
Diferenciální diagnostika tauopatií – klinický pohled
Differential Diagnosis of Tauopathies – a Clinical Approach
Tauopathies are neurodegenerative disorders characterized by accumulation of abnormally modified forms of the tau-protein, predominantly in frontal, temporal and parietal cortical regions, basal ganglia and in the midbrain. Tauopathies are well defined from the molecular biological and biochemical point of view; clinical symptoms may be, however, heterogeneous. Common signs of frontotemporal lobar degenerations include a more or less prominent syndrome of frontotemporal dementia. On the other hand, the clinical picture of frontotemporal dementia may not be caused by a tauopathy only; in many cases the underlying etiopathogenic cause is different. The aim of our review is to classify the relationship between tauopathies and frontotemporal lobar degenerations. This review contains a proposal of a practical approach to refining clinical diagnosis of the different tauopathies.
Key words:
neurodegeneration – tauopathy – frontotemporal lobar degeneration –progressive supranuclear palsy – progressive aphasia – corticobasal syndrome
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Authors:
R. Rusina 1,2; R. Matěj 3,4; E. Růžička 1; J. Roth 1
Authors‘ workplace:
Neurologická klinika a Centrum klinických neurověd, 1. LF UK a VFN v Praze
1; Neurologické oddělení, Thomayerova nemocnice, Praha
2; Oddělení patologie a molekulární medicíny, Thomayerova nemocnice, Praha
3; Ústav patologie, 3. LF UK v Praze
4
Published in:
Cesk Slov Neurol N 2015; 78/111(5): 526-534
Category:
Review Article
doi:
https://doi.org/10.14735/amcsnn2015526
Overview
Tauopatie jsou neurodegenerativní onemocnění, která se vyznačují hromaděním abnormně modifikovaných forem proteinu zvaného tau především v kůře frontálních, temporálních a parietálních laloků, v bazálních gangliích a v mezencefalu. Molekulárně biologicky a biochemicky dobře definovaná skupina chorob má však dosti heterogenní klinické obrazy a je podstatou několika různých neurodegenerativních onemocnění tvořících frontotemporální lobární degenerace. Společným klinickým znakem těchto entit je ve větší či menší míře vyjádřený syndrom frontotemporální demence. Klinické jednotky, jež se řadí mezi klasické frontotemporální demence, však nejsou vždy podmíněny jen tauopatiemi, mnohdy je jejich etiopatogenetická podstata jiná. Účelem tohoto stručného přehledu je utřídění současné úrovně poznání vztahů mezi skupinou tauopatií a frontotemporálních lobárních degenerací. Článek obsahuje i návrh praktického postupu k upřesnění klinické diagnózy jednotlivých nozologických jednotek v rámci tauopatií.
Klíčová slova:
neurodegenerace – tauopatie – frontotemporální lobární degenerace – progresivní supranukleární obrna – progresivní afázie – kortikobazální syndrom
Úvod
Tauopatie tvoří skupinu neurodegenerativních chorob způsobených poruchou intracelulárního metabolizmu proteinu tau [1]. Název tohoto proteinu je odvozen z anglického označení „tubulin associated unit“ a značí, že jde o bílkovinu, která se po fosforylaci váže na tubulin mikrotubulů, startuje jejich polymerizaci a účastní se aktivně intracelulárního transportu. Jakmile je defosforylována, podílí se na stabilizaci mikrotubulů [2].
Ve zdravém mozku se tau protein vyskytuje v šesti izoformách o molekulové hmotnosti 50 – 65 kDa obsahujících 352 – 441 aminokyselin. Je kódován genem MAPT (Microtubulin Associated Unit) na 17. chromozomu. Bílkovina, která fyziologicky vstupuje do interakce s mikrotubuly a má klíčovou roli v intracelulárním transportu, je za patologických okolností hyperfosforylována na specifických vazebných místech. To má za důsledek ukládání obtížně odbouratelných proteinových agregátů ve formě i velmi objemných inkluzí. Konečným důsledkem hromadění abnormálně agregovaného tau proteinu a výpadkem funkce fyziologického tau proteinu je apoptóza postižené buňky [1].
Molekulárně biologicky a biochemicky dobře definovaná skupina tauopatií má dosti heterogenní klinické obrazy a je podstatou několika různých neurodegenerativních onemocnění tvořících skupinu frontotemporálních lobárních degenerací (FTLD). V současnosti jsou FTLD chápány jako skupina onemocnění, která buď spadají mezi tauopatie, nebo v centru jejich patofyziologie stojí jiný protein asociovaný s tvorbou proteinových depozit (např. TDP ‑ 43 či ubikvitin). Klinický obraz tauopatií a non‑tau FTLD se v mnoha případech překrývá a často jsou prakticky jednotlivá onemocnění od sebe klinicky nerozlišitelná a pravou podstatu onemocnění může odhalit až autoptické neuropatologické vyšetření mozku.
Z klinického pohledu lze tauopatie (tab. 1) rozdělit na tři skupiny onemocnění: 1. frontotemporální demence; 2. progresivní afázie; 3. kombinace demence a poruch hybnosti, byť v mnoha případech se klinická symptomatologie překrývá a často závisí hlavně na distribuci depozit hyperfosforylovaného tau proteinu [3 – 6].

