
Molekulárna diagnostika NF1 na Slovensku s využitím analýzy cDNA a MLPA
Molecular Diagnostics of NF1 in Slovakia Using cDNA and MLPA Analysis
Aim:
The aim of our study was to identify causative germline mutation in suspected NF1 patients, in order to help differential diagnostics, as well as to collect as large as possible group of patients for a possible genotype-phenotype correlations.
Material and methods:
Our set of patients consisted of 107 Slovak patients, recruited between 2008 and 2013, who were considered for a diagnosis NF1 by clinical genetics. In order to identify mutations, we employed NF1 cDNA sequencing that enabled us to also detect splicing mutations, as well as MLPA analysis that enables identification of larger deletions.
Results:
By employing the selected methods in our set of 94 unrelated Slovak patients who fulfilled the basic NF1 diagnostic criteria, we uncovered germline mutations in the NF1gene in 83 of them (88.3%). We observed a high proportion of mutations identified in Slovak population only so far (41/83, 49.4%), and we confirmed de novo mutation in 27/42 tested families (64.3%). Genotype-phenotype correlations revealed an increased incidence of optic pathway glioma in patients with a mutation in the 5´end of the NF1 gene.
Conclusion:
Combination of cDNA analysis and MLPA provides an effective method for identification of mutations in the NF1 gene. By employing these methods, we were able to also identify frequently incorrectly classified atypical splicing mutations. Clinical geneticists used our results in the differential diagnosis of the disease, especially in contested cases.
Key words:
NF1 gene – causative germline mutation – RNA splicing – genotype-phenotype correlation – de novo mutations –differential diagnostics
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Authors:
M. Némethová 1,2; A. Bolčeková 3; S. Požgayová 2; D. Ilenčíková 3; A. Hlavatá 3; Ľ. Kádaši 1,4; L. Kovácz 3; A. Zaťková 1
Authors‘ workplace:
Laboratórium genetiky, ÚMFG SAV, Bratislava
1; Laboratórium klinickej a molekulárnej genetiky, II. detská klinika LF UK a DFNsP Bratislava
2; II. detská klinika LF UK a DFNsP Bratislava
3; Katedra molekulárnej biológie, PriF UK, Bratislava
4
Published in:
Cesk Slov Neurol N 2014; 77/110(6): 721-733
Category:
Original Paper
Overview
Cieľ:
Cieľom našej práce bolo identifikovať kauzatívne germinálne mutácie u suspektných NF1 pacientov za účelom diferenciálnej diagnostiky a zozbieranie čo najväčšieho súboru pacientov pre prípadné genotypovo-fenotypové korelácie.
Súbor a metodika:
Náš súbor pozostával zo 107 nezávislych pacientov zo Slovenska, ktorý boli v období rokov 2008-2013 klinickými genetikmi posúdení ako suspektní pre NF1. Na identifikáciu mutácií bolo použité sekvenovanie cDNA NF1 génu, ktoré nám umožnilo úspešnú identifikáciu zostrihových mutácií, ako aj MLPA analýza umožňujúca identifikáciu rozsiahlejších delécií v géne.
Výsledky:
V súbore 94 nepríbuzných slovenských pacientov, ktorí spĺňali základné diagnostické kritériá pre neurofibromatózu, sme za použitia našich metód identifikovali germinálne mutácie v NF1 géne u 83 z nich (88,3 %). Zaznamenali sme vysoký podiel mutácií, ktoré boli zatiaľ identifikované len v slovenskej populácii (41/ 83, 49,4 %), a u 27/ 42 testovaných rodinách sme potvrdili mutáciu de novo (64,3 %). Genotypovo-fenotypové korelácie odhalili zvýšený výskyt očných gliómov u pacientov s mutáciou na 5’ konci NF1 génu.
Závery:
Kombinácia cDNA analýzy a MLPA je efektívnou pre identifikáciu mutácií v NF1 géne. Na základe tejto diagnostickej metódy sme odhalili aj často nesprávne klasifikované netypické zostrihové mutácie. Naše výsledky využili klinickí genetici pri diferenciálnej diagnostike ochorenia, hlavne v sporných prípadoch.
Kľúčová slová:
NF1 gén – kauzatívna germinálna mutácia – RNA zostrih – genotypovo-fenotypové korelácie – de novo mutácie –diferenciálna diagnostika
Úvod
Neurofibromatóza typu 1 (NF1), nazývaná aj Recklinghausenova choroba, je autozomálne dominantné multisystémové ochorenie s incidenciou 1 : 3 500 [1]. Je to heterogénne ochorenie, ale jeho penetrancia je úplná už do ôsmeho roku života [2]. National Institute of Health (NIH) stanovil sedem základných klinických diagnostických kritérií sumarizovaných v tab. 1, z ktorých musí pacient splniť minimálne dve pre stanovenie diagnózy NF1 [3].
![Základné klinické diagnostické kritéria pre NF1 podľa NIH [3].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/a8c4f958907f478cf8b116262b253835.png)
Hlavným rizikom ochorenia je, že u 8-12 % pacientov sa vo veku 20-30 rokov z benígneho plexiformného neurofibrómu vyvinie malígny nádor periférnych nervových puzdier (MPNST) [4]. Ďalšími zriedkavejšími prejavmi spojenými s NF1 môžu byť aj feochromocytóm, astrocytóm, juvenilná myelomonocytická leukémia (JMML) a celková predispozícia k malígnym ochoreniam [5]. Ochorenie vedie aj k postihnutiam ostatných systémov, ako sú kardiovaskulárne a krvné komplikácie, či abnormality v tkanivách pochádzajúcich z neurálnej lišty [6].
NF1 gén a proteín
NF1 gén [7] je lokalizovaný na chromozóme 17q11.2, kde pokrýva približne 280 kb genomickej DNA. Gén pozostáva zo 60 exónov [8-10]. Alternatívnemu zostrihu v rámci NF1 transkriptu podliehajú exóny 9a/ 9br, 23a, 48a [11]. Génový produkt NF1 génu - neurofibromín –je tumor supresorový proteín produkovaný hlavne v melanocytoch CALM (café au lait škvrny), neurónoch, gliových bunkách a Schwanových bunkách [12].
Neurofibromín je negatívnym regulátorom Ras proteínu, a tým aj celej Ras-MAP kinázovej dráhy [13]. Na tejto jeho funkcii sa podieľa Ras-GRD funkčná doména, ktorá je kódovaná exónmi 21-27a NF1 génu [14,15].
Exóny 11-17 kódujú doménu bohatú na cysteín a serín (CSRD), ktorá má tri potenciálne rozpoznávajúce miesta pre cAMP-zavislú proteín kinázu. Táto oblasť NF1 proteínu podlieha fosforylácii proteín kinázami [16]. Bolo zistené, že Ras-GAP aktivita neurofibromínu je regulovaná forsforyláciou CSRD proteín kinázou C [17].
