. Postihuje chlapcov s normálnym vývojom v predchorobí. Vekový priemer začiatku ochorenia je zvyčajne medzi 4. a 10. rokom s najvyšším výskytom okolo 7 ± 1,7 roku života. Začiatok je nenápadný, prítomná je porucha pozornosti a diskrétne zmeny v správaní, ktoré sa hodnotia ako ADHD. Postupne sa objavujú neurologické príznaky a epileptické záchvaty, tie môžu byť prvým príznakom ochorenia a neskôr sa objavujú u všetkých. Ochorenie postupuje rýchlo, vzniká kortikálna slepota, strata sluchu, spastická kvadruparéza so stratou motorických schopností a porucha hltania. Čas od prvých príznakov do plne rozvinutej choroby je krátky, 6 – 12 mesiacov. Chlapci zomierajú do dvoch rokoch od začiatku ochorenia [4,5,15,19,20].
Adrenomyeloneuropatia (AMN)
Je definovaná ako ochorenie dospelých, postihujúce dlhé dráhy miechy a periférne nervy – primárna, postihujúca približne 65 % rizikových mužov a často nie je diagnostikovaná. Začína medzi 20. a 30. rokom života ako pomaly postupujúca progresívna spastická paraparéza, distálne senzorické poruchy, inkontinencia a alopécia. Polyneuropatia nesie znaky prevažne axonálneho, senzorimotorického postihnutia. Popisujú sa zmeny aj na MR mozgu, podľa niektorých prác v 19 – 40 % prípadov a môžu ďalej progredovať tak, ako zmeny u cerebrálnej formy [17,21] .
Adolescentná a adultná cerebrálna forma
Sú podobné detskej cerebrálnej forme, ale priebeh je pomalší [22].
Spinocerebelárna forma
Radí sa ako varianta cerebrálnej AMN [5].
Primárna adrenálna insuficiencia
Addisonova choroba môže vznikať ako forma X ‑ ALD v každom veku a nesúvisí s objavením sa neurologických príznakov. Dochádza k deficitu glukokortikoidov a minerálokortikoidov so zvýšením hodnôt ACTH a znížením kortizolu. Forma primárnej adrenálnej insuficiencie je odhadovaná u 35 % pacientov s X ‑ ALD. Až u 70 % pacientov s X ‑ ALD sa v priebehu ochorenia rozvíja nadobličková nedostatočnosť. Ochorenie sa môže manifestovať hypotenziou a hypoglykémiou pri ľahkých infekciách, pričom adrenálna kríza sa môže končiť u chlapcov s nepoznanou X ‑ ALD fatálne. Vek vzniku je variabilný. Diagnosticky významná je hyperpigmentácia kože. Najmladšie popísané dojča s adrenálnym zlyhaním bolo vo veku šiestich mesiacov [4].
Asymptomatické prípady
Sú známe prípady so zvýšenými hodnotami VLCFA bez neurologickej a nadobličkovej patológie [4].
Heterozygotná forma
Ženy sú prenášačky a v detskom veku nemajú príznaky, ale v dospelosti sa u nich môžu vyvíjať myelopatické a neuropatické zmeny. Viac ako 50 % žien má ťažkosti s chôdzou, pridružujú sa dyzestézie a problémy s močením. Zriedkavý je vývoj adrenálnej insuficiencie a cerebrálneho postihnutia, preto sa vyšetrenie MR a adrenálnych funkcií nevyžaduje. Súhrn v tab. 3 a 4 [22 – 25].
![Fenotyp X-ALD u mužov (upravené podľa [4]).](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/57ca997a74ac3606f3d9bcdb40e6afd0.png)
![Fenotyp X-ALD u žien (upravené podľa [22]).](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/5f1729786052a7c17dc525b9dd43a594.png)
Laboratórne vyšetrenia
Diagnóza ochorenia závisí na dôkaze abnormálne vysokých hladín VLCFA v plazme, v kožných fibroblastoch či iných tkanivách. Pre prax stačí dôkaz vysokých hodnôt v plazme. Muži s ALD majú od prvého dňa života vyššie hodnoty VLCFA, čo je charakteristický znak ochorenia. Nález zvýšených VLCFA nie je špecifický pre X ‑ ADL, je obrazom ochorení s poruchou peroxizómovej beta‑oxidácie. Hladina VLCFA môže byť zvýšená aj u žien. Falošne negatívne výsledky môžu byť v dôsledku hemolýzy a z diétnych dôvodov – ketogénna diéta, preto je odporúčaná DNA analýza [4,25,26].
MR obraz pri X‑adrenoleukodystrofii
Rádiologický obraz X ‑ ALD má pre ochorenie diagnostický význam. Zmeny na MR mozgu môžu mať rozličnú podobu. Najčastejší nález pacientov s cerebrálnou formou je obraz parieto ‑ okcipitálnych lézií BH, hyperintenzívny v T2WI a hypointenzívny v T1WI. Zmeny začínajú v splenium corporis callosi, pričom fibrae arcuatae – U vlákna sú zachované. Väčšina pacientov má v mieste postihnutia dve zóny – prednú, kde je demyelinizácia menej pokročilá, a zadnú, kde je výraznejšie postihnutie. Po podaní kontrastu sa okolo výrazne postihnutej zóny tvorí lem, ktorý oddeľuje menej postihnutú časť od zdravej. Tri zóny na MR sú v zhode s troma zónami, ktoré sa rozpoznávajú histologicky. Vonkajšia je v procese aktívnej demyelinizácie, stredná je v stave výrazného zápalu. Vnútorná zóna je kompletne demyelinizovaná, v nej môžu byť kalcifikáty a dutiny. Lézie sú prevažne symetrické, postupujú frontálne a v konečnom štádiu je prítomná výrazná atrofia. Cerebelum nie je vo včasnom štádiu postihnuté na rozdiel od mozgového kmeňa. Postihnuté sú okcipito ‑ parieto ‑ temporo‑pontínne dráhy, pyramídový trakt, brachium colliculus inferior a superior a lemniscus laterlis. Tento obraz je prítomný u 80 % detských cerebrálnych X ‑ ALD [5,15,22]. Asi 15 % pacientov môže mať obrátený obraz a zmeny začínajú vo frontálnej oblasti so súčasným postihnutím rostrum a genu corpus callosum. Vtedy je postihnutá capsula interna, predné rameno a tr. frontopontinus. Veľmi zriedkavo môže byť v tomto veku postihnutie cerebelárnej BH, stredných cerebelárnych pedunklov a traktov mozgového kmeňa. Raritné je súčasné postihnutie parietálnej aj frontálnej oblasti. U pacientov s typickými zmenami parieto ‑ okcipitálne býva zvyčajne rýchla progresia stavu [5,22,27,28].
U dospelých s AMN nie je postihnutá hlboká BH, ale sú poškodené frontopontínne a kortikospináne vlákna capsula interna a vlákna v mozgovom kmeni, postupne pribúdajú zmeny v cerebelárnych pedunkuloch a slenium corporis callosi. Na MR miechy je prítomná atrofia [5,22,29].
Loes et al [29] vytvorili 34 - bodový skórovací systém na odstupňovanie MR zmien (detailný popis v tab. 5). Číselné ohodnotenie odráža progresiu a presná analýza rozhoduje spolu s klinickým obrazom o indikácii transplantačnej liečby, ktorá stále zohráva podstatnú úlohu v terapii [5,29,30].
![Loes skóre pri X-ALD (upravené podľa [29]).](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/86b390e2a18a2efdb2e99059c06ad30a.png)
Prítomnosť kontrastného enhancementu je dôkazom aktivity ochorenia a v 90 % sa spája s jeho progresiou [27]. FLAIR ukazuje hyperintenzívny signál postihnutých zón. Pri zobrazovaní a posudzovaní aktivity procesu má svoj význam DWI, je totiž citlivejšie ako konvenčné MR a spolu s reštrikciou difúzie poukazuje na aktívny proces demyelinizácie. Prediktívnu hodnotu má tiež MRS. Klesajúci pomer NAA/ cholín je známkou hroziacej demyelinizácie. Pri monitorovaní detských pacientov s asymptomatickou cerebrálnou formou je odporúčaná kombinácia MR a MRS každých šesť mesiacov až do veku 10 rokov. Od 10. do 20. roku, ak je stav stabilný, je vhodné robiť MR jedenkrát za rok [19].
Až u 50 % chorých sa nemusí vyvinúť závažné cerebrálne postihnutie, preto sa neodporúča transplantačná liečba u všetkých pacientov. V prípade progresie nálezu pri hodnote Loes skóre tri a viac bodov je indikovaná urýchlená transplantácia kostnej drene. Liečba transplantáciou nie je indikovaná pri veľmi akútnej forme alebo ťažšom cerebrálnom poškodení – Loes skóre 10 a viac [5,28 – 31].
Hlavné znaky MR obrazu (obr. 1)

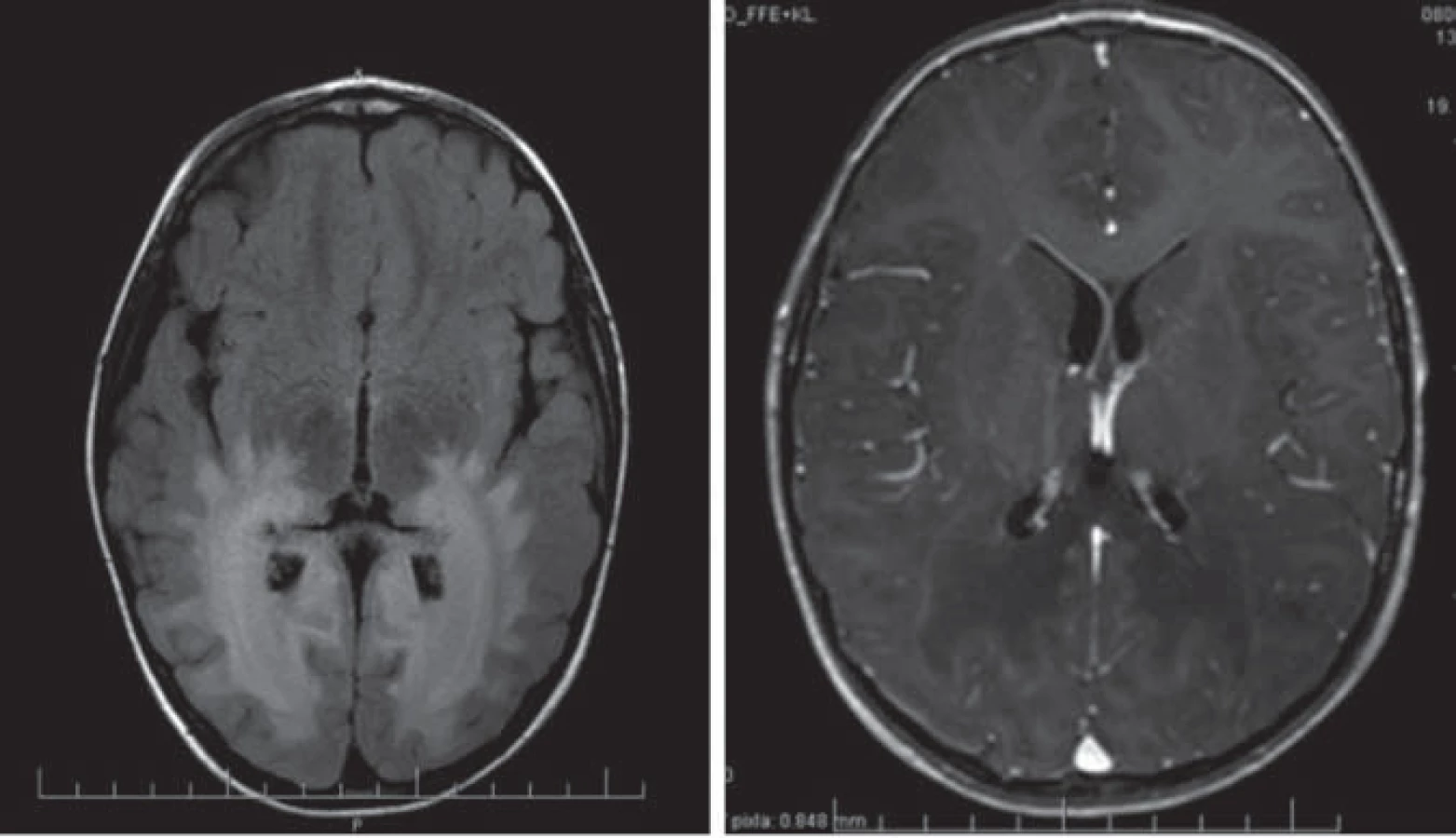
Enhancujúca peritrigonálna demyelinizácia BH, lokalizácia okcipitálne peritirgonálne a v oblasti splenium corporis callosi, typicky nepostihuje U vlákna, lézie sú symetrické, splývavé. Prítomnosť kontrastného enhancementu je dôkazom aktivity ochorenia. Reštrikcia difúzie v DWI poukazuje na aktívny proces demyelinizácie. Pokles NAA v MRS je známkou hroziacej demyelinizácie [5,22].