Syndrom frontotemporální demence se vyznačuje převažujícím postižením frontálních funkcí s časnými poruchami chování, osobnostními změnami a poruchou paměti v důsledku hipokampálních lézí.
Zpočátku izolovaná alterace řeči, která postupně progreduje do obrazu těžké frontální demence, je typická pro primární progresivní afázie [7].
Kombinaci demence a extrapyramidového syndromu nacházíme u kortikobazální degenerace a progresivní supranukleární obrny [8,9].
Z neuropatologického pohledu se tauopatie dělí do tří základních skupin dle počtu opakování vazebného místa pro molekuly mikrotubulů (direct ‑ repeats – R), které mohou být převážně tři (3R, např. FTLD – tau s Pickovými tělísky – tedy Pickova nemoc) nebo čtyři (4R, např. kortikobazální degenerace, progresivní supranukleární obrna anebo nemoc s argyrofilními zrny) a kombinace 3R + 4R tau (např. senilní demence s neuronálními klubky; SDT) či primární věkově vázaná tauopatie (Primary Age ‑ Related Tauopathy; PART) [10].
Správné rozpoznání těchto onemocnění během života pacientů je i v současné době velmi obtížné. U neurodegenerativních onemocnění se klinická diagnóza určuje na úrovni možná („possible“) a pravděpodobná („probable“). Definitivní potvrzení je možné až neuropatologickým vyšetřením mozkové tkáně, přičemž ve významné části případů může být i na renomovaných pracovištích stanovena nesprávná klinická diagnóza [11,12].
Přehled tauopatií a jejich klinických manifestací
Frontotemporální demence (behaviorální varianta)
Syndrom frontotemporální demence se vyznačuje převažujícím postižením frontálních funkcí s časným postižením osobnostních rysů a chování, s apatií a abulií a/ nebo s projevy dezinhibice a ztrátou společenského taktu, a s relativně zachovanou epizodickou pamětí (tab. 2).
![Diagnostická kritéria frontotemporální demence - International Behavioral Variant FTD Criteria Consortium [14].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/d6e143c787601740fe8a55dde2ab86cd.jpg)
Prevalence behaviorální varianty frontotemporální demence (bvFTD) se liší v různých studiích, ve věkové skupině 45 – 65 let se odhaduje na 2,0 – 15,4 na 100 000 obyvatel, nad 65 let se pak blíží 1 % [13].
Důležitý je časový faktor časného rozvoje behaviorálních příznaků u pacientů s bvFTD během posledních tří let. Neuropsychiatrické projevy příznačné pro bvFTD (např. sociálně nevhodné chování, nedodržování společenských norem, impulzivita, apatie, ztráta empatie nebo vřelosti v mezilidských vztazích, kompulzivně ritualistické chování aj.) se nespecificky objevují u různých psychiatrických onemocnění. Pro bvFTD (tab. 2) je charakteristický časný vznik těchto příznaků během prvních tří let rozvoje onemocnění (na rozdíl od stabilních projevů dlouhodobých psychiatrických onemocnění, kde bývají přítomny řadu let až desetiletí) [14].
Historicky se po dlouhou dobu v průběhu 20. století používalo označení Pickova nemoc (pojmenované po Arnoldu Pickovi, význačném pražském neurologovi a psychiatrovi a vlastně i „neuropatologovi“ – v tehdejší době byla neuropatologie považována za klinický obor) pro frontální typ demence s časnými poruchami chování a řeči, a s tzv. Pickovými tělísky (argyrofilní intraneuronální inkluze uložené nejčastěji ve fascia dentata a v pyramidových neuronech hipokampu). V 90. letech 20. století bylo jednoznačně prokázáno, že Pickova tělíska jsou podskupinou depozit tau proteinu, formou se třemi opakováními vazebného místa pro mikrotubuly (3R – viz výše).
Primární progresivní afázie
Jako primární progresivní afázie (PPA) se označuje zpočátku (po dobu nejméně 1 – 2 let trvající) izolovaná alterace řeči, která postupně progreduje do obrazu těžké frontální demence [15]. Na rozdíl od bvFTD zůstává u PPA dlouho zachována soběstačnost a aktivity běžného života, pokud nejsou vázány na užívání řeči (např. telefonování). Ze skupiny PPA (nonfluentní/ agramatická, sémantická a logopenická) se k tauopatiím řadí nonfluentní/ agramatická varianta, která připomíná Brocovu afázii svou nízkou řečovou produkcí [16]. V pozdějších stadiích onemocnění se mohou projevit poruchy chování typické pro behaviorální variantu FTD [7].
Kortikobazální degenerace
Tauopatií s převahou 4R izoformy tau proteinu, která se klinicky projevuje kombinací extrapyramidových symptomů a korové dysfunkce s postižením hlavně v parietální oblasti, je kortikobazální degenerace (CBD) [17]. Termínem kortikobazální syndrom (CBS) (tab. 3) se označuje klinický obraz kortikobazální degenerace bez znalosti neuropatologického podkladu [9].
![Diagnostická kritéria kortikobazálního syndromu – Cambridgeská kritéria [9].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/894d14a761b62604b392c825f04583a0.jpg)
Pro CBS je charakteristický asymetrický parkinsonizmus (bradykineze a rigidita, zpravidla bez třesu), často s dystonickým držením postižené ruky a případně s myoklonickými záškuby, s nápadnou apraxií a/ nebo tzv. syndromem odcizené ruky (alien hand) – abnormální držení v prostoru a ztráta volní kontroly nad pohyby ruky – v kombinaci s poruchou kognice a řeči.