Výsledkom nedostatku neurofibromínu, alebo jeho nesprávnej funkcie v dôsledku mutácie, je spravidla nekontrolovaná bunková proliferácia a tvorba tumorov [18-20].
NF1 mutácie
NF1 pacienti sú väčšinou nositeľmi germinálnej mutácie v NF1 géne v heterozygotnom stave, pričom v 50 % prípadov je to de novo mutácia. V neurofibrómoch a CALM asociovaných s NF1 dochádza k somatickej inaktivácii aj druhej alely NF1 génu [21]. Ochorenie sa teda prejaví po vyradení z funkcie druhej alely somatickou mutáciou [22].
90-95 % germinálnych mutácií predstavujú malé zmeny DNA, tzv. malé mutácie NF1 génu. Ide o substitúcie, malé delécie, či inzercie malého počtu nukleotidov, ktoré vedú k zámene aminokyseliny, priamo k vzniku predčasného stop kodónu, alebo k posunu čítacieho rámca a neskôr tiež k predčasnému stop kodónu [23,24]. Výrazný podiel na NF1 mutáciách predstavujú zostrihové mutácie 29 % [24]. Delécie celého NF1 génu a priľahlých génov (typ I, typ II typ III), nazývané aj mikrodelécie, sa objavujú asi u 5-10 % pacientov [23,25]. Na identifikáciu mutácií sa v súčasnosti využíva viacero metodických postupov založených na sekvenovaní genomickej DNA (gDNA), alebo komplementárnej DNA (cDNA).
Vzhľadom k vysokému podielu zostrihových mutácií v NF1 géne sa odporúčajú hlavne metodiky založené na analýze RNA. Tieto totiž umožňujú rýchlo a efektívne odhaliť i netypické zostrihové mutácie, t.j. mutácie, ktoré sa nachádzajú hlbšie v intronickej oblasti, alebo mutácie v exónoch, ktorých efekt na zostrih mRNA by za použitia DNA analýzy nebol pozorovateľný.
Súbor a metódy
Náš súbor pozostával zo 107 nezávislých pacientov, ktorí boli počas šiestich rokov (2008-2013) zaslaní do nášho laboratória na identifikáciu mutácií v NF1 géne ako súčasť diferenciálnej diagnostiky. U 41 z nich bol v anamnéze uvedený familiárny výskyt ochorenia. Vzorky zaslané do nášho laboratória pochádzali z celého Slovenska a prevažnú väčšinu tvorili detskí pacienti, pričom priemerný vek bol 17,32 rokov. Najmladší pacient mal jeden rok a najstarší 57 rokov.
Nie všetci pacienti jednoznačne spĺňali základné diagnostické kritériá, avšak kompletnú analýzu NF1 génu sme na vyžiadanie a s cieľom overiť novozavádzanú metodiku uskutočnili u všetkých z nich.
Na mutačný skríning NF1 génu sme použili upravený protokol od Messiaenovej a Wimmerovej [24]. Z časti vzorky periférnej krvy pacienta v EDTA sme izolovali DNA pomocou kitu PureGene (Qiagen) a druhú časť sme použili na izoláciu lymfocytov centrifugáciou na ficole, a tieto sme krátkodobo kultivovali. Pred ukončením kultivácie (4-6 hod) sme do kultúry pridali 200 µg/ ml puromycínu, ktorý inaktivuje nonsense mediated mRNA decay (NMD), t.j. mechanizmus degradácie mRNA transkriptov majúcich predčasný terminačný kodón [26]. Použitie puromycínu tak zvyšuje senzitivitu metodiky na mutácie, ktoré vedú k vzniku takýchto transkriptov (mutácie vedúce k vzniku stop kodónu alebo k posunu čítacieho rámca s následným predčasným stop kodónom). Priamo po ukončení kultivácie, alebo najneskôr mesiac po ukončení kultivácie a uskladnení vzoriek pri -80 °C, sme pomocou Trizolu (Invitrogen) izolovali celkovú RNA a pomocou Superscript RT (Invitrogen) pripravili cDNA, podľa protokolu výrobcu. cDNA sme použili na PCR amplifikáciu celej kódujúcej časti NF1 génu v piatich prekrývajúcich sa fragmentoch. Fragmenty sme sekvenovali pomocou 19 špecifických primerov a sekvenačného kitu Big Die® Terminator v3.1 Cycle (Applied Biosystems) a analyzovali na genetickom analyzátore ABI PRISM® 3130xl (Applied Biosystems).
Všetky identifikované mutácie sme overovali na úrovni genomickej DNA (gDNA). Využili sme špecifické primery navrhnuté na základe sekvencií publikovaných v databázach (NCBI, ENSEMBL) (sekvencie primerov sú k dispozícii na vyžiadanie).
Ak sa u pacienta sekvenovaním neodhalila žiadna kauzatívna mutácia, jeho DNA vzorku sme analyzovali pomocou metódy MLPA (Multiple Ligation Dependent Probe Amplification) s NF1 mikrodelečnými MLPA kitmi P122, P081, P082 od firmy MRC Holland, s cieľom identifikovať prípadné rozsiahlejšie delécie. Túto metódu sme použili aj za účelom overovania vnútrogénových delécií odhalených pri sekvenovaní.
Pri analýze mutácií u rodinných príslušníkov pacientov sme amplifikovali a sekvenovali len príslušný exón na úrovni gDNA.
Identifikované mutácie sú popísané podľa konsenzus kritérií a odporučení HGVS (Human Genome Variation Society) [27]. V texte uvádzame staršie konsensus číslovanie exónov NF1 génu, kým v tabuľkách pre komplexnosť uvádzame obe verzie.
Na potvrdenie patogenity novo identifikovaných mutácií sme použili rôzne predikčné programy:
- a) Efekt mutácií meniacich zmysel na štruktúru funkčného NF1 proteínu sme testovali pomocou predikčných programov PolyPhen-2 (Polymorphism Phenotyping v2) [28], SNAP (Screening for NonAcceptable Polymorphisms) [29] a Provean (Protein Variation Effect Analyzer) [30-31].
- b) Na overenie efektu zostrihových mutácií na mRNA zostrih, ktorý sme pozorovali pomocou sekvenovania cDNA, sme použili programy HSF (Human Splicing Finder) [32] a Splice Site Prediction by the Neural Network [33].
Výsledky
Z celkového počtu 107 vzoriek slovenských pacientov zaslaných do nášho laboratória s podozrením na diagnózu NF1 94 spĺňalo základné diagnostické kritériá pre toto ochorenie. Kauzatívna mutácia bola identifikovaná u 83/ 94 (88,3 %) pacientov, 43 žien a 40 mužov. U 73/ 83 (88 %) pacientov bola mutácia potvrdená priamym sekvenovaním. Išlo o tzv. malé mutácie, z ktorých najčastejšie boli posunové mutácie 43 % (36/ 83), nasledovali zostrihové mutácie 17 % (14/ 83), nezmyselné mutácie 13 % (11/ 83), mutácie meniace zmysel 12 % (10/ 83) a in frame delécie 3 % (2/ 83). U zvyšných 10 pacientov (10/ 83, 12 %) boli pomocou analýzy MLPA identifikované rozsiahle vnútrogénové mutácie u piatich (5/ 83, 6 %) a delécie celého génu tiež u piatich pacientov (5/ 83, 6 %). Zastúpenie jednotlivých typov mutácií je znázornené v grafu 1.