Diferenciálna diagnostika podľa MR obrazu
Alexandrova choroba pre frontálnu lokalitu a kontrastný enhancement, MLD, PVL, hypoglykemická encefalopatia, ochorenia BH so zvýšeným laktátom [5,11, 13,22,28,32,33].
Liečba
Pre ochorenie nie je kauzálna liečba. Dostupná je:
- symptomatická terapia u pacientov s plne rozvinutou neurologickou symptomatológiou;
- hormonálna substitúcia; kortikoidy sú nevyhnutné a život zachraňujúce u pacientov s adrenálnou nedostatočnosťou;
- transplantácia kostnej drene je prospešná pre pacientov s X ‑ cerebrálnou ALD detského a adolescentného veku s MR nálezom aktívnych zmien a miernym klinickým obrazom. Pri pokročilom postihnutí liečba neprináša očakávané zlepšenie. Zákrok je závažný, a preto nie je odporúčaný pre pacientov bez neurologickej symptomatológie;
- diétna reštrikcia a inhibícia endogénnej syntézy VLCFA Lorenzovým olejom (zmesi glyceryl trioleate a glyceryl trierucate v pomere 4 : 1) u asymptomatických pacientov na oddialenie neurologického postihnutia;
- sľubnou je perspektíva génovej terapie pomocou autológnej transplantácie s geneticky opravenými vlastnými kmeňovými bunkami a tiež transplantácia oligodendrocytov ako i pluripotentných kmeňových buniek na reparovanie poškodených tkanív [5,34 – 36].
Klinický a rádiologický obraz metachromatickej leukodystrofie
Metachromatická leukodystrofia (MLD) je lyzozómová sfingolipidóza s autozómovo recesívnou dedičnosťou, spôsobená deficitom enzýmu arylsulfatázy A. Sulfatid je prítomný vo veľkom množstve v myelíne a je pravdepodobne zodpovedný za správnu organizáciu paranodálneho axogliálneho spojenia a správne usporiadanie iónových kanálov tejto oblasti. Deficit arysulfatázy A spôsobuje poruchu v odbúravaní sulfatidu. Výskyt ochorenia je 1 : 100 000 až 1 : 40 000. Gén pre arylsulfatázu A ARSA je lokalizovaný na chromozóme 22q13.3 [5,12,37,38].
Klinické príznaky a laboratórne vyšetrenia
MLD je heterogénne ochorenie s rozdielnym klinickým obrazom v závislosti od veku nástupu. Podľa autorov Gieselmann a Krägeloh ‑ Mann [39] je navrhnuté delenie na: neskorú infantilnú formu – začína pred tretím rokom, juvenilnú formu – medzi tretím až 16. rokom, a adultnú formu – po 16. roku [5,39,40].
Neskorá infantilná forma
Začína sa medzi prvým a druhým rokom, je uvádzaná ako najčastejšia. Vznikajú abnormálne pohybové vzorce s rekurváciou kolien pri chôdzi, charakteristické sú známky spasticity s hypo ‑ až areflexiou, nasleduje zhoršenie kognície a jemnej motoriky, pridáva sa porucha zraku zapríčinená optickou atrofiou, nystagmus a epileptické záchvaty. Pacienti umierajú v decerebračnom stave [5,41].
Juvenilná forma
Začína zhoršovaním školského prospechu, neadekvátnym správaním alebo psychiatrickými príznakmi. U väčšiny pacientov je len ľahké postihnutie koordinačných schopností, znížené reflexy, postihnutie periférnych nervov s pozitívnym Babinskim príznakom bez spastickej postúry. Pomalý priebeh neskôr strieda závažné zhoršenie s jednoznačnou spasticitou.
Adultná forma
Pri adultnej forme MLD je klinický priebeh pomalší, ochorenie je charakterizované postupným rozpadom intelektových schopností, emočnou instabilitou, psychiatrickou symptomatológiou (bludy a halucinácie), čo môže viesť k mylnej diagnóze psychózy (schizofrénia, depresia). Neskôr pribúda spastická kvadruparéza s ataxiou a extrapyramídovými prejavmi. Periférna neuropatia často chýba. Prehlbuje sa demencia, pacienti strácajú kontakt s okolím. Vo finálnom vegetatívnom štádiu sú v dekortikačnej alebo decerebračnej postúre [5,39,40,42,43].
Špecifické nálezy: postihnutie periférneho nervového systému. Pre včasné formy ochorenia je neuropatia takmer pravidlom, adultné formy môžu mať kondukčné skúšky v norme [5]. Periférna neuropatia vedie k spomaleniu rýchlosti vedenia, predĺženiu latencie, zníženiu amplitúdy [44], senzorické vlákna sú ťažšie postihnuté ako motorické [45]. Neskorá detská forma môže mať uvedené zmeny prítomné už vo včasnom štádiu, ale u niektorých pacientov môžu aj chýbať [46]. Léziu v CNS ukazujú patologické nálezy BAEP [39]. MLD je neurologické ochorenie, ale niektorí pacienti trpia už v mladosti cholecystolitiázou a polypmi žlčníka, ktoré sú s najväčšou pravdepodobnosťou spôsobené postihnutím autonómneho nervového systému [47]. Celkové bielkoviny v likvore sú zvýšené u neskorej infantilnej, veľmi často aj pri juvenilnej forme, zriedka však u pacientov s adultnou formou [5].
U všetkých pacientov s MLD je prítomná zvýšená exkrécia sulfatidov v moči. Aktivita arylsulfatázy A je nízka s výnimkou poruchy aktivačného faktoru sapozínu B, kedy môže byť normálna. Stanovuje sa v leukocytoch periférnej krvi a z kožných fibroblastovh. Genetické vyšetrenie potvrdí mutácie v géne ARSA [5,38,39]. Hladina zníženej aktivity arylsulfatázy A nedokazuje MLD, keďže až 0,2 – 0,5 % zdravej populácie má pseudodeficit. Pre diagnózu je rozhodujúci dôkaz vysokého obsahu sulfatidu v moči, ktorý odlíši deficit od pseudodeficitu a tiež deficit sapozínu B [48].
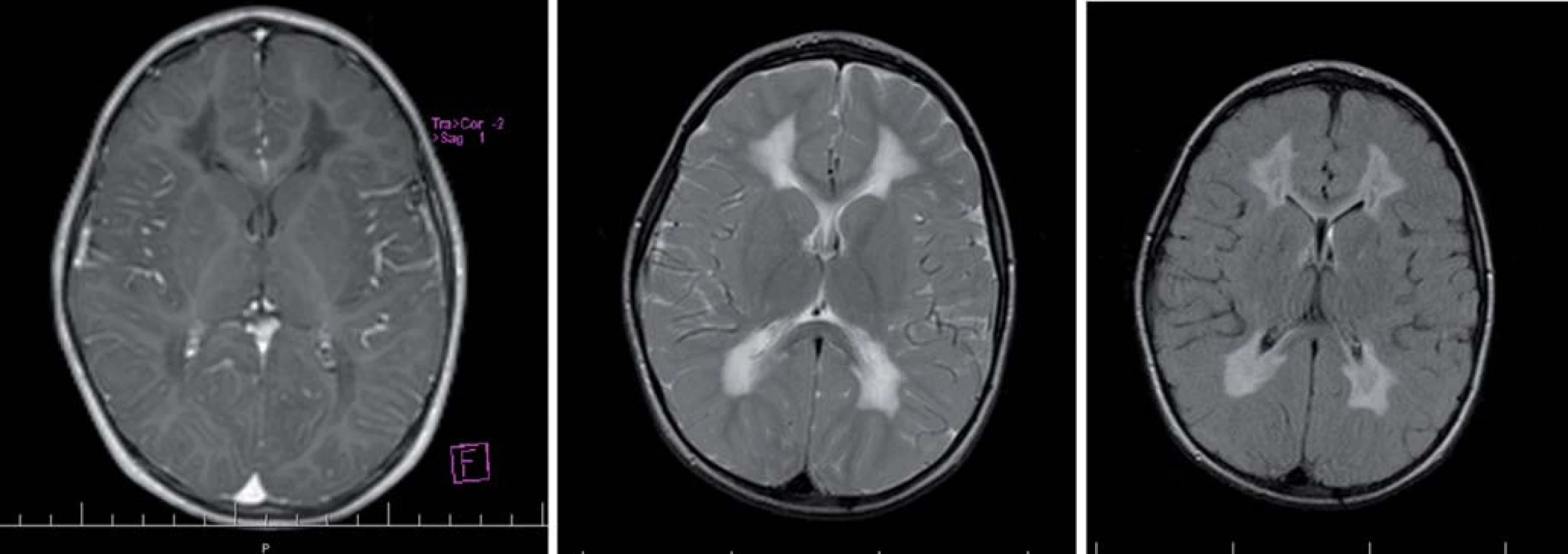
MR obraz pri metachromatickej leukodystrofii
U pacientov s neskorou infantilnou formou sú prítomné už vo včasnom štádiu ochorenia symetrické splývavé zmeny vysokého signálu v T2WI a FLAIR a hyposignálu na T1WI obrazoch, vznikajúcich najprv periventrikulárne. Neskôr sa zmena signálu rozširuje do vnútra hemisfér a cerebrálnej BH. U vlákna – fibrae arcuatae nie sú typicky postihnuté, môžu byť zasiahnuté v neskorších štádiách ochorenia. V abnormálne zmenenej BH sú vidieť pruhy nižšej intenzity, uložené radiálne. Corpus callosum je postihnuté hneď od začiatku, zatiaľ čo subkortikálne oblasti sú zmenené až v pokročilom štádiu. Demyelinizačné zmeny je vidieť aj na zadných rohoch capsula interna, descendentných pyramídových dráhach a cerebelárnej BH. Abnormálna BH nie je kontrastná [5,22,38,39,49].