Obraz CBS nemusí být podmíněn pouze depozity proteinu tau (pak se jedná o CBD), ale podobné projevy může vyvolat řada jiných neurodegenerativních procesů, v první řadě fokální varianta Alzheimerovy nemoci (AD) [18,19] nebo varianta progresivní supranukleární obrny (PSP s kortikobazálním syndromem; PSP ‑ CBS [20]), vzácněji se CBS může objevit i u demence s Lewyho tělísky (DLB), Creutzfeldtovy ‑ Jakobovy nemoci, FTLD nebo dokonce u Parkinsonovy nemoci (PD).
Progresivní supranukleární obrna (Steeleův- Richardsonův- Olszewskiho syndrom)
PSP se řadí do skupiny tauopatií s převahou 4R izoformy patologicky změněného tau proteinu [8]. Typický klinický obraz (tab. 4) je tvořen kombinací hlavních projevů: 1. parkinsonský syndrom (PS) s axiální převahou, rigiditou trupu a poměrně často i hyperextenčním postavením hlavy a šíje; 2. postupně se horšící porucha chůze s posturální instabilitou a častými pády, typicky s retropulzí; 3. supranukleární porucha sdružených (konjugovaných) pohybů očí, především vertikálních a specificky pro PSP směrem dolů, se zachovanými okulocefalickými reflexy; 4. dysexekutivní syndrom až do stupně demence s výraznými projevy frontálního postižení, s těžkými motorickými i řečovými perseveracemi, sníženou plynulostí řeči (verbální fluencí) a nápadnou apatií [21,22].
![Diagnostická kritéria progresivní supranukleární obrny – kritéria NINDS-SPSP (National Institute of Neurological Disorders and Stroke and Society for Progressive Supranuclear Palsy) [37].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/406bff4380b1c461ac3c9aed7add9de8.jpg)
PSP je klinicky velmi heterogenní jednotka s variantami, které se liší v klinickém obrazu, v rychlosti rozvoje a v délce přežití; mají rovněž různé typy neuropatologického postižení [23,24].
Richardsonův syndrom (PSP ‑ RS) je nejčastější forma s „klasickým“ obrazem PSP odpovídajícím historickému popisu [25] – kombinace axiální rigidity, supranukleární pohledové obrny, pseudobulbárního syndromu, častých pádů a těžké subkortikální demence.
PSP s parkinsonizmem (PSP ‑ P) se vyznačuje pomalejší progresí s delším přežitím, počátečním asymetrickým PS s přechodným efektem levodopy, proto může být v počátečních stadiích obtížně odlišitelná od PD [26]. Instabilita s pády a demence vznikají až v pozdní fázi onemocnění.
U PSP s akinezí a freezingem chůze (PSP ‑ Pure Akinesia with Gait Freezing; PSP ‑ PAGF) je hlavním příznakem porucha iniciace chůze a zárazy (freezing) při chůzi a jiných činnostech (řeč, psaní) [27,28].
PSP s kortikobazálním syndromem (PSP -Corticobasal Syndrome; PSP ‑ CBS): vzácná kombinace progresivní asymetrické dystonie doprovázené někdy PS, dále je přítomna apraxie a parietální syndrom (končetinová apraxie, příznak cizí ruky, kortikální porucha čití nebo akalkulie) [20].
PSP s progresivní nonfluentní afázií (PSP ‑ Progressive Non ‑ Fluent Aphasia; PSP-PNFA): překrývá se s nonfluentní/ agramatickou variantou progresivní afázie v kombinaci s parkinsonizmem [29].
Nemoc s argyrofilními zrny
Po roce 2000 bylo popsáno onemocnění, které se neuropatologicky projevuje depozity patologické formy tau proteinu rovněž s převahou 4R izoformy patologicky změněného tau proteinu ve formě tzv. argyrofilních zrn, jež dala tomuto onemocnění pojmenování [30,31]. Na definici a klinických kritériích tohoto onemocnění se v současné době intenzivně pracuje [30].
Dědičné formy
Vzhledem k poměrně častému familiárnímu výskytu FTLD (až 30 – 50 % pacientů může mít rodinnou zátěž s postižením některého z příbuzných prvního stupně) se předpokládá dosti silný vliv genetických faktorů, které však ještě nejsou jednoznačně objasněny, jedná se nejspíše o multifaktoriální typ dědičnosti. U některých familiárních forem tauopatií je zjištěn autozomálně dominantní přenos onemocnění se specifickou mutací genu pro tau protein (MAPT) na dlouhém raménku chromozomu 17 (tzv. familiární tauopatie, demence frontálního typu s parkinsonizmem s mutací v genu MAPT). Přehled mutací je pravidelně aktualizován ve volně přístupné databázi (http:/ / www.molgen.ua.ac.be/ FTDMutations/ default.cfm?MT=0&ML=2&Page=FTD). Z hlediska dosud známých patogenních mutací jsou PSP, AGD a CBD považovány za sporadická onemocnění, i když i zde byly popsány jednotlivé rodiny s patogenními mutacemi v genu MAPT.