U 11/ 94 pacientov spĺňajúcich diagnostické kritéria sme ani sekvenovaním ani MLPA neidentifikovali žiadnu kauzatívnu NF1 mutáciu (tab. 2c).
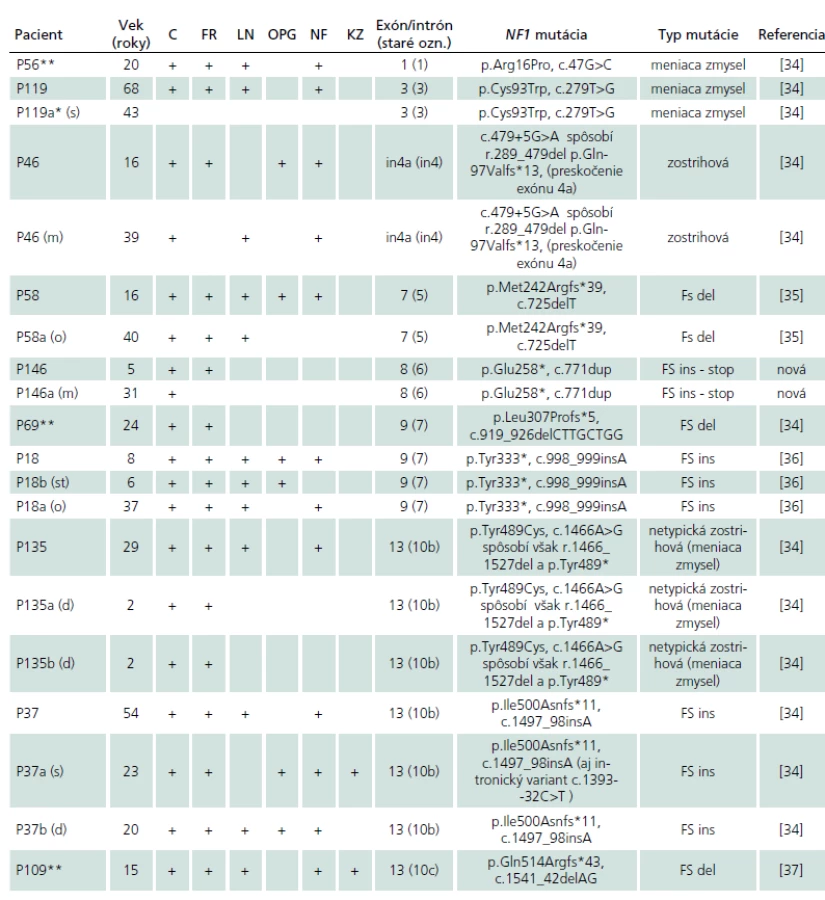


U 41 rodín bol v anamnéze uvádzaný familiárny výskyt NF1, avšak šesť z nich pochádzalo práve zo skupiny pacientov bez identifikovanej NF1 mutácie. Zo zvyšných 35 rodín v dvoch rodinách sme na základne genetickej analýzy u príbuzných familiárny výskyt nepotvrdili (P21 a P41, tab. 2b) a v šiestich rodinách sme nemali k dispozícii DNA rodinných príslušníkov. Na základe identifikácie identickej NF1 mutácie sme teda familiárny výskyt NF1 potvrdili celkovo v 27 slovenských rodinách. V súlade s publikovanými výsledkami sme aj v týchto rodinách pozorovali vysokú fenotypovú variabilitu NF1 znakov (tab. 2a).
V 42 rodinách sme testovali prítomnosť identifikovanej NF1 mutácie u oboch rodičov pacienta. V 27 (64,3 %) z nich bol potvrdený vznik mutácie de novo, vo zvyšných 15 rodinách sme potvrdili familiárny výskyt ochorenia (tab. 2b).



Z 83 mutácií identifikovaných v našom súbore bolo 41 (49,4 %) mutácií nových, identifikovaných zatiaľ len v slovenskej populácii. Tridsať osem z nich sme prezentovali v práci Nemethova et al [34] a tri sú prvý krát popísané v tejto práci. Dve z nových mutácií sú duplikácie: v exónoch 6(c.771dupT, p.Glu258*, pacient P146) a 16 (c.2598_2601dup, p.Gly868Hisfs*6, pacient P144), a tretia je delécia v exóne 19b (c.3258_65del, p.Pro1087Argfs*16, pacient P140) (tab. 2a, b).
V našom súbore sme pozorovali tiež prítomnosť troch rekurentných mutácií, z ktorých každá bola pozorovaná v rovnakej pozícii nezávisle u dvoch nepríbuzných pacientov: v exónoch 12a (c.1756_59delACTA, p.Thr586Valfs*18, P87, P118), 22 (c.3826C>T, p.Arg1276*, P54, P61) [33] a 10c (c.1541_42delAG, p.Gln514Argfs*43, P109, P129) (tab. 2a, b).
Netypické zostrihové mutácie
Zo 14 zostrihových mutácií bolo deväť (64,3 %) typických, postihujúcich klasické konsenzus oblasti, a päť (35,7%) bolo netypických. Dve netypické zostrihové mutácie sa nachádzali hlbšie v intoronickej oblasti. U pacienta P83 to je malá substitúcia v pozícii -19 intrónu 9, ktorá spôsobí aktiváciu kryptického akceptorového miesta v tomto intróne a inzerciu 17 nukleotidov do mRNA transkriptu a následný posun čítacieho rámca. V prípade pacienta P90, v dôsledku delécie 20 nukleotidov zahŕňajúcich aj akceptorové zostrihové miesto intrónu 28, dochádza k delécii exónu 29 alebo k delécii exónov 29 a 30 naraz, čo vedie ku vzniku predčasného terminačného kodónu a ku skráteniu NF1 proteínu (tab. 2b).