MRS pri MLD ukazuje zvýšené hodnoty Cho a redukciu NAA v sivej a BH je ako neurálnou a axonálnou stratou, mIns stúpa so vznikajúcou gliózou [5,39]. Pri juvenilnom type s neskorším začiatkom môžu prevažovať frontálne zmeny a u včasného skôr okcipitálne, ale vždy je postihnutie difúzne a fibrae arcutae sú zachované. Typicky prítomné radiálne pásy – tigrovitý vzhľad vnútri abnormálnej BH nemusia byť u všetkých pacientov a strácajú sa v pokročilom štádiu. Niektorí pacienti majú postihnutie BH cerebela. Lézie po kontraste neenhancujú. Postihnutie sivej hmoty je nezávažné [37].
Hlavné znaky MR obrazu (obr. 2)
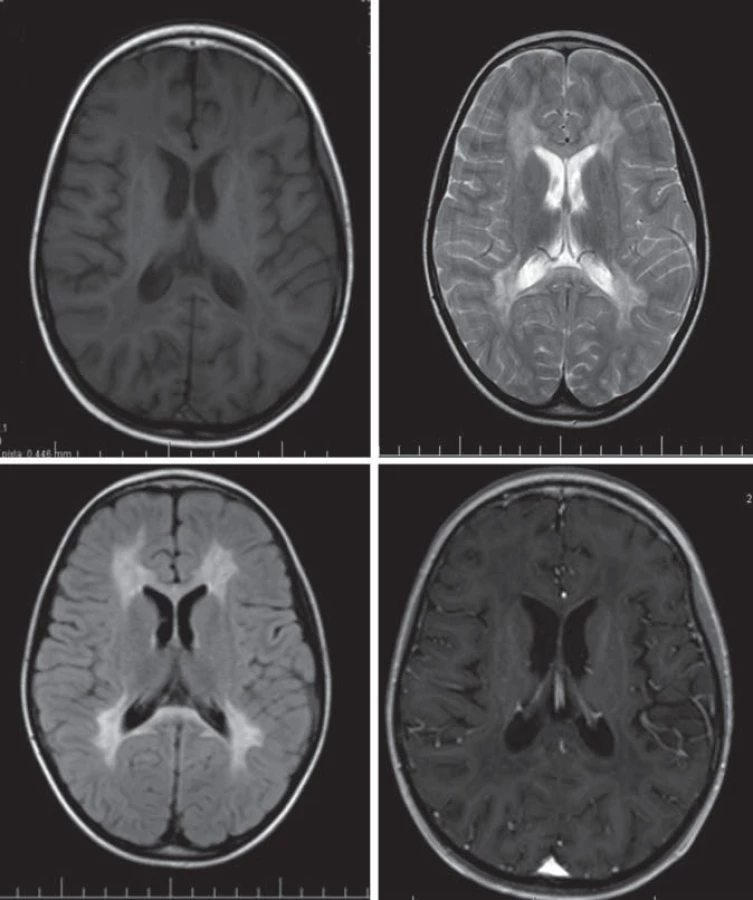
Splývavý motýlí vzhľad zvýšeného signálu T2WI, uloženie je v periventrikulárnej a hlbokej cerebrálnej BH, na začiatku bez postihnutia neskôr s postihnutím U vláken. Zmeny sú nekontrastné.
Diferenciálna diagnostika MR nálezu
Krabbeho globoidná leukodystrofia, X ‑ ALD, difúzne zápalové postihnutie typu ADEM, kongenitálna CMV, PVL, PMD.
Najťažšie je rozlíšenie Krabbeho choroby, ktorej obraz je veľmi podobný, niekedy môže mať aj radiálne žíhanie. Nápomocné je natívne CT. Pri globoidnej leukodystrofii je zvýšený signál thalamu, corona radiata, capsula interna a cerebela, ktorý sa pri MLD nikdy nevyskytuje. Kontrastné MR nám pomôže odlíšiť zápalové postihnutie a cerebrálnu formu X ‑ ALD, kde lézie jasne enhancujú [5,22,28,29,32].
Liečba
Existuje veľa pokusov zmeniť prirodzený priebeh MLD. Transplantácia kostnej drene sa používa u stále rastúceho počtu pacientov s cieľom korigovať hladinu arylsulfatázy A, ako aj zastaviť progresiu [40]. Výsledok závisí od štádia ochorenia v čase transplantácie. Prospešná je transplantácia v presymptomatickom štádiu u detí s neskorou infantilnou formou alebo po včasnom objavení príznakov pri juvenilnej forme. Pri trvaní ochorenia viac ako 6 – 12 mesiacov sa u detí s juvenilnou formou vôbec neodporúča. Pri správnom načasovaní transplantácie sa popisuje stabilizácia stavu a zlepšenie MR nálezu [5,39]. Ďalšia možnosť je enzýmová terapia. V roku 2011 boli prvýkrát zverejnené výsledky I/ II fázy klinického skúšania intratékálneho podávania rekombinantnej arylsulfatázy A u 13 detí [50]. Génová liečba za pomoci vektora zabezpečuje dostatočnú tvorbu enzýmu, prvé pokusy práve prebiehajú u pacientov s neskorou infantilnou a juvenilnou formou [51,52].
Klinický a rádiologický obraz Krabbeho globoidnej leukodystrofie
Krabbeho globoidná leukodystrofia (GLD) je autozómovo recesívne lyzozómové ochorenie postihujúce BH centrálneho a periférneho nervového systému. V USA a Európe je odhadovaná incidencia 1 : 100 000. Názov globoidná je odvodený z názvu globoidnej bunky, čo sú abnormálne „upratovacie“ bunky. Príčinou ochorenia je deficit galaktocerebrozidázy (GALC), ktorý vedie k nahromadeniu galaktozylceramidu. Galaktozylceramid a sulfatid sú dva hlavné glykosfingolipidy myelínu. Sú syntetizované počas aktívnej myelinizácie a správny pomer týchto lipidov je nutný pre stabilný myelín. Globoidné bunky obsahujú vysokú koncentráciu galaktozylceramidu, v patogenéze je však pravdepodobne najdôležitejší toxický vplyv psychozínu (galaktozilpsychozín), ktorý inhibuje proteín kinázu C, čím narúša činnosť peroxizómov, aktivuje fosfolipázu A2, ktorá indukuje apoptózu mnohých typov buniek vrátane oligodendrocytov a Schwannových buniek. Normálny mozog obsahuje minimálne množstvo psychozínu, pri deficite GALC (GALC hydrolyzuje psychozín) sa koncentrácia v BH mozgu a periférnych nervoch zväčšuje 10 - až 20 - násobne. Gén GALC, kódujúci galaktocerebrozidázu, je na chromozóme 14q3.1 [53 – 55].
Klinické príznaky a laboratórne vyšetrenia
Rozlišujeme niekoľko foriem GLD:
- kongenitálna,
- včasná infantilná – medzi prvým a šiestym mesiacom, je považovaná za klasický a najčastejší typ,
- neskorá infantilná – medzi šiestym mesiacom a tretím rokom,
- juvenilná medzi štvrtým a 10. rokom,
- adolescentná – adultná od 10. roku [4].
Včasná infantilná forma
Je charakterizovaná tromi štádiami: prvé – celková iritabilita, s obdobiami kriku, zvlášť pri kŕmení a pestovaní, prítomná je extrémna citlivosť na svetlo a hluk, hypertonus končatín, zastavenie vývoja, epizódy vysokej teploty bez infekcie. V druhom štádiu vzniká opistotonus, myoklónie končatín, kontinuálne zvýšená teplota a regres vo všetkých schopnostiach. V treťom štádiu je decerebrácia bez schopnosti pohybu, hypotónia, kachexia a cerebrálna hyperpyrexia. Priemerný vek vzniku je okolo štvrtého mesiaca života a smrť nastáva v prvom roku.
Neskorá infantilná a juvenilná forma
Ďalšie formy ako neskorá infantilná a juvenilná majú v popredí problémy s videním, ataxiu, regres, spastickú parézu, parestézie a zmiešanú senzorimotorickú demyelinizačnú periférnu neuropatiu [56]. Pri juvenilnej forme sa u detí vyvíja strata zraku spolu s hemiparézou, ataxiou a psychomotorickým regresom s možným rýchlym nástupom. Niektorí pacienti sú bez výraznejšej zmeny dlhé obdobie, u iných je neustála progresia a postupný vývoj do vegetatívneho stavu. Priebeh včasnej infantilnej formy je typický, avšak priebeh neskorej infantilnej formy varíruje a zdá sa, že je ovplyvnená vonkajšími faktormi (infekcia, traumatické poškodenie mozgu). Sú uvádzané zriedkavé formy periférnej neuropatie ako izolovaný príznak neskorej infantilnej formy [4,57,58].
Adolescentá a adultná forma
Môže sa prejaviť stratou manuálnych zručností, pálením, parestéziami a slabosťou končatín často bez intelektuálneho zhoršenia; alebo postupným psychickým a fyzickým zhoršovaním [54]. Pri včasnej infantilnej forme je výrazne zvýšená hodnota celkových bielkovín v likvore, ešte pred vznikom príznakov, hodnota bielkovín v likvore je nižšia u neskoršej infantilnej formy. Významnú cenu pri vyšetrení majú znížené rýchlosti vedenia. Hoci viac ako 99 % pacientov s Krabbeho chorobou má mutáciu v GALC géne, je niekoľko prípadov podobných globoidnej leukodystrofii spôsobených mutáciou sapozínu A, možno i C [53,59]. Oba sú nízkomolekulové aktivačné proteíny, potrebné pre pôsobenie GALC. Diagnostickou metódou je meranie enzýmovej aktivity galaktocerebrozidázy v leukocytoch periférnej krvi alebo v kožných fibroblastoch. Enzýmovú diagnostiku môžu komplikovať prípady s pseudodeficitom GALC. Od roku 2006 je možné skríningové enzýmové vyšetrenie zo suchej kvapky krvi tandemovou hmotnostnou spektroskopiou [54,60].
MR obraz pri Krabbeho globoidnej leukodystrofii
V prípade klasického typu so včasným začiatkom sú významné charakteristické znaky na natívnom CT mozgu [61]. V prvom štádiu je zvýšenie denzity thalamov, corona radiata, menej často zadného ramienka capsula interna, spôsobené hromadením globoidných buniek a glie. V druhom a treťom štádiu sú symetrické periventrikulárne arey zníženej denzity BH hemisfér a corpus medullare cerebela.
Vo včasnom štádiu je na MR zvýšený signál v T2WI periventrikulárne so zachovanými U vláknami, prítomné sú radiálne usporiadané pásy, ktoré však nie sú podmienkou, postihnuté je aj corpus callosum, pyramídový trakt, zadné ramienko capsula interna, cerebelárna biela hmota, hilus a nucleus dentatus [62]. Vo včasnom štádiu skorej infantilnej formy pre nedostatočnú myelinizáciu môže byť MR klamlivo normálne, pomocou je CT [5,61]. V prípade neskorej infantilnej a juvenilnej formy sú prítomné na CT hyperdenzity thalamov, BG a okcipitálnych lalokov. Na MR sa zobrazia splývavé lézie zvýšenej intenzity s maximom v parieto ‑ okcipitálnej periventrikulárnej BH postupujúce temporálne s postihnutím corpus callosum. U vlákna sú zachované, postihnutá je posteriorná časť internej kapsuly a pyramídový trakt v mozgovom kmeni. V pokročilom štádiu sa rozširujú a prehlbujú zmeny v BH a nastupuje globálna atrofia s rozšírením komôr a subarachnoidálnych priestorov [5]. V prípade adultných foriem je dlhý čas MR normálny, neskôr sa môžu objaviť zmeny pyramídovej dráhy posteriornej časti capsula interna a mozgového kmeňa symetricky alebo asymetricky. Variabilne sa môžu objaviť jemné zmeny ukazujúce enhancement v hraničných zónach postihnutého a zdravého tkaniva hlbokej BH, prevažne však enhancement nie je prítomný [62].