Stručný přehled typických nálezů pomocných vyšetření u tauopatií
Neuropsychologie
Frontotemporální demence (behaviorální varianta)
V časných stadiích bvFTD mohou být výsledky běžných neuropsychologických testů ještě bez přesvědčivého nálezu. Je proto důležité hodnotit chování pacienta, tedy pátrat po časných behaviorálních příznacích evokujících bvFTD (tab. 2) a administrovat testy citlivé na frontální funkce – např. Wisconsinský test třídění karet (Wisconsin Card Sorting Test; WCST), test Londýnské věže (Tower Of London; TOL), Stroopův test; test cesty (Trail Making Test; TMT).
Testování tak prokáže především postižení exekutivních funkcí (zhoršená schopnost plánování, úsudku, uvažování, řešení problémů, abstrakce a mentální flexibilita). Často nacházíme impulzivitu, nepozornost, perseverativní odpovědi a během testování pacienti opakovaně porušují pravidla zadání testů.
Vyšetření paměti nachází alteraci pracovní paměti a pozornosti. Epizodická paměť je narušena především v oblasti výbavnosti, nápověda tedy zlepšuje výkon.
V oblasti zrakově‑prostorových funkcí se objevují potíže v plánování kresby a jsou přítomny perseverace.
Nonfluentní/ agramatická varianta progresivní afázie
Neuropsychologické vyšetření potvrdí přítomnost poruchy řeči – řeč je obtížná, těžkopádná s gramatickými chybami a obtížným vybavováním slov. Pacienti mají problémy s hledáním slov (anomie) a vyskytují se fonemické parafázie, gramatické struktury jsou také narušeny, podobně jako prozódie. Porozumění je oslabeno na úrovni komplexnějších vět, ale zachováno na úrovni slov. Poznávání objektů zůstává dlouho nedotčeno.
Progresivní supranukleární obrna (Steeleův‑ Richardsonův‑ Olszewskiho syndrom)
Z psychologického hlediska se jedná o tzv. fronto‑subkortikální dysfunkci, pro kterou jsou charakteristické změny v kognici a chování projevující se pomalým zpracováním a vyhledáváním informací, nízkou plynulostí řeči (verbální fluencí), palilalií, oslabenou pracovní pamětí, problémy v shiftingu (ve schopnosti změnit nastavení myšlenkového procesu), rigiditou myšlení, poruchou úsudku a plánování.
Typický je bradypsychizmus, stereotypie, imitační a utilizační chování a perseverační tendence (příznak potlesku – „applause sign“ nebo „clapping test“ – neschopnost zastavit tleskání po pokynu zopakovat trojí tlesknutí examinátora).
Z neuropsychiatrických symptomů dominuje apatie (91 % všech pacientů), dále může být přítomna dezinbice, deprese, anxieta a iritabilita. U některých subtypů převažuje apraxie nebo porucha řeči nad exekutivním profilem (PSP ‑ CBS, PSP ‑ PNFA).
Kortikobazální degenerace
Pro pacienty s kortikobazálním syndromem je typické časné postižení zrakově‑prostorových funkcí a těžká apraxie ideomotorická (potíže napodobit činnost, i když představa o ní je zachována) i konstrukční. V pokročilé fázi onemocnění se objevuje i apraxie oblékání a ideatorní apraxie. Porucha řeči zahrnuje nonfluentní afázii s dysartrií. V pozdějších fázích se rozvíjí typický frontální dysexekutivní syndrom s poklesem slovní plynulosti (verbální fluence), zpomalením psychomotorického tempa, nedostatečným náhledem, vázne kognitivní odhad i schopnost abstrakce. Často bývá přítomna deprese, apatie, iritabilita či v menší míře agitovanost a dezinhibice.
Zobrazovací vyšetření
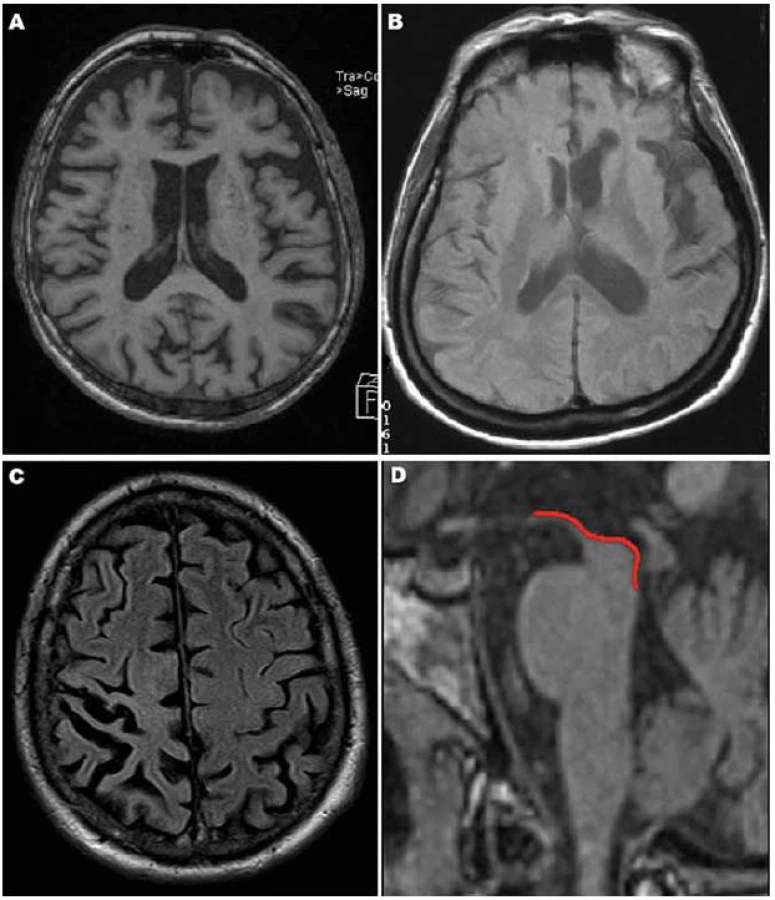
Nejpřínosnějším zobrazením pro diagnostiku tauopatií je MR mozku s nálezem fokální atrofie (obr. 1, 2), kterou je však nutno vždy interpretovat v klinickém kontextu. Radioizotopové vyšetření (SPECT nebo PET) může přinést doplňující informace, zejména v případech, kdy míra atrofie není příliš přesvědčivá.