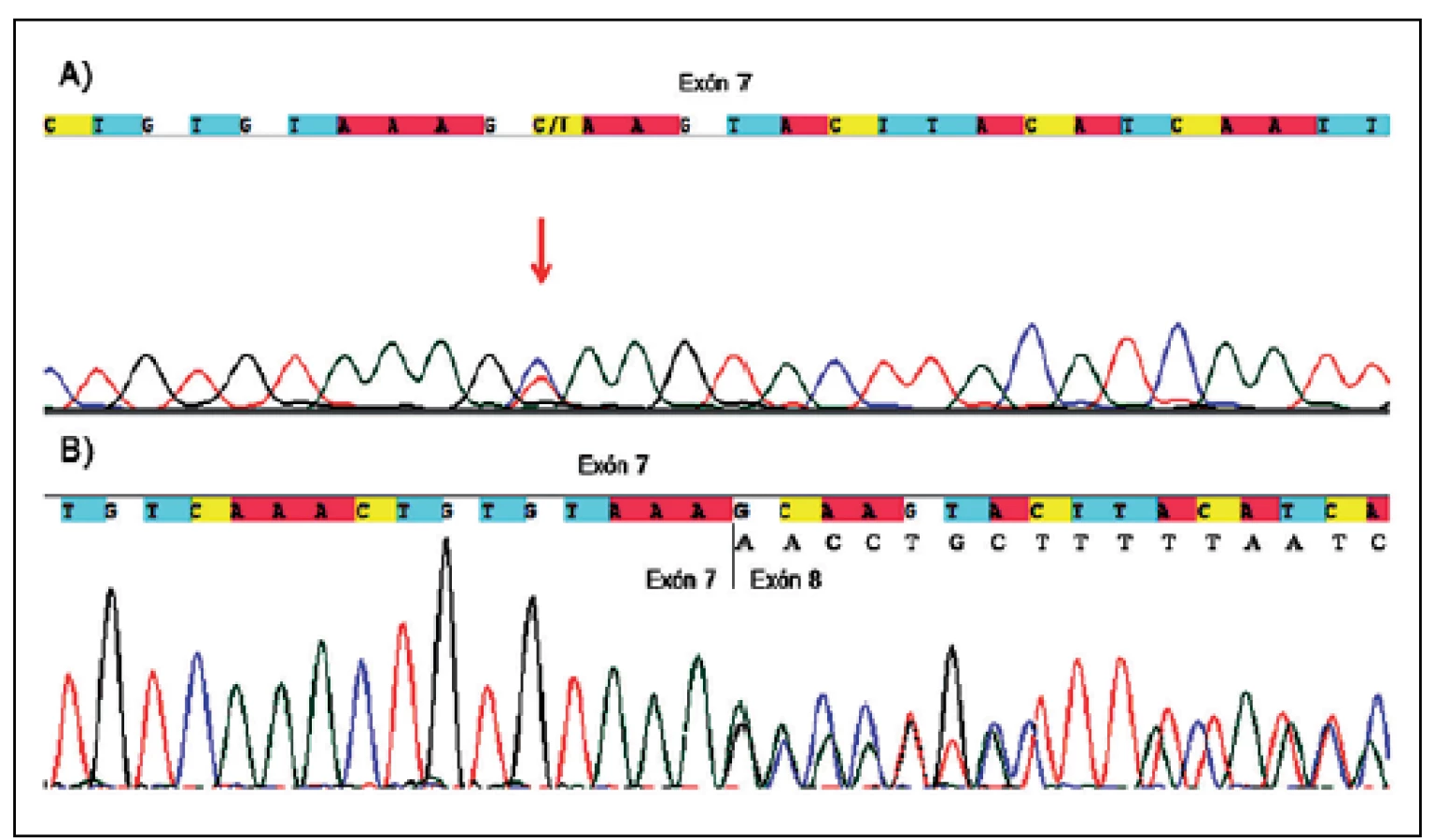
Ďalšie netypické zostrihové mutácie by boli bez analýzy cDNA popísané len ako meniace zmysel. Ide konkrétne o mutácie v exóne 7 (c.989C>T , p.Ala330Val, P68) (obr. 1a) a v exóne 10b (c.1466A>G, p.Tyr489Cys, P135), pri ktorých však došlo k aktivácií kryptického donorového zostrihového miesta, čo v prvom prípade viedlo k delécii časti exónu 7 (p.Ala330_Lys354del, r.988_1062 del 75bp) (obr. 1b) a v druhom k delécii časti exónu 10b a následnému vzniku predčasného stop kodónu hneď po tyrozíne v pozícii 489 (p.Tyr489*, r.1466_1527 del 62bp).
Mutácia c.4268A>G (p.Lys1423Arg) v exóne 24 bola v literatúre tiež popísaná ako mutácia meniaca zmysel [34]. U pacienta P139 (tab. 2b) sme však pozorovali preskočenie exónu 24 (r.4111_4269del147, p.Val1371_Lys1423del), pravdepodobne v dôsledku oslabenia donorového miesta a zrušenia exonického enhancerového miesta v dôsledku uvedenej substitúcie (predikcie pomocou Human Splicing Finder).

Nepatogénne varianty
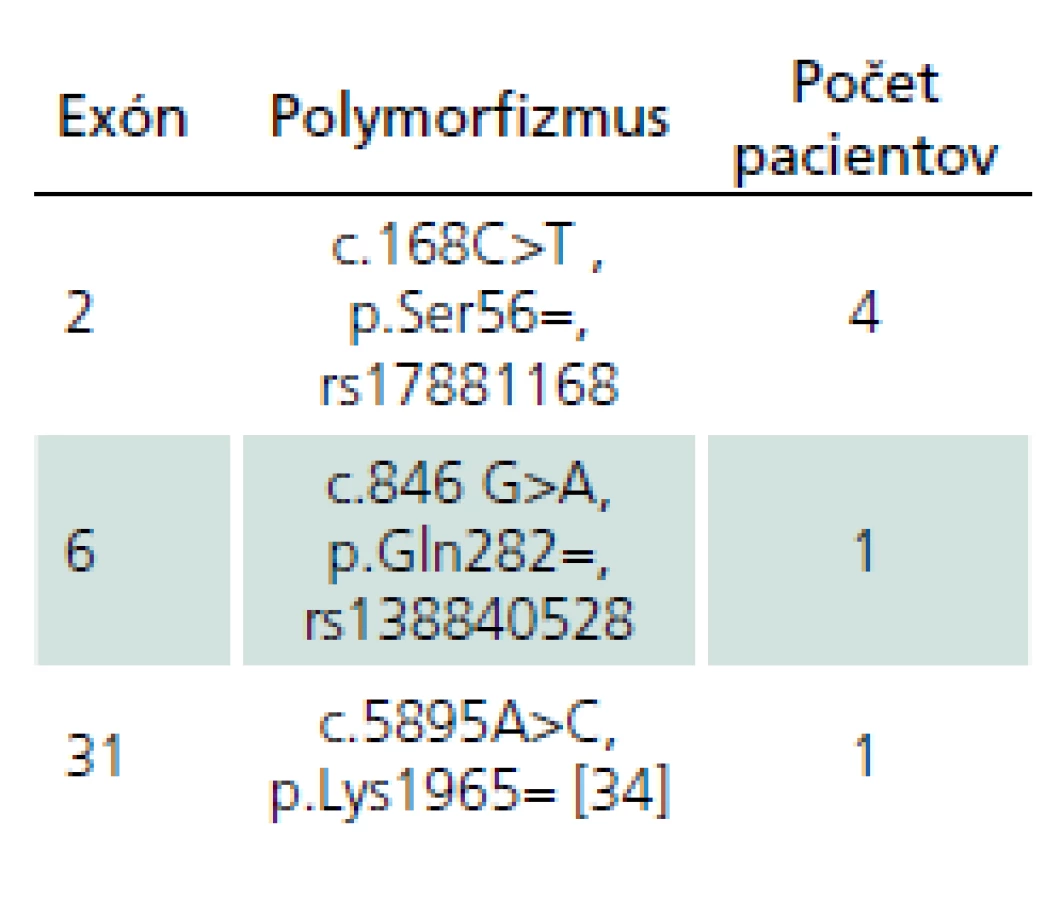
Okrem kauzatívnych mutácií sme u šiestich pacientov pozorovali aj prítomnosť tzv. tichých variantov v exónoch, ktoré sumarizujeme v tab. 3. U žiadneho z nich nebol zatiaľ popísaný vplyv na patogenézu NF1.