Hlavné znaky MR obrazu (obr. 3)
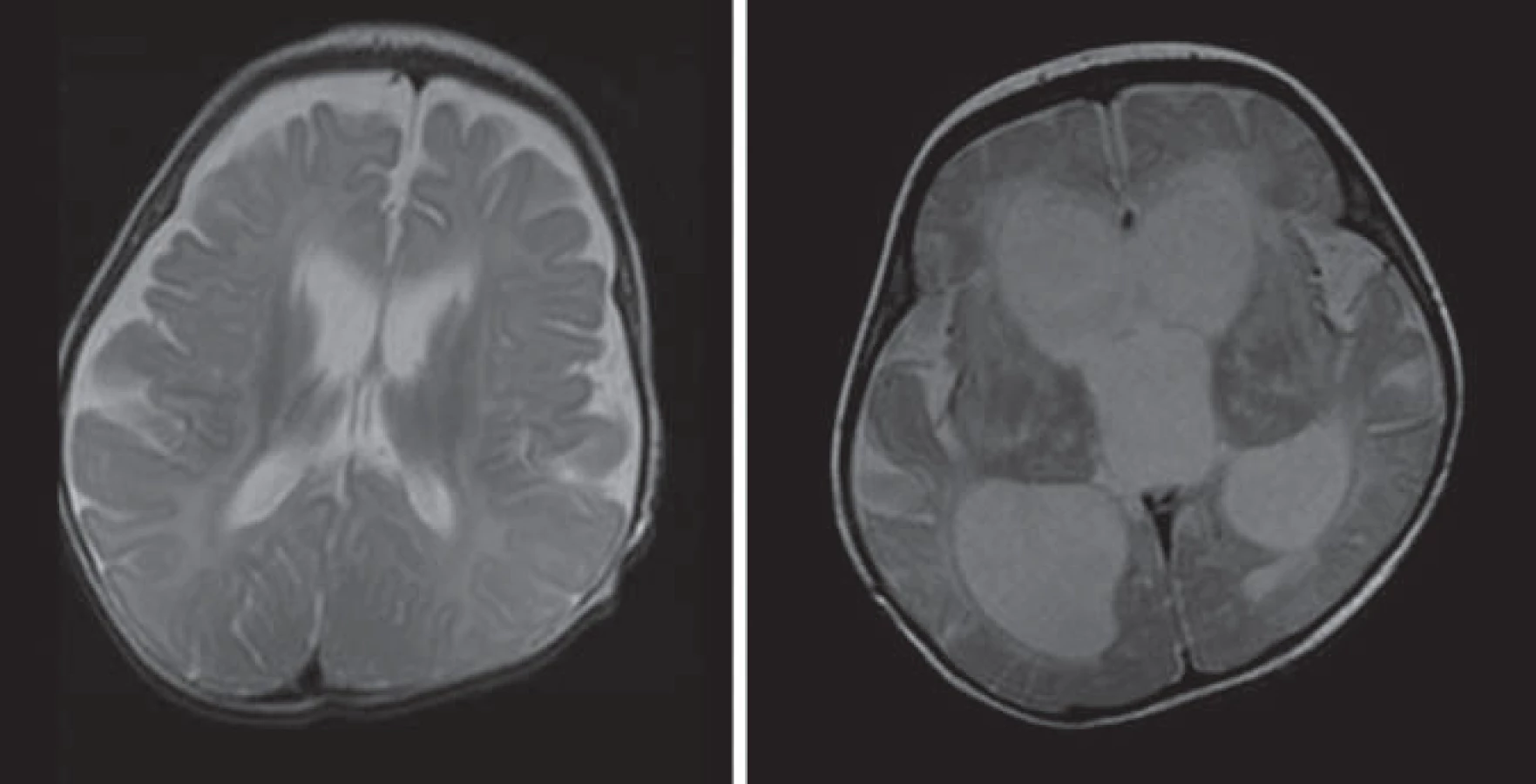
Hyperdenzita thalamov, BG, corona radiata, pyramídovej dráhy na CT. Splývavé lézie zvýšenej intenzity signálu na T2WI a FLAIR parietookcipitálej periventrikulárnej BH s postihnutím corpus callosum, zmeny sú nekontrastné [5,28,30].
Diferenciálna diagnostika MR nálezu
MLD, včasný začiatok GM1, GM2 (podoba pre hyperdenzita thalamov a bazálnych ganglií na CT), X ‑ cerebrálna adrenoleukodystrofia (okcipitálne zmeny pri neskoršom začiatku globoidnej leukodystrofie), neuronálna ceroid lipofuscinóza a staus marmoratus (hypoxické postihnutie novorodencov v oblasti BG a thalamov) [5,15,46,63,64].
Liečba
Ako úspešná liečba sa ukazuje transplantácia krvotvorných buniek asymptomatických pacientov pri infantilnej forme a pri včasných príznakoch foriem s neskorým začiatkom. Transplantácia u pacientov s rozvinutou klinickou symptomatológiou, ako u iných leukodystrofií je spojená s vysokou mortalitou a morbiditou. Iná liečba GLD nie je dostupná, takže je potrebná včasná diagnóza. Neskoršie štádiá je možné liečiť len symptomaticky [54].
Klinický a rádiologický obraz Canavanovej choroby
Canavanovej choroba je autozómovo recesívne ochorenie zapríčinené deficitom aspartoacylázy, ktorý spôsobuje spongiformnú degeneráciu BH mozgu. Deficit enzýmu vedie k inhibícii hydrolýzy kyseliny N ‑ acetylaspartovej a hromadeniu metabolitu N ‑ acetylaspartátu vo vysokom množstve v moči, plazme, likvore, mozgu a je zároveň hlavným diagnostickým znakom ochorenia. Gén pre aspartoacylázu ASPA je lokalizovaný na krátkom ramene chromozómu 17p13 – ter. Epidemiologicky ide o zriedkavé panetnické ochorenie s najväčším výskytom 1 : 5 600 u aškenázskych Židov [65–67].
Klinický obraz a laboratórne vyšetrenia
Rozlišujú sa tri formy ochorenia: kongenitálna, infantilná a juvenilná. Najčastejšia je infantilná forma. Pre ochorenie je typická makrocefalia s obrazom výraznej axiálnej hypotónie. Deti sa vyvíjajú do šiesteho mesiaca normálne, následne vzniká výrazná hypotónia, chudobná spontánna motorická aktivita, problémy s kŕmením, poruchy videnia, prítomný je zjavný regres schopností, neskôr sa objavuje spasticita končatín, ale axiálne hypotónia pretrváva. Choreoatetóza, tonické, myoklonické záchvaty, autonómne krízy s vracaním a hyperpyrexia s poruchou termoregulačného centra sú ďalšie sprievodné prejavy choroby. Dĺžka života je približne štyri až 10 rokov. Kongenitálna forma sa prejavuje výraznou hypotóniou, neschopnosťou sať, prehĺtať a dráždivosťou. Deti umierajú v prvých dňoch a týždňoch. Juvenilná forma sa prejavuje vo veku, kedy je už vyvinutá chôdza a vývoj sa vždy oneskoruje [5,65,68]. Ojedinele sa objavili aj prípady Canavanovej choroby s miernejším priebehom [69]. Diagnóza je založená na náleze zvýšených hodnôt N ‑ acetylaspartátu v moči a plazme. Celkové bielkoviny v likvore môžu byť elevované. V EEG je zvyčajne nález difúzne pomalej aktivity. Zrakové evokované potenciály majú patologický nález alebo sú nevýbavné [5,68,70].
N ‑ acetylaspartát je tiež zvýšený v plazme a likvore, ale na stanovenie diagnózy je postačujúci nález v moči. Exkrécia N ‑ acetylaspartátu býva zvýšená rádovo na 1 440,5 ± 873,1 mmol/ mol kreatinínu v porovnaní s kontrolami, kde sú hodnoty 23,5 ± 16,1 mmol/ mol kreatinínu. Určenie aktivity enzýmu nie je pre diagnózu rozhodujúce [67,68].
MR obraz pri Canavanovej leukodystrofii
U detí s Canavanovej chorobou sú prítomné difúzne zmeny zvýšeného signálu v T2WI lokalizované subkortikálne cerebrálne a cerebelárne. Na T1WI sú zmeny hyposignálne. Postihnutie je symetrické, difúzne. Centrálne časti, ako periventrikulárna BH a corpus callosum zostávajú dlho zachované. V priebehu času vzniká atrofia, o čom svedčí rozširujúci sa subarachnoidálny priestor a komorový systém. Prítomný je typický obraz s bilaterálnym postihnutím thalamov a globus pallidus, mozgového kmeňa a cerebela [5,66,71].
Na DWI sa na začiatku ochorenia ukazuje reštrikcia difúzie (zvýšený signál so znížením hodnôt ADC) a v neskoršom štádiu zvýšenie difúzie (znížený signál na DWI a zvýšenie hodnôt ADC) [5,28]. Na MRS je prítomný vysoký pík NAA, hodnoty cholínu sú znížené, čo poukazuje na hypomyelinizáciu. U mnohých ostatných ochorení je NAA normálne alebo znížené, ide teda o špecifický marker ochorenia [5,71].Vo včasnom veku môže byť ale nález nevýrazný a často interpretovaný ako normálny [72].
Hlavné znaky MR obrazu (obr. 4)
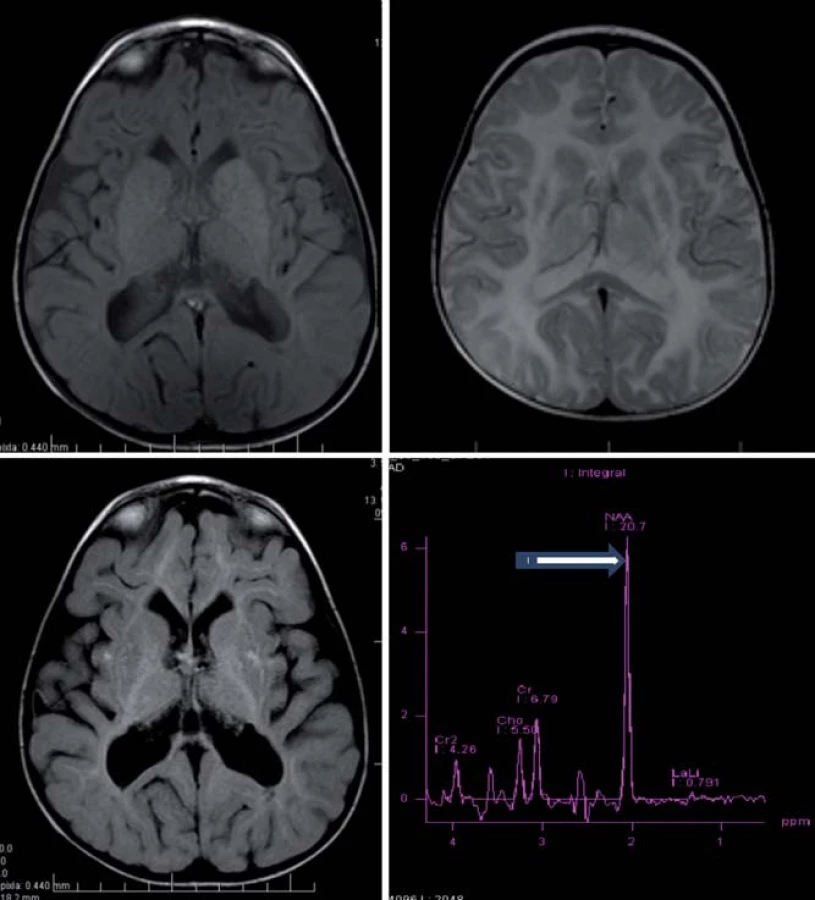
Megalencefalia s difúznym zvýšením signálu v T2WI a so včasným postihnutím U vláken, hyperintenzívne lézie globus pallidus a thalamu pri zachovanom nucleus caudatus a putamen [5,28].