Frontotemporální demence (behaviorální varianta)
Atrofie mozku u FTD je na MR nejvíce vyjádřena ve frontálních oblastech (obr. 1A), v menší míře pak temporálně (zejména temporopolárně, dorzální část bývá ušetřena). Typická je markantní asymetrie v atrofii a – ve srovnání s jinými demencemi – nápadně pokročilý stupeň atrofie, především u tauopatií [32].
SPECT nachází hypoperfúzi především frontálně, mnohdy asymetrickou, částečně i v temporální krajině. PET může v časné fázi onemocnění nalézt hypometabolizmus předního cingula, frontální krajiny a inzuly.
Nonfluentní/ agramatická varianta progresivní afázie
MR i SPECT nachází asymetrické postižení (atrofii/ hypoperfúzi) s převahou v zadní fronto ‑ inzulární krajině v dominantní hemisféře (obr. 1B), obraz je dosti heterogenní [33].
Kortikobazální degenerace
V časném stadiu bývá MR normální, později se rozvíjí fokální asymetrická atrofie především v parietální oblasti (obr. 1C), někdy i frontálně.
SPECT prokáže v typických případech hypoperfúzi především parietálně, mnohdy asymetrickou, částečně i ve frontální krajině.
Progresivní supranukleární obrna (Steeleův‑ Richardsonův‑ Olszewskiho syndrom)
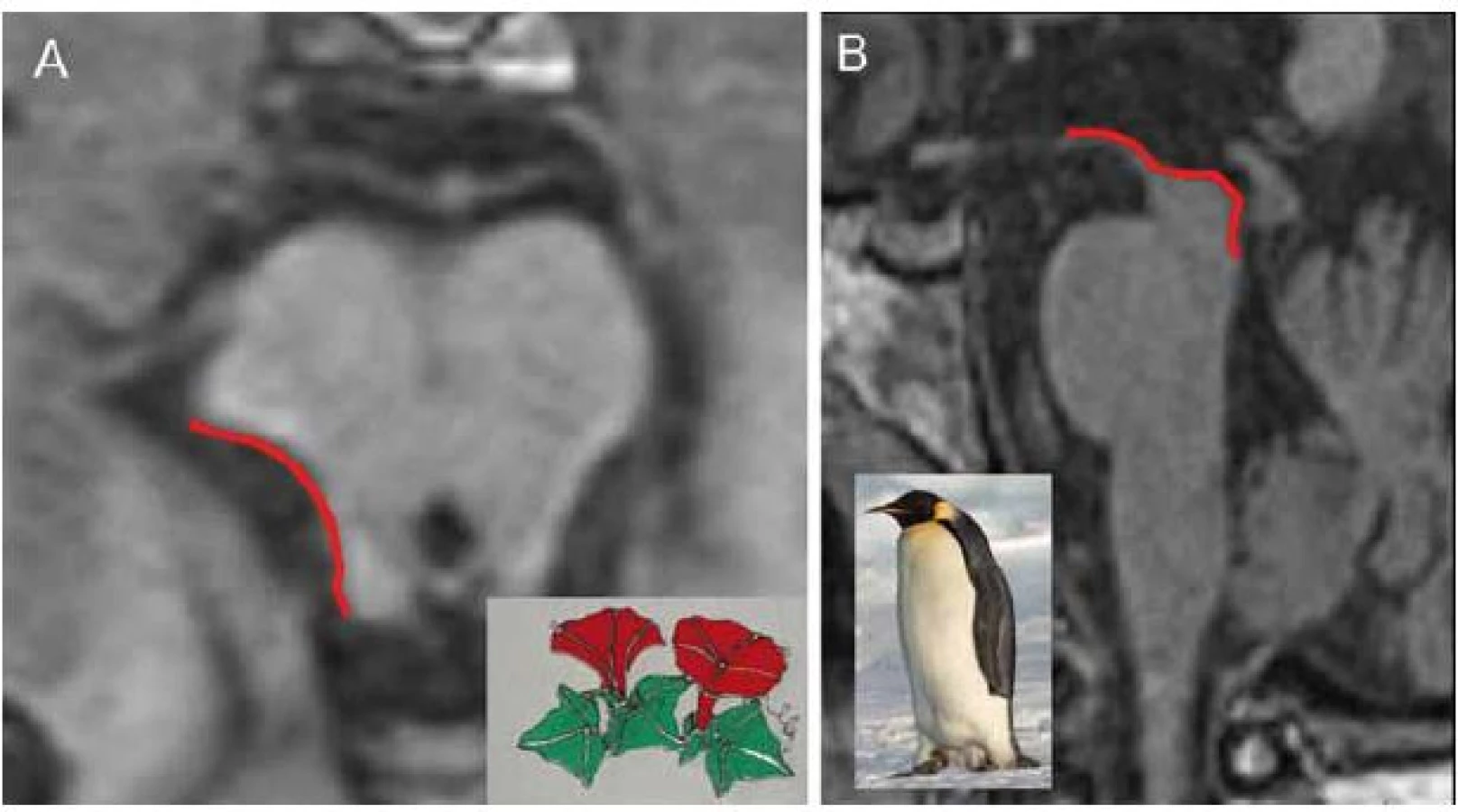
Typickým nálezem u PSP je nález atrofie mezencefalu (obr. 1D). V axiálním řezu konkavita zevního okraje tegmenta mezencefalu je nazývána příznakem myšáka Mickeyho („Mickey Mouse sign“) či svlačce („morning glory sign“) (obr. 2A). Na sagitálním řezu kombinace relativně zachovaného pontu s těžkou atrofií středního mezencefalu (oploštění až konkavita dorza mezencefalu) připomíná svým vzhledem tučňáka („standing penguin silhouette sign“) nebo kolibříka („hummingbird sign“) (obr. 2B).
FDG ‑ PET obvykle nachází hypometabolizmus thalamu a frontální krajiny (PSP ‑ RS) nebo putamen (PSP ‑ P). Tento nález je nicméně nespecifický, nejedná se o rutinní diagnostickou metodu pro PSP. Perfúzní SPECT může nalézt nespecifickou hypoperfúzi s frontální převahou.
Receptorový SPECT s radioaktivně značeným ligandem dopaminového transportéru (DaTSCAN, ioflupan) prokáže u PSP úbytek presynaptických dopaminergních zakončení ve striatu. K odlišení od PD je nutné prokázat i postižení postsynaptické za použití např. jodbenzamidu (derivátu neuroleptik), který se vychytává na postsynaptických D2 dopaminových receptorech striata. Toto vyšetření se však rutinně v České republice neprovádí.
Biochemie mozkomíšního moku