V dvoch rodinách sme identifikovali aj prítomnosť intronických variantov. V prvej z nich (P37) bola prítomná posunová mutácia c.1497_98insA (p.Ile500Asnfs*11) u otca, dcéry a syna, pričom syn bol nositeľom aj intronického variantu c.1393-32C>T (rs2905876) (tab. 2a) [34].
V druhej rodine (P41) popri mutácii meniacej zmysel v exóne 28, c.4981T>C (p.Cys1661Arg), bol u pacientky identifikovaný aj variant c.5205+23T>C (rs9894648) v intróne 28, ktorý bol však pozorovaný aj u jej matky, ktorá nebola nositeľkou patogénnej mutácie (tab. 2b).
Zastúpenie základných klinických znakov
V našom súbore sme zo základných klinických znakov u 83 nepríbuzných pacientov zachytili až 100% (83/ 83) výskyt café au lait škvŕn a u 89 % (74/ 83) freckling. Ďalšie diagnostické znaky sa vyskytovali menej často: Lischove noduly 32,5 % (27/ 83), očný glióm 36 % (30/ 83), neurofibrómy 60 % (50/ 83), kostné zmeny 5 % (4/ 83).
Fenotypovo-genotypové korelácie
Pri fenotypovo genotypových porovnaniach sme zistili, že pacienti s mutáciou meniacou zmysel mali vyšší výskyt Lischových nodulov (50 %, 5/ 10) a neurofibrómov (80%, 8/ 10) v porovnaní s pacientami s mutáciou spôsobujúcou zmenu dĺžky proteínu (posunové mutácie, nezmyselné mutácie, delécie celého génu alebo jeho časti), kde sa Lischove noduly vyskytovali u 28,8 % (21/ 73) pacientov a neurofibrómy u 54,8 % (40/ 73).
Očný glióm bol najmenej často prítomný u pacientov s mutáciou meniacou zmysel (2/ 10; 20%). Jeho výskyt je však vyšší u pacientov s mutáciami na 5’ konci NF1 génu, t.j. v prvých 16 exónoch. V našom súbore pacientov ide zatiaľ o 12/ 21 (57,1 %) takýchto prípadov [59].
NF1 pacienti bez mutácie
U 11/ 94 pacientov spĺňajúcich diagnostické kritéria sme ani sekvenovaním ani MLPA neidentifikovali žiadnu kauzatívnu NF1 mutáciu. Dvaja z nich mali znaky segmentálnej NF1, u ktorých pravdepodobne došlo k mutácii v somatických bunkách.
Zastúpenie analyzovaných NF1 pacientov z jednotlivých krajov Slovenska
Pacienti, prípadne vzorky pacientov s podozrením na neurofibromatózu typu 1, k nám prichádzajú zo všetkých krajov Slovenska, pričom prevažovali pacienti zo západného Slovenska (graf 2). Na grafu je znázornená aj početnosť pozitívnych a negatívnych výsledkov pacientov v rámci jednotlivých krajov.

Diskusia
V našej práci sme ukázali, že nami zvolená analýza cDNA je efektívna metóda identifikácie mutácií v tak veľkom géne, ako je NF1. V súbore 94 pacientov, ktorí spĺňali NIH diagnostické kritériá, sme mutáciu úspešne identifikovali u 83 z nich (88,3 %). Takmer polovica (41/ 83) identifikovaných NF1 mutácií bola zatiaľ popísaná len v slovenskej populácii.
Vďaka našej metóde sme správne odhalili a klasifikovali až päť netypických zostrihových mutácií. Podiel netypických zostrihových mutácií z celkového počtu zostrihových zmien v našom súbore je 35,7 % (5/ 14), a je teda vyšší než 30 % uvádzaných v práci Messiaenovej a Wimmerovej [60], kde charakterizujú doposiaľ najväčší súbor NF1 pacientov.
V našej populácii u 83 nepríbuzných pacientov sú najčastejšími posunové (43 %), zostrihové (17 %) mutácie a nezmyselné mutácie (13 %), ktoré však v práci Messiaenovej a Wimmerovej [24] predstavovali 26, 29 a 23 %. Rozdiel sme pozorovali aj vo frekvencii výskytu rozsiahlych vnútrogénových delécií. V našom súbore predstavovali až 6 % v porovnaní s 2,5 % vo vyššie uvedenej štúdii.
V porovnaní so štúdiou z Českej republiky z roku 2007 [61], v ktorej analyzovali 22 nepríbuzných pacientov, sme pozorovali približne rovnaké zastúpenie jednotlivých druhov mutácií. Len jedna mutácia bola pozorovaná v oboch populáciách, konktrétne posunová mutácia v exóne 4b (c.499_502delTGTT, p.Cys167Glnfs*10) identifikovaná u pacienta P12.
Podiely jednotlivých typov mutácií v našom súbore sme porovnali aj s inými menšími súbormi, u ktorých však tiež boli pozorované rozdielne frekvencie v porovnaní so súborom 2 900 NF1 pacientov Messiaenovej a Wimmerovej [24]. Potvrdilo sa nám však pozorovanie, že u slovenských pacientov je tendencia k vyššiemu podielu malých inzercií a delécií (43 %) a nižšiemu podielu nezmyselných mutácií (13 %) v porovnaní so zodpovedajúcimi podielmi 18-27,5 % a 23-37 % uvádzanými v citovaných prácach [23,48,52,62-66]. Pozorované odlišnosti môžu teda predstavovať odchýlku spôsobenú malým počtom pacientov v našom súbore, alebo odrážajú skutočne rozdielne mutačné spektrum NF1 génu v slovenskej populácii. Tieto pozorovanie je potrebné overiť na väčšom súbore.
V NF1 géne neboli popísané žiadne hot spot oblasti, mutácie sú distribuované po celej dĺžke génu. Za hlavnú funkciu NF1 proteínu ako negatívneho regulátora Ras onkogénu sú zodpovedné hlavne domény CSRD a Ras-GRD, preto sme predpokladali akumuláciu mutácií práve v nich. V našom súbore boli mutácie v CSRD a Ras-GRD prítomné u 36,1 % (30/ 83) pacientov. Tieto domény pokrývajú približne 1/ 3 (33,3 %) celej sekvencie NF1 génu (17/ 58 exónov exprimovaných v leukocytoch), nejde teda o štatisticky významný výsledok.