Diferenciálna diagnostika MR nálezu
MSUD, Alexandrova choroba, leukoencefalopatia so subkortikálnymi cystami, kongenitálna muskulárna dystrofia, PKAN, MLD [4,5,13,15,28,32,63,64].
Liečba
Pre ochorenie nie je známa špecifická liečba. Pri typickom ochorení je prognóza nepriaznivá. Väčšina detí umiera v prvej dekáde [5]. Sú popisované pokusy o génovú terapiu použitím adeno‑asociovaného vírusu 2 (AAV ‑ 2) ako vektora pre aspartoacylázu. Liečba bola aplikovaná podľa dostupných dát u 10 detí, z ktorých sedem nevytvorilo protilátky proti AAV ‑ 2. Enzýmová liečba rekombinantnou aspartoacylázou je v štádiu skúšania na zvieracích modeloch [73,74].
Klinický a rádiologický obraz Pelizaeus‑ Merzbacherovej choroby
Pelizaeus ‑ Merzbacherova choroba (PMD) je zriedkavé X ‑ viazané neurologické ochorenie postihujúce myelinizáciu CNS, spôsobené zmenami PLP1 génu na chromozóme Xq22, ktorý kóduje proteolipidový proteín. Incidencia ochorenia nie je presne známa; odhaduje sa na 1 : 400 000. Pacient s PMD má mozog príliš „ľahký“ k veku a má známky difúznej atrofie postihujúcej obe hemisféry, obzvlášť kmeň a cerebelum. Mikroskopické skúšky dokazujú stratu myelínu vo všetkých častiach CNS. Patologický obraz je rovnaký u všetkých typov PMD, varíruje len intenzita postihnutia v závislosti od závažnosti neurologického obrazu. Najťažšie zmeny, až absencia myelínu sú u vrodenej formy PMD [5,75,76].
Klinický obraz a laboratórne vyšetrenia
Zvyčajne sa delí na tri typy: klasický typ I, vrodený typ II a mierny typ III.
Klasický typ I
Začína sa v prvom roku života. Postihuje takmer výlučne chlapcov. Začína sa nystagmoidnými očnými pohybmi, ktoré sú opisované ako „tancujúce“ alebo „túlavé“ pohyby. U niektorých detí vzniká stridor (z obrny laryngeálnych abduktorov alebo v dôsledku malácie). Prítomné je vývojové oneskorovanie, spomalenie rastu hlavy a záchvaty. V neskoršom veku sa pridružuje kvadruparéza, ataxia, extrapyramídové príznaky, dystónia a choreoatetóza. Časté je postihnutie vízu spolu s optickou atrofiou. Nystagmoidné pohyby postupne miznú. Sociálny kontakt je pomerne dobre zachovaný a jeho úroveň prevyšuje pohybové schopnosti. Isté zlepšovanie stavu je možno očakávať do 10. až 12. roku, potom nastáva trvalé zhoršovanie motorické aj mentálne. Smrť nastáva v mladom alebo strednom veku v dôsledku interkurentného ochorenia [5,76,77].
Vrodený typ II
Nazývaný aj Seitelbergerov typ, je ešte zriedkavejší a ide o ťažkú formu PMD. Manifestuje sa v novorodeneckom a včasnom dojčenskom veku. Už v novorodeneckom veku je hypotónia, problémy s kŕmením, neprítomnosť primitívnych reflexov a stridor. Vznikajú nystagmoidné očné pohyby a extrapyramídové hyperkinézy, pridáva sa epilepsia, spasticita, ataxia a optická atrofia, prehlbuje sa mikrocefália. Dieťa nerobí žiadne vývojové pokroky a smrť nastáva v prvých rokoch [5].
Mierny typ III
Definuje sa príznakmi medzi neonatálnym a klasickým typom, je menej progresívny. Pacienti môžu s pomocou chodiť a môžu mať známky demyelinizačnej polyneuropatie [5]. Niekedy majú ťažkosti aj matky chlapcov a sestry pacientov s klasickou PMD. Prítomná je mierna spasticita, dysfunkčný mechúr, zmeny osobnosti a polyneuropatia. Mutácie PLP1 génu vedú k širokému spektru chorôb od vrodenej PMD cez klasickú PMD až k čistej X ‑ viazanej spastickej paraplégii typ 2 (SPG2). X ‑ SPG2 a PMD sú alelicky podmienené [5,76,78].
Rutinné a metabolické vyšetrenia nie sú pre diagnózu prínosom. Kondukčné štúdie sú pri PMD zväčša normálne, ale u niektorých pacientov (napr. s null mutáciou) je mierna multifokálna demyelinizačná periférna neuropatia [79].
Pomocou je BAEP, kde býva prítomná len I. a II. vlna, pričom ostatné chýbajú (známka poruchy mozgového kmeňa). ERG sú normálne, VEP a SSEP sú neprítomné alebo abnormálne s predĺženými latenciami, abnormálnou vlnou a nízkou amplitúdou. Definitívna diagnóza je však založená na DNA analýze mutácie génu PLP1 [5,80].
MR obraz pri Pelizaeus‑ Merzbacherovej chorobe
MR vysoko podporuje diagnózu a často jasne ukazuje zastavenie myelinizácie. Medzi množstvom myelínu a klinickým stavom pacientov je súvislosť. V prípade vrodenej PMD nie je vidno žiaden myelín. Myelinizované štruktúry majú v T2WI nízku intenzitu a vysoký signál v T1WI, preto je na MR v T2WI vysoký signál všetkých nemyelinizovaných štruktúr, v T1WI je to naopak signál nízkej intenzity. Pri klasickej forme je vysoký signál prítomný v mozgovom kmeni, cerebelárnej BH, v zadnom ramene capsula interna, thalame a globus palidus, malá časť myelínu je priamo periventrikulárne ako časť corona radiata, v subkortikálnej BH pre ‑ a postcentrálneho gýru, v periventrikulárnej časti radiatio optica. Nález zodpovedá obdobiu prvých mesiacov života dieťaťa, ale nezodpovedá aktuálnemu veku pacienta. Cerebrálna BH je redukovaná v objeme, s miernym rozšírením komorového systému a tenkým corpus callosum, kortexom a rozšíreným subarachnoideálnym priestorom. U dojčiat v prvých mesiacoch života nie je možné rozpoznanie ochorenia inak ako opakovaním MR. Chlapci vo veku, kde sa predpokladá ukončená myelinizácia väčšiny vlákien, s opísaným obrazom zastavenej myelinizácie, sú vysoko suspektní z Pelizaeus ‑ Merzbacherovej choroby [4,5,81 – 83]. MRS cerebrálnej BH je normálna. Niekedy sa nájde vyšší pík NAA. U niektorých žien nosičiek sú zmeny na MR podobné ako u postihnutých chlapcov [5,28].
Hlavné znaky MR obrazu (obr. 5)
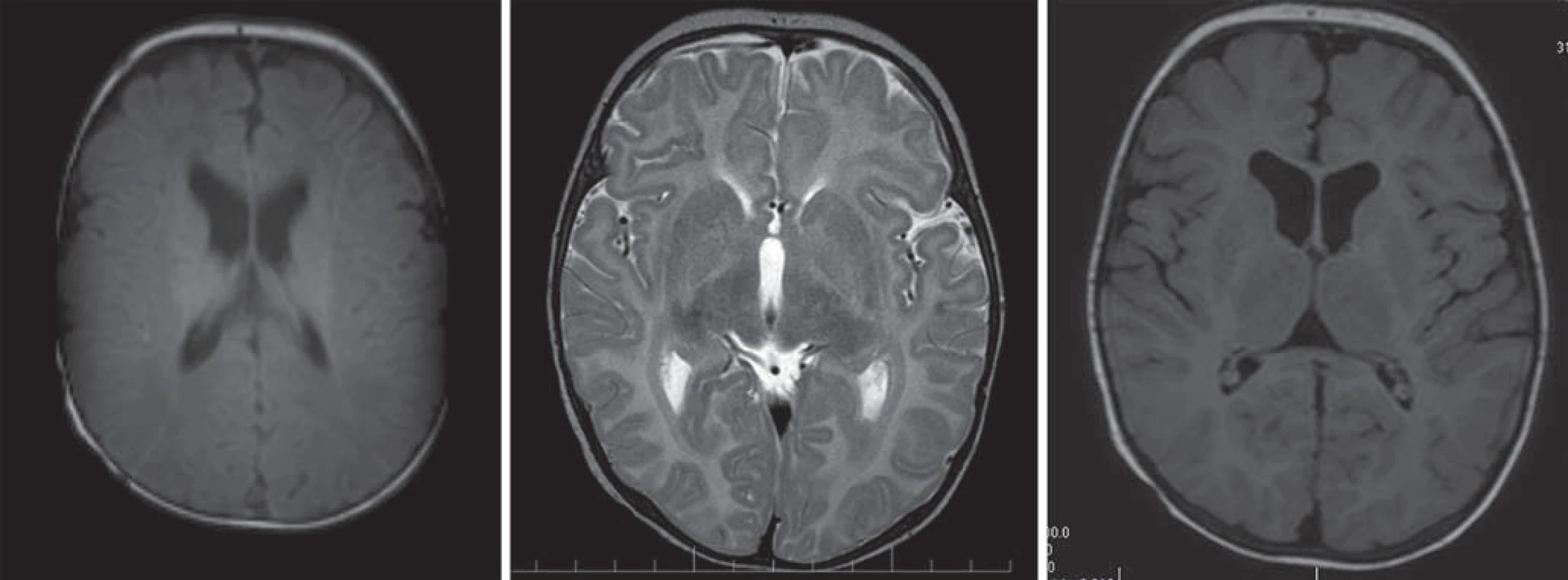
Chlapci s obrazom zastavenej myelinizácie s chýbajúcim myelínom na T1WI a T2WI. Cerebrálna hmota je redukovaná v objeme, rozšírený komorový systém, tenké corpus callosum, tenký kortex, rozšírený subarachnoideálny priestor [4,5,28].
Diferenciálna diagnostika MR nálezu
Pelizaeus ‑ Merzbacher „Like Disease“ (PMDL) [77], ťažká asfyxia, kongenitálna infekcia, pri opakovanom MR je vidieť postup myelinizácie. DNA „repair disorders“ a sialidóza, GM1, GM2 je ďalšia skupina ochorení s hypomyelinizáciou. Cocayneov syndróm má okrem hypomyelinizácie depozity kalcia v bazálnych gangliách [5,13,20,32,83].
Liečba
Okrem podpornej liečby nie je v dnešnej dobe známa efektívna pomoc pre PMD a SPG2. Sľubnou stratégiou sa ukazuje transplantácia buniek tvoriacich myelín [5].