V mozkomíšním moku u tauopatií lze najít zvýšenou hladinu celkového, méně často i fosforylovaného, tau proteinu.
Návrh diagnostického postupu při zvažování klinické diagnózy taupatie
Klinická diagnóza tauopatií vychází z klinického obrazu, kognitivního profilu a MR obrazu. Jednotlivé nálezy mohou být nespecifické nebo neúplné a až jejich souhrnné zhodnocení umožní stanovit klinický závěr. Definitivní potvrzení je možné teprve neuropatologickým vyšetřením při autopsii (mozková biopsie se u neurodegenerativních onemocnění rutinně neprovádí). Při splnění specifických diagnostických kritérií pro jednotlivá onemocnění (tab. 1) může být klinický závěr značně relevantní.
Tauopatie jsou – ve srovnání s jinými neurodegeneracemi, jako např. AD nebo PD – relativně vzácné a mohou mít velmi různorodý obraz. Proto bývají mnohdy rozpoznány pozdě nebo diagnostikovány nesprávně. V následujícím textu a v algoritmu (schéma 1) navrhujeme praktický postup zaměřený k upřesnění klinické diagnózy jednotlivých nozologických jednotek v rámci tauopatií. Cílem tohoto sdělení je usnadnit klinickým neurologům, psychiatrům a geriatrům časné rozpoznání tauopatií, nikoliv nabízet zevrubný návod k diferenciální diagnostice demencí obecně.
Námi navrhovaný algoritmus zohledňuje, co z klinických údajů a z pomocných vyšetřovacích metod svědčí pro přítomnost tauopatie a co proti ní. V dalším textu uvádíme komentáře k diagnostickému algoritmu (schéma 1). Nejprve se zmíníme o základních premisách a poté budou komentovány některé klíčové aspekty klinické, neuropsychologické a MR. Pořadí jednotlivých kroků jsme vybírali i podle prevalence jednotlivých onemocnění (např. DLB – synukleinopatie, nepatří mezi tauopatie) je mnohem častější než PSP (tauopatie).

Úvodní kroky a základní předpoklady
Prvním krokem při diagnostice tauopatií je vyloučit jiný než neurodegenerativní původ obtíží, zejména rozsáhlé vaskulární změny v bílé hmotě hemisfér, dále encefalitidy, paraneoplastické syndromy, intoxikace, tumory, metabolické změny nebo zánětlivé postižení v rámci autoimunitních nebo systémových onemocnění.
Tauopatie se projevují především postižením kognitivních funkcí a/ nebo parkinsonizmem. Navrhovaný algoritmus (schéma 1) vychází ze dvou možných přístupů – kognitivního (převažující klinickou manifestací u daného pacienta je demence, následně se další úvahy liší podle toho, zda je či není přítomen PS) a motorického (v popředí je parkinsonizmus, ke kterému se může nebo nemusí přidávat i kognitivní deteriorace).