V literatúre sa uvádza, že až 50 % NF1 mutácií vzniká de novo [21]. V našom súbore 42 analyzovaných rodín je to až 64,3 %.
V prípade NF1 dôležitú skupinu mutácií predstavujú zostrihové mutácie. V našom súbore zo 14 takýchto mutácií bolo deväť (64,3 %) typických, postihujúcich klasické konsenzus oblasti, a päť (35,7 %) netypických. Dve netypické zostrihové mutácie sa nachádzali hlbšie v intoronickej oblasti a zvyšné tri postihovali exóny, pričom bez analýzy cDNA by boli popísané len ako meniace zmysel. Naša metodika však správne odhalila ich efekt na zostrih. Napríklad mutácia v exóne 24, c.4268A>G, je v databázach uvádzaná ako klasická mutácia meniaca zmysel (p.Lys1423Arg) (P139) [56]. Analýza našou metodikou však jednoznačne ukazuje, že táto jednonukleotidová zámena ovplyvňuje zostrih a vedie k vynechaniu exónu 24 z mRNA (p.Val1371_Lys1423del) [34].
Vo všeobecnosti pri NF1 nie sú známe jednoznačné fenotypovo-genotypové korelácie. Zvýšený výskyt očného gliómu u pacientov nesúcich germinálnu mutáciu na 5’ konci NF1 génu (exóny 1-16), ktorý sme pozorovali v našom súbore [59], koreluje s výsledkami nedávno publikovanej práce Sharifa et al [35].
Ďalšou možnou koreláciou zistenou na menšom súbore pacientov je zvýšená prítomnosť mozgových gliómov a MPNST u pacientov so zostrihovou mutáciou [36], ktoré v prípade NF1 predstavujú takmer 1/ 3 všetkých mutácií [60].
Výskumy tiež ukázali, že NF1 pacienti s mikrodeléciou v NF1 géne majú vyššie riziko vzniku MPNST ako pacienti s inou mutáciou [41], čo sa však v našom súbore zatiaľ nepotvrdilo. Rizikovým vekom pre vznik MPNST je 20-30 rokov. Štyri pätiny pacientov s mikrodeléciou z nášho súboru sú mladší, čiže je ešte možné, že v rizikovom období sa u nich MPNST vyvinie.
Za predpokladu, že incidencia ochorenia je 1 : 3 500 [1] a je rovnomerne rozšírené na Slovensku, naša záchytnosť pacientov s NF1 je ešte príliš nízka. V našej práci budeme pokračovať s cieľom vytvoriť čo najväčšiu spoločnú databázu NF1 pacientov, aby bolo možné hodnotiť fenotypovo-genotypové korelácie v čo najväčšom súbore pacientov.
Použitím našich metód sme u 11 pacientov, ktorí jednoznačne spĺňali NF1 diagnostické kritériá, nenašli patogénne NF1 mutácie. Celkovo je naša metodika efektívna a záchytnosť mutácií 88,3 % je vysoká. Je však stále možné, že uvedení pacienti sú nositeľmi mutácie, ktorá sa nachádza v promótorovej oblasti, či v časti NF1 génu, ktorý nebol dostatočne pokrytý našimi PCR primermi, alebo mutácia nebola našimi metódami zachytená. U dvoch z týchto pacientov (P36, P59) klinické znaky naznačujú, že môže ísť o segmentálnu neurofibromatózu, u ktorej mutácie nie sú prítomné v bunkách lymfocytov, ale len v bunkách postihnutého segmentu. Tieto sme však nemali k dispozícii pre analýzu prípadných somatických NF1 mutácií.
Analýza NF1 génu bola robená u všetkých pacientov, ktorých vzorka bola doručená na naše pracovisko. U 13 pacientov, ktorí v skutočnosti nespĺňali základné klinické kritéria, sme sa tiež rozhodli analýzu uskutočniť. Jednak kvôli požiadavke klinických genetikov za účelom diferenciálnej diagnostiky, jednak kvôli testovaniu metodiky v čase jej zavádzania. Nakoľko NF1 mutácia u týchto pacientov s nejasným fenotypom nebola identifikovaná, diagnóza klasickej NF1 bola vylúčená a pokračujú analýzy s cieľom prehodnotiť alternatívne diagnózy, ako sú NF2, schwanomatóza, Noonan syndróm, Leopard syndróm, McCune-Albright syndróm, Legius syndróm [40], kranio-facio-kutánny syndróm (CFC) a Costello syndróm [42,43].
V tejto skupine pacientov boli zatiaľ geneticky potvrdené dva prípady schwanomatózy (mutácia v géne INI1) a jeden pacient s Legius syndrómom (mutácia v géne SPRED1). Na základe klinických znakov bola u troch pacientov stanovená diagnóza McCune Albright syndróm.
NF1 si vyžaduje v prvom rade dôkladné úvodné klinické vyšetrenie a zhodnotenie diagnostických kritérií. Ak je pacient vyšetrovaný v ranom detskom veku, nie vždy sú u neho vyvinuté už všetky NF1 znaky, nakoľko 100% penetrancia ochorenia je do ôsmeho roku života [2]. Práve v týchto prípadoch, ako aj v prípade netypických NF1, u ktorých klinické znaky nie sú jednoznačné, testovanie prítomnosti NF1 mutácie napomôže potvrdeniu diagnózy, a tým aj výberu vhodného terapeutického postupu.
Metodika je pomerne nákladná a časovo náročná, nemala by teda nahrádzať klinické vyšetrenie, ani zhodnotenie alternatívnych diagnóz u nejasných prípadov. Je potrebné v každom prípade individuálne zvážiť tieto zdanlivo protikladné argumenty, či pacienta na mutácie analyzovať, alebo nie. Ide o pomerne novú diagnostiku u nás, ktorú sme zaviedli do praxe i v novo vzniknutom a jedinom Centre komplexnej starostlivosti o deti s neurofibromatózou, ktoré vzniklo pod záštitou II. detskej kliniky LF UK a DFNsP v Bratislave. Naša skúsenosť ukazuje, že mutačná analýza môže efektívne usmerniť proces diferenciálnej diagnostiky NF1 na Slovensku.
Mgr. Andrea Zaťková, PhD.
Laboratórium genetiky
ÚMFG SAV
Vlárska 5
833 34 Bratislava
e-mail: andrea.zatkova@savba.sk
Prijato k recenzii: 12. 5. 2014
Prijato do tlače: 28. 8. 2014
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Sources
1. Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol 2009; 61(1): 1-14. doi: 10.1016/ j.jaad.2008.12.051.
2. DeBella K, Szudek J, Friedman JM. Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics 2000; 105(3 Pt 1):608-614.
3. Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol 1988; 45(5): 575-578.
4. Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet 2002; 39(5): 311-314.
5. Cichowski K, Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell 2001; 104(4): 593-604.
6. Brannan CI, Perkins AS, Vogel KS, Ratner N, Nordlund ML, Reid SW et al. Targeted disruption of the neurofibromatosis type 1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev 1994; 8(9): 1019-1029.