Klinický a rádiologický obraz Alexandrovej choroby
Alexandrova choroba je zriedkavé ochorenie CNS s odhadovaným výskytom 1 : 100 000. Ochorenie je spôsobené mutáciou v géne GFAP lokalizovanom na chromozóme 17q21, ktorý kóduje kyslý gliový vláknový proteín (GFAP). Prípady sa zachytili zväčša sporadicky ako mutácie de novo, väčšinou na alelách paternálneho pôvodu (infantilné a juvenilné formy), ale sú publikované i zriedkavé prípady rodín s dominantnou formou dedičnosti. V dôsledku zmien GFAP dochádza k poruche astrocytu. V dokázaných prípadoch sa v rodinách zachytila dominantná forma dedičnosti. Hoci funkcia GFAP nie je úplne objasnená, je gliový proteín pravdepodobne zapojený do riadenia tvaru, pohybu a funkcie astroglie. Hrá tiež dôležitú úlohu vo fungovaní ďalších buniek, vrátane oligodendrocytu a podieľa sa na dlhodobej údržbe myelínu [5,84 – 86].
Klinický obraz a laboratórne vyšetrenia
Alexandrova choroba sa zvyčajne delí na tri formy. Vekové rozloženie u rôznych autorov varíruje.
Infantilná forma
Podľa autorov van der Knaap a Valk začína medzi šiestym mesiacom a štvrtým rokom, podľa Russo et al medzi nutlým a druhým rokom.
Juvenilná forma
Začína medzi štvrtým a 14., podľa Russo et al od druhého do 12. roku.
Adultná forma
Začína od 14. roku vyššie, podľa Russo et al od 12. roku [5,87].
Neonatálna forma je zriedkavá a môže mať klinickú podobu refraktérnych záchvatov a leukodystrofických zmien na MR [88]. Novšia klasifikácia podľa Messinga et al [84] uvažuje iba o dvoch typoch: včasnom (od 0 do 12 rokov) a neskorom (od 12 a viac rokov). Infantilný typ je podľa literatúry častejší, adultný je však často nediagnostikovaný [84]. Prejavuje sa zaostávaním psychomotorického vývoja, záchvatmi a rastúcou makrocefáliou, je prítomný kvadruspastický syndróm s bulbárnymi a pseudobulbárnymi príznakmi, vracaním, extrapyramídové príznaky, abnormálne pohyby očí a nystagmus. Dôsledkom sú apnoické ataky a chronická hypoventilácia. U niektorých je obraz zvýšeného intrakraniálneho tlaku. Juvenilná forma má bohatú a variabilnú symptomatológiu. Ochorenie môže mať fluktuujúci priebeh, čím je začiatok skorší, tým je priebeh progresívnejší. V úvode sú záchvaty a zhoršenie vývoja, nastupujú bulbárne a pseudobulbárne príznaky, zhoršenie reči, dysartria, problémy s hltaním, chrapot, apnoické ataky, záchvaty ranného vracania. Pridáva sa spasticita, cerebelárna ataxia, zmeny správania a kognitívna deteriorácia. Trvanie je do niekoľko mesiacov až rokov [5,82,84,89,90].
Klinický obraz adultnej formy sa zvyčajne začína po 14. roku života alebo neskôr. Klasické príznaky sú prítomné len zriedkavo. Namiesto toho sú skoro vždy prítomné bulbárny/ pseudobulbárny syndróm, cerebelárna ataxia, spasticita a demencia. Prídavné príznaky sú palatálny myoklonus, nystagmus a iné abnormálne očné pohyby, spánkové apnoe, autonómna dysfunkcia (hypo ‑ a hypertermia), oneskorenie alebo urýchlenie sexuálnej zrelosti. Palatálny myoklonus je u pacientov s adultnou formou veľmi častým príznakom a pri jeho výskyte sa odporúča vyšetrovať Alexandrovu chorobu [85,91 – 93].
Pri stanovení diagnózy nie sú nápomocné bežné laboratórne vyšetrenia. Nález celkových bielkovín v likvore je normálny, prítomné sú šokové proteíny („alfa B crystalline“ a HSP 27), ktoré sa môžu vyskytovať aj v iných podmienkach. Na EEG sú zachytené vysokovoltové vlny nad frontálnymi regiónmi [5].
Prítomnosť Rosenthalových vlákien z biopsie mozgu je hlavným znakom Alexandrovej choroby. Vlákna sú eozinofilné, ľahko postrehnuteľné svetelným aj elektrónovým mikroskopom. Názov „vlákna“ nevystihuje dostatočne ich vzhľad a štruktúru, nie sú vláknami svojím vzhľadom, ale môžu byť amorfné, ovoidné s priemerom od 10 do 40 µm a dĺžkou až do 100 µm. Tvoria predovšetkým zhluky v okolí jadra astrocytu a potom pri vzrastajúcom počte migrujú. Tieto astrocytové inklúzie sa nachádzajú v subpiálnej, perivaskulárnej a periventrikulárnej oblasti CNS, v oblastiach, kde za normálnych okolností je vysoký obsah GFAP. Pri včasnom začiatku je zhlukovanie sústredené do rostrálnych častí CNS a v prípadoch s neskorším začiatkom môžu byť v cerebele, mozgovom kmeni a mieche. GFAP je jediný gén, mutáciou ktorého sa dáva do súvisu s Alexandrovou chorobou [84,90,93 – 96].
MR obraz pri Alexandrovej chorobe
Podľa autorov van der Knaap a Valk je u detí päť MR kritérií Alexandrovej choroby [5]:
- rozsiahle symetrické lézie s predilekciou frontálne;
- prítomnosť periventrikulárneho lemu zníženého signálu na T2WI a zvýšeného na T1WI prevažne okolo frontálnych rohov;
- abnormality BG a thalamu (variabilný signál na T2WI môže byť zvýšený alebo znížený);
- postihnutie kmeňa, zvlášť stredného mozgu a miechy v T2WI hyperintenzívne a enhancujúce;
- kontrastný enhancement jednej alebo viacerých nasledujúcich štruktúr: periventrikulárne, BH frontálnych lalokov, optická chiazma, fornix, BG, thalamus, nucleus dentatus, cerebelárny kortex, kmeň.
Adultná forma má postihnutia mozgového kmeňa, cerebela a miechy v rôznej kombinácii. Nálezy pripomínajú mnohopočetný glióm, sú často asymptomatické a kontrastné. Niektorí pacienti mali len atrofiu dolného kmeňa a hornej časti miechy. Typický MR obraz zahŕňa prednostné postihnutie frontálnych oblastí s diencefalickými štruktúrami a kmeňom spolu s kontrastným enhancementom [97,98]. Podľa MR môžeme s vysokou pravdepodobnosťou usudzovať na Alexandrovu chorobu (obr. 6).
![Juvenilná forma Alexandrovej choroby.
Obr. 6a) Na T2WI s hyperintenzívnymi zmenami BH frontálne obojstranne, BG sú tmavé a atrofické, viditeľný je tenký periventrikulárny lem nízkej intenzity (šípky).
Obr. 6b) Po podaní kontrastu je vidieť enhancement na povrchu cerebela a v nucleus dentatus [100].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/a6facfa339609c3863ae9ff5fe509e65.jpg)
Diferenciálna diagnóza MR nálezu
X ‑ ALD, Canavanovej choroba, MLD, megalencefalická leukoencefalopatia so subkortikálnymi cystami, glutarová acidúria typ I, mukopolysacharidóza [5,13,15,22,28,32,99].
Liečba
Dostupná terapia dnes neexistuje. Súčasná liečba je zameraná na symptómy. Možnosti kauzálnej liečby závisia od detailnejšieho objasnenia spojenia medzi mutáciou GFAP a klinickými prejavmi poruchy [5,85].
Záver
Leukoencefalopatie predstavujú širokú skupinu hereditárnych a získaných ochorení s rozličným priebehom. Spája ich obraz rozsiahlych difúznych zmien BH na MR, ktorý zodpovedá sekčným nálezom, preto má MR významné miesto pri diagnostike a rozpoznávaní zmien. Ťažký klinický priebeh v závislosti na veku a primárne postihnutie myelínu a s myelínom súvisiacich buniek definuje pojem leukodystrofie, kde sú na MR v T2WI hypersignálne zmeny splývavého charakteru, môžu byť kontrastné alebo nekontrastná a na T1WI je signál variabilný (hyposignálny pri demyelinizačných a izo ‑ alebo hypersignálny pri hypomyelinizačných leukodystrofiách, súhrnný prehľad v tab. 2).
V súčasnom období sú pre niektoré leukodystrofie isté terapeutické možnosti, podmienkou je však včasná diagnóza. Pokročilé štádium a výrazný rozsah zmien BH nie je vhodný na transplantačnú ani genetickú liečbu, preto je nevyhnutné leukodystrofie včas rozpoznať. MR mozgu zohráva nezastupiteľnú úlohu v tomto procese. V niektorých prípadoch leukopatií je MR diagnostickou metódou napr.: X‑adrenoleukodystrofia, Canavanovej choroba, Alexandrova choroba, MSUD, periventrikulárna leukomalácia. V iných prípadoch diagnózu vysoko podporuje ako pri metachromatickej leukodystrofii a zasa inde môže správne nasmerovať laboratórne vyšetrenia a urýchliť diagnostický proces: hypomyelinizačné leukodystrofie (PMD, PMD like).
V práci sme načrtli schému delenia leukopatií, podali prehľad „nových“ leukodystrofií a možností ich laboratórneho vyšetrenia. Podrobnejšie sme rozpracovali klinický a MR obraz „klasických“ leukodystrofií (tab. 6) s cieľom urýchliť diagnostický proces a pokúsiť sa o vhodnú terapiu.
![Súhrnný prehľad MR zmien „klasických“ leukodystrofií (upravené podľa [28] a [5]).](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/ee7e9c12f570fd5546ab4649cfb76dfe.png)
MUDr. Miriam Kolníková, PhD.
Klinika detskej neurológie
LF UK a DFNsP
Limbova 1
833 40 Bratislava
e-mail: kolnikova@dfnsp.sk
Prijato k recenzii: 6. 6. 2014
Prijato do tlače: 1. 9. 2014
Recenzenti
MUDr. Martin Hřebíček, Ph.D.
MUDr. Josef Kraus, CSc.
prof. MUDr. Zdeněk Seidl, CSc.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Miriam Kolníková, PhD. (1964)

V rokoch 1982 až 1988 absolvovala štúdium na LF UK v Bratislave (odbor všeobecné lekárstvo). V letoch 1988 až 1991 pracovala ako sekundárna lekárka na Detskom oddelení v Trenčíne, v roku 1991 atestácia z pediatrie (I. st.). Od roku 1996 pracuje na Klinike detskej neurológie (1998 – atestácia z detskej neurológie). Od nástupu na kliniku je súčasťou pracovnej skupiny zameranej na dedičné metabolické poruchy pri Detskej fakultnej nemocnici v Bratislave pod vedením prim. MUDr. Behúlovej, PhD. Je členka Slovenskej pediatrickej, neurologickej a biochemickej spoločnosti. V apríli 2014 obhájila doktorandskú prácu s témou leukodystrofie detského veku. Je autorka alebo spoluautorka 13 prác v domácich a zahraničných časopisoch, aktívne sa účastní mnohých domácich a zahraničných kongresov, kde bolo prezentovaných viac ako 75 jej odborných prác vo forme prednášok alebo posterov.
Sources
1. Ambler Z, Bednařík J, Růžička E. Klinická neurologie I. část obecná. 2nd ed. Praha: Triton 2008.
2. Ganong FG. Review of Medical Physiology. 16th ed. New York: Norwalk Appleton & Lange 1993.
3. Klika E, Vacek Z, Dvořák M, Kapeller K. Histológia. 1st ed. Martin: Osveta 1988.
4. Raymond GV, Eichler F, Fatemi A, Naidu S. Leukdystrophies. London: Mac Keith Press 2011.
5. van der Knaap MS, Valk J. Magnetic Resonance of Myelination and Myelin Disorderes. 3rd ed. Berlin Heidelberg New York: Springer 2005.