Klinické aspekty
Neurologické vyšetření je zaměřeno především na hybnost, svalový tonus a okulomotoriku.
Hlavní hybné poruchy
PS je tvořen kombinací hypokineze, rigidity, třesu a posturální instability.
Důležitým diferenciálně diagnostickým kritériem odlišujícím tauopatie od PD je neodpovídavost nebo pouze přechodné trvání odpovědi na léčbu levodopou (tj. nedochází ke zlepšení parkinsonských příznaků nebo je zlepšení jen přechodné (trvající méně než 12 měsíců) při podávání levodopy v dávce nejméně 250 mg třikrát denně po dobu alespoň dvou měsíců).
Držení těla, chůze, stabilita
Specifické a nápadné držení těla mívají pacienti s PSP. Převažuje u nich axiální rigidita extenčního typu, až s nápadnou hyperextenzí hlavy a šíje (retrocollis). Tento projev však nemusí být vždy vyjádřen, relativně často lze u PSP pozorovat i běžné flekční držení, typické pro PS.
Chůze je u PSP kromě poruchy iniciace a zárazů (freezingu) charakteristická hypokinetickou chůzí s výraznou posturální instabilitou. Na rozdíl od jiných příčin této tzv. frontální poruchy (apraxie) chůze není instabilita provázena „strachem z pádu“ s krátkými krůčky a rozšiřováním základny, ale pacient s PSP padá bez jakýchkoli obranných reflexů, dozadu ihned po povstání ze sedu („příznak rakety“) nebo kterýmkoli směrem v průběhu chůze.
Pády jsou jedním z prvních projevů PSP a dochází k nim nejčastěji ze všech chorob doprovázených PS. Brýlový hematom, modřiny v obličeji, četné zhmožděniny na končetinách jsou při vyšetření nápadně často přítomny.
Dystonie a myoklonus
Jednostranná končetinová dystonie je charakteristickým příznakem CBD v časných a středních stadiích nemoci, s progresí nemoci může být přítomna oboustranně. Dystonie se vyskytuje obvykle akrálně, postihuje především prsty a předloktí, aktivuje se pohybem. U CBD se může objevit též fokální myoklonus horní končetiny, charakteristicky s aktivací pohybem či náhlým senzorickým podnětem.
U PSP se kromě extenční rigidity či dystonie šíje může objevit blefarospazmus, obvykle se složkou apraxie otevírání víček či s retrakcí horních víček. Také výraz obličeje bývá často změněn dystonickým stahem periorálního svalstva, což v kombinaci s parézou pohledu propůjčuje obličeji zvláštní strnulý, „užaslý“ výraz s velmi řídkým mrkáním a otáčením celé hlavy s očima upřenýma před sebou za hlasovým nebo zvukovým podnětem (zdravý jedinec se při pohledu do strany nejprve podívá očima a teprve pak, pokud to nestačí, otočí i celou hlavu).
Okohybné poruchy
Charakteristická pro PSP je porucha sdružených očních pohybů, především ve vertikálním směru (specificky u PSP vázne pohled dolů, zatímco izolovaná paréza pohledu vzhůru může být projevem ischemického postižení mezencefalu), později i do stran. Postiženy jsou nejdříve volní sledovací pohyby (s dlouho zachovaným „automatickým“ sledováním pohybujícího se předmětu v zorném poli).
Sakadické pohyby jsou zpomalené a hypometrické. Pro supranukleární ráz pohledové parézy svědčí výbavnost okulocefalických reflexů při pasivních pohybech hlavou. V pokročilých stadiích je přítomna kompletní zevní oftalmoplegie.
Hlavní poruchy kognitivních funkcí
V časné fázi kognitivní deteriorace je pro správnou diagnózu neurodegenerací rozhodující určit domény nebo kognitivní funkce, které jsou postiženy časně a predominantně (např. epizodická paměť u AD, zrakově‑prostorové funkce u DLB, exekutivní funkce u PSP nebo FTD, apraxie u CBD).
V pokročilých stadiích demence bývá kognitivní profil nespecifický s těžkým postižením všech domén, výrazným frontálním útlumem a behaviorálními projevy (behaviorální a psychologické symptomy u demencí, BPSD).
Důležité je upřesnění anamnestických údajů nejen od pacienta, ale také od rodinných příslušníků či pečovatele. Zaměřujeme se na podrobný průběh onemocnění, dobu trvání obtíží, schopnost orientace, rychlost horšení a na informace o poruchách chování a osobnostních změnách. Důležité je i posouzení míry soběstačnosti u běžných denních činností.
Fluktuace kognitivních funkcí není typická pro tauopatie, ale je užitečným diferenciálně diagnostickým příznakem umožňujícím DLB. Pozornost, bdělost a kognitivní stav může kolísat v řádu minut, hodin nebo dní, někdy bývá dezorientace, spavost, jindy pacienti prázdně hledí před sebe, jindy se vyjadřují nekoherentně, nebo nekomunikují, apod.