7. Viskochil D, Buchberg AM, Xu G, Cawthon RM, Stevens J, Wolff RK et al. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell 1990; 62(1): 187-192.
8. Cawthon RM, O‘Connell P, Buchberg AM, Viskochil D, Weiss RB, Culver M et al. Identification and characterization of transcripts from the neurofibromatosis 1 region: the sequence and genomic structure of EVI2 and mapping of other transcripts. Genomics 1990; 7(4): 555-565.
9. Wallace MR, Marchuk DA, Andersen LB, Letcher R,Odeh HM, Saulino AM et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science 1990; 249(4965): 181-186.
10. Li Y, O‘Connell P, Breidenbach HH, Cawthon R, Stevens J, Xu G et al. Genomic organization of the neurofibromatosis 1 gene (NF1). Genomics 1995; 25(1): 9-18.
11. Skuse GR, Cappione AJ. RNA processing and clinical variability in neurofibromatosis type I (NF1). Hum Mol Genet 1997; 6(10): 1707-1712.
12. Stocker KM, Baizer L, Coston T, Sherman L, Ciment G. Regulated expression of neurofibromin in migrating neural crest cells of avian embryos. J Neurobiol 1995; 27(4): 535-552.
13. Williams VC, Lucas J, Babcock MA, Gutmann DH,Korf B, Maria BL. Neurofibromatosis type 1 revisited. Pediatrics 2009; 123(1): 124-133. doi: 10.1542/ peds.2007-3204.
14. Martin GA, Viskochil D, Bollag G, McCabe PC, Crosier WJ, Haubruck H et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 1990; 63(4): 843-849.
15. Xu GF, O‘Connell P, Viskochil D, Cawthon R, Robertson M, Culver M et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 1990; 62(3): 599-608.
16. Izawa I, Tamaki N, Saya H. Phosphorylation of neurofibromatosis type 1 gene product (neurofibromin) by cAMP-dependent protein kinase. FEBS Lett 1996; 382(1-2): 53-59.
17. Mangoura D, Sun Y, Li C, Singh D, Gutmann DH,Flores A et al. Phosphorylation of neurofibromin by PKC is a possible molecular switch in EGF receptor signaling in neural cells. Oncogene 2006; 25(5): 735-745.
18. Korf BR, Rubenstein AE. Neurofibromatosis: a handbook for patients, families, and health care professionals. 2nd ed. New York: NY Thieme Medical Publishers 2005.
19. Viskochil D. Genetics of neurofibromatosis 1 and the NF1 gene. J Child Neurol 2002; 17(8): 562-570.
20. Yunoue S, Tokuo H, Fukunaga K, Feng L, Ozawa T,Nishi T et al. Neurofibromatosis type I tumor suppressor neurofibromin regulates neuronal differentiation via its GTPase-activating protein function toward Ras. J Biol Chem 2003; 278(29): 26958-26969.
21. Radtke HB, Sebold CD, Allison C, Haidle JL, Schneider G. Neurofibromatosis type 1 in genetic counseling practice: recommendations of the National Society of Genetic Counselors. J Genet Couns 2007; 16(4): 387-407.
22. Kern SE, Winter JM. Elegance, silence and nonsense in the mutations literature for solid tumors. Cancer Biol Ther 2006; 5(4): 349-359.
23. Kluwe L, Siebert R, Gesk S, Friedrich RE, Tinschert S, Kehrer-Sawatzki H et al. Screening 500 unselected neurofibromatosis 1 patients for deletions of the NF1 gene. Hum Mutat 2004; 23(2): 111-116.
24. Messiaen L, Wimmer K. Mutation analysis of the NF1 gene by cDNA-based sequencing of the coding region. In: Cunha KSG, Geller M (eds). Advances in neurofibromatosis research. New York: Nova Science 2012 : 89-108.
25. Pasmant E, Sabbagh A, Spurlock G, Laurendeau I, Grillo E, Hamel MJ et al. NF1 microdeletions in neurofibromatosis type 1: from genotype to phenotype. Hum Mutat 2010; 31(6): E1506-E1518. doi: 10.1002/ humu.21271.
26. Chang YF, Imam JS, Wilkinson MF. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem 2007; 76 : 51-74.
27. Den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat 2000; 15(1): 7-12.
28. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7(4): 248-249. doi: 10.1038/ nmeth0410-248.
29. Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res 2007; 35(11): 3823-3835.
30. Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PloS One 2012; 7(10): e46688. doi: 10.1371/ journal.pone.0046688.
31. Choi Y. A fast computation of pairwise sequence alignment scores between a protein and a set of single-locus variants of another protein. In: Proceedings of the ACM Conference on bioinformatics, computational biology and biomedicine (BCB‚12). ACM, New York, NY, USA 2012 : 414-417.
32. Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M et al. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 2009; 37(9): e67. doi: 10.1093/ nar/ gkp215.
33. Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol 1997; 4(3): 311-323.
34. Nemethova M, Bolcekova A, Ilencikova D, Durovcikova D, Hlinkova K, Hlavata A et al. Thirty-nine novel neurofibromatosis 1 (NF1) gene mutations identified in Slovak patients. Ann Hum Genet 2013; 77(5): 364-379. doi: 10.1111/ ahg.12026.
35. Sharif S, Upadhyaya M, Ferner R, Majounie E, Shenton A, Baser M et al. A molecular analysis of individuals with neurofibromatosis type 1 (NF1) and optic pathway gliomas (OPGs), and an assessment of genotype-phenotype correlations. J Med Genet 2011; 48(4): 256-260. doi: 10.1136/ jmg.2010.081760.
36. Alkindy A, Chuzhanova N, Kini U, Cooper DN, Upadhyaya M. Genotype-phenotype associations in neurofibromatosis type 1 (NF1): an increased risk of tumor complications in patients with NF1 splice-site mutations? Hum Genomics 2012; 6 : 12. doi: 10.1186/ 1479-7364-6-12.
37. Robinson PN, Buske A, Neumann R, Tinschert S, Nurnberg P. Recurrent 2-bp deletion in exon 10c of the NF1 gene in two cases of von Recklinghausen neurofibromatosis. Hum Mutat 1996; 7(1): 85-88.
38. Park VM, Pivnick EK. Neurofibromatosis type 1(NF1): a protein truncation assay yielding identification of mutations in 73% of patients. J Med Genet 1998; 35(10): 813-820.
39. Fahsold R, Hoffmeyer S, Mischung C, Gille C, Ehlers C, Kücükceylan N et al. Minor lesion mutational spectrum of the entire NF1 gene does not explain its high mutability but points to a functional domain upstream of the GAP-related domain. Am J Hum Genet 2000; 66(3): 790-818.
40. Viskochil D, Carey JC. Alternate and related forms of the neurofibromatoses In: Huson SM, Hughes RA (eds). The Neurofibromatoses a clinical and pathogenetic overview. London: Chapman Hall 1994 : 445-474.