6. Havrdová E a kolektív. Neuroimunologie. Praha: Maxdorf 2001.
7. Kolníková M, Sýkora P. Leukoencefalopatie – zriedkavé, ale závažné ochorenia bielej hmoty mozgu v detskom veku. Pediatr Prax 2011; 12(1): 27 – 28.
8. Perlman SJ, Mar S. Leukodystrophies. Adv Exp Med Biol 2012; 724 : 154 – 171. doi: 10.1007/ 978 ‑ 1 ‑ 4614 ‑ 0653 ‑ 2_13.
9. Powers JM. The Leukodystrophies: overview and classification. In: Lazzarini RA (ed). Myelin Disorders and Classification. London: Elsevier/ Academic Press 2004 : 663 – 690.
10. Kohlschütter A, Bley A, Brockmann K, Gärtner J, Krägeloh ‑ Mann I, Rolfs A et al. Leukodystrophies and other genetic metabolic leukoencephalopathies in children and adults. Brain Dev 2010; 32(2): 82 – 89. doi: 10.1016/ j.braindev.2009.03.014.
11. Kohlschütter A, Eichler F. Childhood leukodystrophies: a clinical perspective. Expert Rev Neurother 2011; 11(10): 1485 – 1496. doi: 10.1586/ ern.11.135.
12. Ambler Z, Bednařík J, Růžička E a kolektiv. Klinická neurologie II. část speciální. 1st ed. Praha: Triton 2010.
13. Vanderver A, Tonduti D, Schiffmann R, Schmidt J,Van der Knaap MS. Leukodystrophy Overview. Seattle: University of Washington 2014.
14. Kemp S, Berger J, Aubourg P. X‑linked adrenoleukodystrophy: clinical, metabolic, genetic and pathophysiological aspects. Biochim Biophys Acta 2012; 1822(9): 1465 – 1474. doi: 10.1016/ j.bbadis.2012.03.012.
15. Kolníková M, Sýkora P, Petrovič R, Fischerová M, Chandoga J. X ‑ viazaná adrenoleukodystrofia. Cesk Slov Neurol N 2013; 76/ 102(2): 197 – 202.
16. Powers JM, DeCiero DP, Cox C, Richfield EK, Ito M,Moser AB et al. The dorsal root ganglia in adrenomyeloneuropathy: neuronal atrophy and abnormal mitochondria. J Neuropathol Exp Neurol 2001; 60(5): 493 – 501.
17. Powers JM, DeCiero DP, Ito M, Moser AB, Moser HW. Adrenomyeloneuropathy: a neuropathologic review featuring its noninflammatory myelopathy. J Neuropathol Exp Neurol 2000; 59(2): 89 – 102.
18. Powers JM, Pei ZT, Heinzer AK, Deering R, Moser AB, Moser HW et al. Adreno‑leukodystrophy: oxidative stress of mice and men. J Neropathol Exp Neurol 2005; 64(12): 1067 – 1079.
19. Kemp S, Berger J, Aubourg P. X‑linked adrenoleukodystrophy: clinical, metabolic, genetic and pathophysiological aspects. Biochim Biophys Acta 2012; 1822(9): 1465 – 1474. doi: 10.1016/ j.bbadis.2012.03.012.
20. Moser H, Dubey P, Fatemi A. Progress in X‑linked adrenoleukodystrophy. Curr Opin Neurol 2004; 17(3): 263 – 269.
21. Kumar AJ, Köhler W, Kruse B, Naidu S, Bergin A, Edwin D et al. MR findings in adult onset adrenolekodystrophy. AJNR Am J Neuroradiol 1995; 16(6): 1227 – 1237.
22. Barcovich AJ. Pediatric Neuroimaging, 4th Ed. Philadelphia: Lippincton Williams and Wilkins 2000.
23. Jangouk P, Zackowski KM, Naidu S, Raymond GV.Adrenoleukodystrophy in female heterozygotes: underrecognized and undertreated. Mol Genet Metab 2012; 105(2): 180 – 185. doi: 10.1016/ j.ymgme.2011.11.001.
24. Moser HW, Raymond GV, Dubey P. Adrenoleukodystrophy: new approaches to a neurodegenerative disease. JAMA 2005; 294(24): 3131 – 3134.
25. Kemp S, Wanders R. Biochemical aspects of X‑linked adrenoleukodystrophy. Brain Pathol 2010; 20(4): 831 – 837. doi: 10.1111/ j.1750 ‑ 3639.2010.00391.x.
26. Chandoga J, Petrovič R, Futas J, Ďurovčíková D, Štofko J, Jančo S et al. X ‑ viazaná adrenoleukodystrofia – najčastejšia dedičná metabolická porucha peroxizómov. Neurol Prax 2006; 2 : 84 – 89.
27. Melhem ER, Loes DJ, Georgiades CS, Raymond GV, Moser HW. X‑linked adrenoleukodystrophy: the role of contrast ‑ enhanced MR imaging in predicting disease progression. AJNR Am J Neuroradiol 2000; 21(5): 839 – 844.
28. Osborn AG, Salzman KL, Barkovich AJ et al. Diagnostic Imaging: Brain. 2nd ed. Salt Lake City: Amirsys Publishing 2010.
29. Loes DJ, Fatemi A, Melhem ER, Gupte N, Bezman L,Moser HW et al. Analysis of MRI patterns aids prediction of progression in X‑linked adrenoleukodystrophy. Neurology 2003; 61(3): 369 – 374.
30. Seidl Z, Vaněčková M, Vítek T, Kron M, Dvořáková L, Zeman J. X‑adrenoleukodystrofie ‑ hodnocení lézí mozku modalitou magnetické rezonance pomocí „Loes score“. Ces Radiol 2007; 61(3): 275 – 278.
31. Moser H, Dubey P, Fatemi A. Progress in X‑linked adrenoleukodystrophy. Curr Opin Neurol 2004; 17(3): 263 – 269.
32. Yang E, Prabhu SP. Imaging manifestations of the leukodystrophies, inherited disorders of white matter. Radiol Clin North Am 2014; 52(2): 279 – 319. doi: 10.1016/ j.rcl.2013.11.008.
33. Kang EG, Jeon SJ, Choi SS, Song CJ, Yu IK. Diffusion MR imaging of hypoglycemic encephalopathy. AJNR Am J Neuroradiol 2010; 31(3): 559 – 564. doi: 10.3174/ ajnr.A1856.
34. Baumann M, Korenke GC, Weddige ‑ Diedrichs A, Wilichowski E, Hunneman DH, Wilken B et al. Haematopoietic stem cell transplantation in 12 patients with cerebral X‑linked adrenoleukodystrophy. Eur J Pediatr 2003; 162(1): 6 – 14.
35. Moser HW, Raymond GV, Kohler W, Sokolowski P,Hanefeld F, Korenke GC et al. Evaluation of the preventive effect of glecerol trioleate ‑ trieurcutae („Lorenzo’s oil“) therapy in X‑linked adrenoleukodystrophy: results of two concurrent trials. Adv Exp Med Biol 2003; 544 : 369 – 387.
36. Cartier N, Hacein‑Bey ‑ Abina S, Bartholomae CC,Veres G, Schmidt M, Kutschera I et al.: Hematopoetic stem cell gene therapy with a lentiviral vector in X‑linked adrenoleukodystrophy. Science 2009; 326(5954): 818 – 823. doi: 10.1126/ science.1171242.
37. Groeschel S, Dali CI, Clas P, Bohringer J, Duno M, Krarup C et al. Cerebral gray and white matter changes and clinical course in metachromatic leukodystrophy. Neurology 2012; 79(16): 1662 – 1670. doi: 10.1212/ WNL.0b013e31826e9ad2.
38. Groeschel S, Kehrer C, Engel C, Dali CI, Bley A, Steinfeld R et al. Metachromatic leukodystrophy: natural course of cerebral MRI changes in relation to clinical course. J Inherit Metab Dis 2011; 34(5): 1095 – 1102. doi: 10.1007/ s10545 ‑ 011 ‑ 9361 ‑ 1.
39. Gieselmann V, Krägeloh ‑ Mann I. Metachromatic leukodystrophy. In: Raymond GV, Eichler F, Fatemi A,Naidu S (eds). Leukdystrophies. London: Mac Keith Press 2011 : 130 – 155.
40. Gieselmann V, Krägeloh ‑ Mann I. Metachromatic Leukodystrophy – An Update. Neuropediatrics 2010; 41(1): 1 – 6. doi: 10.1055/ s ‑ 0030 ‑ 1253412.
41. Gieselmann V. Metachromatic leukodystrophy: genetics, pathogenesis and therapeutic options. Acta Paediatr Suppl 2008; 97(457): 15 – 21. doi: 10.1111/ j.1651 ‑ 2227.2008.00648.x.
42. Kehrer C, Blumenstock G, Gieselmann V, Krägeloh ‑ Mann I. German leukonet; The natural course of gross motor deterioration in metachromatic leukodystrophy. Dev Med Child Neurol 2011; 53 : 850 – 855. doi: 10.1111/ j.1469 ‑ 8749.2011.04028.x.
43. von Figura K, Gieselmann V, Jaeken J. Metachromatic leukodystrobphy. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds). The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw Hill 2001 : 3695 – 3724.
44. Cameron CL, Kang PB, Burns TM, Darras BT, Jones HR jr. Multifocal slowing of nerve conduction in metachromatic leukodystrophy. Muscle Nerve 2004; 29(4): 531 – 536.
45. Bindu PS, Mahadevan A, Taly AB, Christopher R, Gayathri N, Shankar SK. Peripheral neuropathy in metachromatic leucodystrophy. A study of 40 cases from south India. J Neurol Neurosurg Psychiatry 2005; 76(12): 1698 – 1701.
46. Brown FR 3rd, Shimizu H, McDonald JM, Moser AB, Marquis P, Chen WW et al. Auditory evoked brainstem response and high‑performance liquid chromatography sulfatide assay as early indices of metachromatic leukodystrophy. Neurology 1981; 31(8): 980 – 985.
47. Ries M, Deeg KH. Polyposis of the gallbladder associated with metachromatic leukodystrophy. Eur J Pediatr 1993; 152(5): 450 – 451.
48. Rafi MA, Coppola S, Liu SL, Rao HZ, Wenger DA. Disease ‑ causing mutations in cis with the common arylsulfatase A pseudodeficiency allele compound the difficulties in accurately identifying patients and carriers of metachromatic leukodystrophy. Mol Genet Metab 2003; 79(2): 83 – 90.
49. Eichler F, Grodd W, Grant E. Sessa M, Biffi A, Bley A et al. Metachromatic leukodystrophy: a scoring system for brain MR imaging observations. Am J Neuroradiol 2009; 30(10): 1893 – 1897.
50. Dali CI, Barton NW, Hanson LG, Krägeloh ‑ Mann I, Groeschel S, Kraraup C et al. Outcome of Phase I/ II Trial of intravenous Enzyme Replacement with Recombinant Human Arylsulfatase A (rhASA) in Children with Metachromatic Leukodystrophy (MLD). Eur J Paediatr Neurol 2011; 15 : 22.
51. Batzios SP, Zafeiriou DI. Developing treatment options for metachromatic leukodystrophy. Mol Genet Metab 2012; 105(1): 56 – 63.
52. Biffi A, Aubourg P, Cartier N. Gene therapy for leukodystrophies. Hum Mol Genet 2011; 20(1): 42 – 53. doi: 10.1093/ hmg/ ddr142.
53. Suzuki K. Globoid cell leukodystrophy (Krabbe’s disease): update. J Child Neurol 2003; 18(9): 595 – 603.
54. Wenger DA. Krabbe disease. In: Raymond GV, Eichler F, Fatemi A, Naidu S (eds). Leukdystrophies. London: Mac Keith Press 2011 : 90 – 105.
55. Formichi P, Radi E, Battisti C, Pasqui A, Pompella G, Lazzerini PE et al. Psychosine‑induced apoptosis and cytokine activation in immune peripheral cells of Krabbe patients. J Cell Physiol 2007; 212(3): 737 – 743.
56. Siddiqi ZA, Sanders DB, Massey JM. Peripheral neuropathy in Krabbe disease: electrodiagnostic findings. Neurology 2006; 67(2): 263 – 267.
57. Lyon G, Hagberg B, Evrard P, Vanier M, Allaire C, Pavone L et al. Symptomatology of late onset Krabbe’s leukodystrophy: the European experience. Dev Neurosci 1991; 13 : 240 – 244.
58. Tada K, Taniike M, Ono J, Tsukamoto H, Inui K, Okada S. Serial magnetic resonance imaging studies in a case of late onset globoid cell leukodystrophy. Neuropediatrics 1992; 23(6): 306 – 309.
59. Spiegel R, Bach G, Sury V, Mengistu G, Meidan B,Shalev S et al. A mutation in the saposin A coding region of the prosaposin gene in an infant presenting as Krabbe disease: first report of saposin A deficiency in humans. Mol Genet Metab 2005; 84(2): 160 – 166.
60. De Gasperi R, Gama Sosa MA, Sartorato EL, Battistini S, MacFarlane H, Gusella JF et al. Molecular heterogeneity of late ‑ onset forms of globoid ‑ cell leukodystrophy. Am J Hum Genet 1996; 59(6): 1233 – 1242.
61. Sasaki M, Sakuragawa N, Takashima S, Hanaoka S,Arima M. MRI and CT findings in Krabbe disease. Pediatr Neurol 1991; 7(4): 283 – 288.
62. Loes DJ, Peters C, Krivit W. Globoid cell leukodystrophy: distinguishing early onset from late ‑ onset disease using a brain MR imaging scoring method. Am J Neuroradiol 1999; 20(2): 316 – 323.
63. Costello DJ, Eichler AF, Eichler FS. Leukodystrophies: classification, diagnosis, and treatment. Neurologist 2009; 15(6): 319 – 328. doi: 10.1097/ NRL.0b013e3181b287c8.
64. Phelan JA, Lowe LH, Glasier CM. Pediatric neurodegenerative white matter processes: leukodystrophies and beyond. Pediatr Radiol 2008; 38(7): 729 – 749. doi: 10.1007/ s00247 ‑ 008 ‑ 0817 ‑ x.
65. Matalon R. Canavan disease: diagnosis and molecular analysis. Genet Test 1997; 1(1): 21 – 25.
66. Matalon R, Matalon KM. Canavan disease prenatal diagnosis and genetic counseling. Obstet Gynecol Clin North Am 2002; 29(2): 297 – 304.
67. Matalon RM, Matalon KM. Spongy degeneration of the brain, Canavan disease: biochemical and molecular findings. Front Biosci 2000; 5: D307 – D311.
68. Kimberlee M, Matalon R. Canavan disease. In: Raymond GV, Eichler F, Fatemi A, Naidu S (eds). Leukodystrophies. London: Mac Keith Press 2011 : 156 – 169.
69. van der Knaap MS, Barth PG, Vrensen GF, Valk J. Histopathology of an infantile ‑ onset spongiform leukoencephalopathy with a discrepantly mild clinical course. Acta Neuropathol 1996; 92(2): 206 – 212.
70. Fernandes J, Saudubray JM, Berghe G,Walter JH. Diagnostika a léčba dědičných metabolických poruch. 4th ed. Praha: Triton 2008.
71. Sreenivasan P, Purushothaman KK. Radiological clue to diagnosis of Canavan disease. Indian J Pediatr 2013; 80(1): 75 – 77. doi: 10.1007/ s12098 ‑ 012 ‑ 0794 ‑ 9.
72. McAdams HP, Geyer CA, Done SL, Deigh D. CT and MR imaging of Canavan disease. Am J Neuroradiol 1990; 11(2): 397 – 399.
73. Leone P, Shera D, McPhee SW, Francis JS, Kolodny EH, Bilaniuk LT et al. Long‑term follow‑up after gene therapy for Canavan disease. Sci Transl Med 2012; 4(165): 165 – 173. doi: 10.1126/ scitranslmed.3003454.
74. Zano S, Malik R, Szucs S, Matalon R, Viola RE. Modification of aspartoacylase for potential use in enzyme replacement therapy for the treatment of Canavan disease. Mol Genet Metab 2011; 102(2): 176 – 180. doi: 10.1016/ j.ymgme.2010.10.012.
75. Boespflug ‑ Tanguy O. Inborn errors of brain myelin formation. Handb Clin Neurol 2013; 113 : 1581 – 1592. doi: 10.1016/ B978 ‑ 0 ‑ 444 ‑ 59565 ‑ 2.00027 ‑ 7.
76. Nave KA, Dhaunchak AS. Pelizaeus ‑ Merzbacher disease: genetic model and mechanisms. In: Raymond GV, Eichler F, Fatemi A, Naidu S (eds). Leukodystrophies. London: Mac Keith Press 2011 : 170 – 187.
77. Hobson GM, Garbern JY. Pelizaeus ‑ Merzbacher disease, Pelizaeus ‑ Merzbacher‑like disease 1, and related hypomyelinating disorders. Semin Neurol 2012; 32(1): 62 – 67. doi: 10.1055/ s ‑ 0032 ‑ 1306388.
78. Kibe T, Miyahara J, Yokochi K, Iwaki A. A novel PLP mutation in a Japanese patient with mild Pelizaeus ‑ Merzbacher disease. Brain Dev 2009; 31(3): 248 – 251. doi: 10.1016/ j.braindev.2008.08.001.
79. Shy ME, Hobson G, Jain M, Boespflug ‑ Tanguy O, Garbern J, Sperle K et al. Schwann cell expression of PLP1 but not DM20 is necessary to prevent neuropathy. Ann Neurol 2003; 53(3): 354 – 365.
80. Seeman P, Kršek P, Naměstková K, Malíková M, Belšan T, Prošková M. Pelizaeus ‑ Merzbacher’s Disease (PMD) – Detection of the most frequent mutation of the Proteolipid protein gene in Czech patients and famillies with the classical form of PMD. Cesk Slov Neurol N 2003; 66 : 95 – 104.
81. Caro PA, Marks HG. Magnetic resonance imaging and computed tomography in Pelizaeus ‑ Merzbacher disease. Magn Reson Imaging 1990; 8(6): 791 – 796.
82. Perlman SJ, Mar S. Leukodystrophies. Adv Exp Med Biol 2012; 724 : 154 – 171. doi: 10.1007/ 978 ‑ 1 ‑ 4614 ‑ 0653 ‑ 2_13.
83. Steenweg ME, Vanderver A, Blaser S, Bizzi A, de Koning TJ, Mancini GM et al. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain 2010; 133(10): 2971 – 2982. doi: 10.1093/ brain/ awq257.
84. Messing A, Brenner M, Feany MB, Nedergaard M, Goldman JE. Alexander disease. J Neurosci 2012; 32(15): 5017 – 5023. doi: 10.1523/ JNEUROSCI.5384 ‑ 11.2012.
85. Messing A, LaPash D, CM Hagemann TL. Strategies for treatment in Alexander disease. Neurotherapeutics 2010; 7(4): 507 – 515. doi: 10.1016/ j.nurt.2010.05.013.
86. Li R, Johnson AB, Salomons GS, van der Knaap MS, Rodriguez D, Boespflug ‑ Tanguy O et al. Propensity for paternal inheritance of de novo mutations in Alexander disease. Hum Genet 2006; 119(1 – 2): 137 – 144.
87. Russo LS jr, Aron A, Anderson PJ. Alexander’s disease: a report and reappraisal. Neurology 1976; 26(7): 607 – 614.
88. Singh N, Bixby C, Etienne D, Tubbs RS, Loukas M. Alexander’s disease: reassessment of a neonatal form. Childs Nerv Syst 2012; 28(12): 2029 – 2031. doi: 10.1007/ s00381 ‑ 012 ‑ 1868 ‑ 8.
89. Brenner M, Goldman JE, Quinlan R, Messing A. Alexander disease: a genetic disorder of astrocytes. In: Parpura V, Haydon P (eds). Astrocytes in Physiology of the Nervous System. New York: Springer 2009 : 591 – 648.
90. Flint D, Brenner M. Alexander disease. In: Raymond GV, Eichler F, Fatemi A, Naidu S (eds). Leukdystrophies. London: Mac Keith Press 2011 : 106 – 129.
91. Balbi P, Seri M, Ceccherini I, Uggetti C, Casale R, Fundarò C et al. Adult ‑ onset Alexander disease: report on a family. J Neurol 2008; 255(1): 24 – 30.
92. Howard KL, Hall DA, Moon M, Agarwal P, Newman E, Brenner M. Adult ‑ onset Alexander disease with progressive ataxia and palatal tremor. Mov Disord 2008; 23(1): 118 – 122.
93. Pareyson D, Fancellu R, Mariotti C, Romano S, Salmaggi A, Carella F et al. Adult ‑ onset Alexander disease: a series of eleven unrelated cases with review of the literature. Brain 2008; 131(9): 2321 – 2331. doi: 10.1093/ brain/ awn178.
94. Li R, Johnson AB, Salomons G, Goldman JE, Naidu S,Quinlan R et al. Glial fibrillary acidic protein mutations in infantile, juvenile, and adult forms of Alexander disease. Ann Neurol 2005; 57(3): 310 – 326.
95. Wippold FJ 2nd, Perry A, Lennerz J. Neuropathology for the neuroradiologist: Rosenthal fibers. Am J Neuroradiol 2006; 27(5): 958 – 961.
96. Sawaishi Y. Review of Alexander disease: beyond the classical concept of leukodystrophy. Brain Dev 2009; 31(7): 493 – 498. doi: 10.1016/ j.braindev.2009.03.006.
97. Romano S, Salvetti M, Ceccherini I, De Simone T, Savoiardo M. Brainstem signs with progressing atrophy of medulla oblongata and upper cervical spinal cord. Lancet Neurol 2007; 6(6): 562 – 570.
98. Huttner HB, Richter G, Hildebrandt M, Blümcke I, Fritscher T, Brück W et al. Acute onset of fatal vegetative symptoms: unusual presentation of adult Alexander disease. Eur J Neurol 2007; 14(11): 1251 – 1255.
99. Schiffmann R, van der Knaap MS. Invited article: an MRI‑based approach to the diagnosis of white matter disorders. Neurology 2009; 72(8): 750 – 759.
100. van der Knaap MS, Naidu S, Breiter SN, Blaser S, Stroink H, Springer S et al. Alexander disease: diagnosis with MR imaging. AJNR Am J Neuroradiol 2001; 22(3): 541 – 552.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2014 Issue 5
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Memantine Eases Daily Life for Patients and Caregivers
Most read in this issue
- Česká tréninková verze Montrealského kognitivního testu (MoCA‑ CZ1) k časné detekci Alzheimerovy nemoci
- Leukodystrofie – klinické a rádiologické aspekty
- Bariéry nervového systému za fyziologických a patologických stavů
- Chirurgická léčba supratentoriálních kortiko‑ subkortikálních kavernomů