41. De Readt T, Brems H, Wolkenstein P, Vidaud D, Pilotti S, Perrone F et al. Elevated risk for MPNST in NF1 microdeletion patients. Am J Hum Genet 2003; 72(5): 1288-1292.
42. Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, Cruz MS et al. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science 2006; 311(5765): 1287-1290.
43. Stevenson DA, Swensen JJ, Viskochil DH. Neurofibromatosis type 1 and other syndromes of the Ras Pathway. In: Kaufman D et al (eds). Neurofibromatoses. Basel: Karger 2008 : 32-45.
44. Tinschert S, Naumann I, Stegmann E, Buske A, Kaufmann D, Thiel G et al. Segmental neurofibromatosis is caused by somatic mutation of the neurofibromatosis type 1 (NF1) gene. Eur J Hum Genet 2000; 8(6): 455-459.
45. Lee MJ, Su YN, You HL, Chiou SC, Lin LC, Yang CC et al. Identification of forty-five novel and twenty-three known NF1 mutations in Chinese patients with neurofibromatosis type 1. Hum Mutat 2006; 27(8): 832.
46. Abernathy CR, Colman SD, Kousseff BG, Wallace MR. Two NF1 mutations: frameshift in the GAP-related domain, and loss of two codons toward the 3‘ end of the gene. Hum Mutat 1994; 3(4): 347-352.
47. Osborn MJ, Upadhyaya M. Evaluation of the protein truncation test and mutation detection in the NF1 gene: mutational analysis of 15 known and 40 unknown mutations. Hum Genet 1999; 105(4): 327-332.
48. Wimmer K, Yao S, Claes K, Kehrer-Sawatzki H, Tinschert S, De Raedt T et al. Spectrum of single - and multiexon NF1 copy number changes in a cohort of 1,100 unselected NF1 patients. Genes Chromosomes. Cancer 2006; 45(3): 265-276.
49. Upadhyaya M, Spurlock G, Monem B, Thomas N, Friedrich RE, Kluwe L et al. Germline and somatic NF1 gene mutations in plexiform neurofibromas. Hum Mutat 2008; 29(8): E103-E111. doi: 10.1002/ humu.20793.
50. Ars E, Kruyer H, Morell M, Pros E, Serra E, Ravella Aet al. Recurrent mutations in the NF1 gene are common among neurofibromatosis type 1 patients. J Med Genet 2003; 40(6): e82.
51. Heim RA, Kam-Morgan LN, Binnie CG, Corns DD,Cayouette MC, Farber RA et al. Distribution of 13 truncating mutations in the neurofibromatosis 1 gene. Hum Mol Genet 1995; 4(6): 975-981.
52. Griffiths S, Thompson P, Frayling I, Upadhyaya M.Molecular diagnosis of neurofibromatosis type 1 : 2 years experience. Fam Cancer 2007; 6(1): 21-34.
53. Wimmer K, Roca X, Beiglbock H, Callens T, Etzler J,Rao AR et al. Extensive in silico analysis of NF1 splicing defects uncovers determinants for splicing outcome upon 5‘ splice-site disruption. Hum Mutat 2007; 28(6): 599-612.
54. General information. LOVD Gene homepage [on-line]. Available from URL: https:/ / grenada.lumc.nl/ LOVD2/ mendelian_genes/ home.php?select_db=NF1.
55. Origone P, Bellini C, Sambarino D, Banelli B, Morcaldi G, La RC et al. Neurofibromatosis type 1 (NF1): Identification of eight unreported mutations in NF1 gene in Italian patients. Hum Mutat 2003; 22(2): 179-180.
56. Han SS, Cooper DN, Upadhyaya MN. Evaluation of denaturing high performance liquid chromatography (DHPLC) for the mutational analysis of the neurofibromatosis type 1 (NF1) gene. Hum Genet 2001; 109(5): 487-497.
57. Bottillo I, Ahlquist T, Brekke H, Danielsen SA, van den Berg E, Mertens F et al. Germline and somatic NF1 mutations in sporadic and NF1-associated malignant peripheral nerve sheath tumours. J Pathol 2009; 217(5): 693-701. doi: 10.1002/ path.2494.
58. Mattocks C, Baralle D, Tarpey P, French-Constant C,Bobrow M, Whittaker J. Automated comparative sequence analysis identifies mutations in 89% of NF1 patients and confirms a mutation cluster in exons 11-17 distinct from the GAP related domain. J Med Genet 2004; 41(4): e48.
59. Bolcekova A, Nemethova M, Zatkova A, Hlinkova K, Pozgayova S, Hlavata A et al. Clustering of mutations in the 5‘ tertile of the NF1 gene in Slovakia patients with optic pathway glioma. Neoplasma 2013; 60(6): 655-665. doi: 10.4149/ neo_2013_084.
60. Messiaen LM, Wimmer K. NF1 mutational spectrum. In: Kaufman D et al (eds). Neurofibromatoses. Basel: Karger 2008 : 63-77.
61. Bendova S, Krepelova A, Petrak B, Kinstova L, Musova Z, Rausova E et al. Novel mutations in the NF1 gene in Czech patients with neurofibromatosis type 1. J Mol Neurosci 2007; 31(3): 273-279.
62. Cnossen MH, van der Est MN, Breuning MH, van Asperen CJ, Breslau-Siderius EJ, van der Ploeg AT et al. Deletions spanning the neurofibromatosis type 1 gene: implications for genotype-phenotype correlations in neurofibromatosis type 1? Hum Mutat 1997; 9(5): 458-464.
63. Lázaro C, Gaona A, Ainsworth P, Tenconi R, Vidaud D, Kruyer H et al. Sex differences in mutational rate and mutational mechanism in the NF1 gene in neurofibromatosis type 1 patients. Hum Genet 1996; 98(6): 696-699.
64. Messiaen LM, Callens T, Mortier G, Beysen D, Vandenbroucke I, Van RN et al. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum Mutat 2000; 15(6): 541-555.
65. Rasmussen SA, Colman SD, Ho VT, Abernathy CR,Arn PH, Weiss L et al. Constitutional and mosaic large NF1 gene deletions in neurofibromatosis type 1. J Med Genet 1998; 35(6): 468-471.
66. Valero MC, Martin Y, Hernandez-Imaz E, Marina Hernandez A, Melean G, Valero AM et al. A highly sensitive genetic protocol to detect NF1 mutations. J Mol Diagn 2011; 13(2): 113-122. doi: 10.1016/ j.jmoldx.2010.09.002.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2014 Issue 6
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Memantine Eases Daily Life for Patients and Caregivers
Most read in this issue
- Diazepam i. m. – nejčastěji užívaný, ale nevhodný lék ke zvládání akutní úzkosti, agitovanosti a agresivity
- Střelné poranění hlavy replikou historické zbraně – patofyziologie a popis kazuistiky
- Test neverbální fluence – Five Point Test: normativní data pro dospělé
- Adultní forma Pompeho nemoci