Významným diferenciálně diagnostickým znakem je časový faktor. Pokud prvním známkám demence předchází několikaletý rozvoj klinicky jasné PD, jedná se o demenci při PD (PD‑D). U DLB demence předchází nebo se objevuje souběžně do jednoho roku s rozvojem motorických projevů parkinsonizmu. Tauopatie se vyznačují různou mírou kombinace parkinsonských a kognitivních projevů, jejich kombinace v čase může být souběžná nebo mírně oddálená, ale výrazně se liší od klasického průběhu pomalu progredujícího typického parkinsonizmu s pozdním nástupem demence u PD ‑ D.
Paměť
Postižení epizodické paměti (události, jež pacient prožil), která je vázána na hipokampální struktury, je typickým projevem AD. U frontotemporálních demencí jsou časně postiženy přední temporální laloky a temporoparietální pomezí, které mají vztah k sémantické paměti (všeobecné deklarativní informace, pojmy, zeměpisné znalosti aj.) a bývá výrazně narušena pracovní paměť (umožňuje uchovávat informace po dobu několika sekund a je závislá na funkci dorzolaterálního prefrontální kortexu).
Exekutivní funkce
Exekutivní funkce zahrnují schopnost plánovat, rozhodovat, cílevědomě jednat a řešit problémy. Pro fungování exekutivních funkcí je důležitý prefrontální kortex. Časné a výrazné postižení exekutivních funkcí a změny osobnosti jsou typické pro bvFTD.
Řeč a jazyk
Časným a dlouho izolovaným postižením řeči se vyznačují primární progresivní afázie. Významným diferenciálně diagnostickým rysem je plynulost (fluence) řeči. Prvky řeči, které umožňují rozlišení mezi subtypy PPA, jsou produkce řeči, porozumění a repetice.
Nonfluentní/ agramatická varianta (tauopatie) má výrazně narušenou produkci řeči, u sémantické varianty převažuje těžká porucha porozumění, zatímco pro logopenickou variantu je příznačné výrazné narušení produkce řeči a především repetice delších vět.
Pro řeč u PSP je charakteristická hypokinetická dysartrie se spastickými prvky (neplynulá, zpomalená a setřená artikulace) s dysfonií rázu zhrubělého šeptavého hlasu. Specifické poruchy řeči mohou napomoci odlišení různých příčin PS už od počátečních stádií onemocnění [33]. Porucha artikulace bývá provázena poruchou polykání pro tekutiny i pro tuhá sousta.
Zrakově‑prostorové funkce
Jsou závislé na činnosti parietálního, okcipitálního a frontálního laloku.
Pokud se pacientovi (např. při kresbě krychle) nedaří kresba spontánně, požádáme jej o kopii podle předlohy. Jestliže se kresba ani potom nezdaří, můžeme uvažovat o konstrukční apraxii, pokud se kvalita kresby naopak kopií zlepší, jedná se o exekutivní postižení.
K posouzení ideomotorické apraxie lze předvést smysluplná gesta (např. pozdravit, předvést stopování), a nedaří‑li se to, pak gesta napodobit podle examinátora. Užitečné je také prověření imitace speciálních gest bez významu a testování ideatorní apraxie zahrnuje úlohy zaměřené na předvedení činnosti s předměty denní potřeby (např. hřeben, klíče aj.).
Výrazné postižení zrakově‑prostorových funkcí nacházíme u DLB a u některých pacientů s bvFTD, typická je časná apraxie u CBS. Naproti tomu u PSP nebo u PPA mohou být zrakově‑prostorové funkce dlouhodobě relativně zachovány.
Magnetická rezonance
Tauopatie nemají specifický diagnostický marker ani na MR (ve smyslu případných změn signálu), ani v mozkomíšním moku (např. u PSP bývá hladina tau i fosfo ‑ tau proteinů výrazně zvýšena, ale může být dlouho normální i ve velmi rozvinuté fázi onemocnění). Proto je nutné vždy interpretovat MR nález v klinickém kontextu.
Typická je frontální převaha atrofie u bvFTD (obr. 1A), asymetrická atrofie dominantní hemisféry u primárních progresivních afázií (obr. 1B), nebo atrofie parietální u CBD (obr. 1C). U PSP nacházíme atrofii mezencefala, nejzřetelnější při zobrazení v transverzální a sagitální rovině (obr. 1D).
Situaci však komplikuje skutečnost, že radiolog nemusí (zejména pokud nemá k dispozici relevantní klinické informace na žádance) fokální atrofii zhodnotit a explicitně uvést v popisu vyšetření. Je proto vhodné při podezření na tauopatie explicitně radiologa informovat, pokládat cílené otázky a nespokojit se jen s popisem, ale prohlédnout si obrazovou dokumentaci i osobně.
Atypické projevy a překrývání
V poslední době se ukazuje, že společný výskyt více markerů definujících různé neurodegenerace není vzácný; je naopak poměrně častý, a to zejména úměrně se stoupajícím věkem [34].
Můžeme se tak setkat s typickým nálezem určitého onemocnění provázeným ohraničenými depozity jiného onemocnění, které však není plně rozvinuté (souběžná neurodegenerativní patologie, např. v terénu potvrzené Pickovy nemoci se vyskytnou depozita alfa‑synukleinu v amygdale) – nebo i s opravdovou komorbiditou, současným výskytem dvou odlišných nozologických jednotek (např. Pickova nemoc a AD [35], nebo PSP v kombinaci s AD [36]).
Překrývání neurodegenerací může být příčinou rychlejšího průběhu i někdy velmi atypických klinických obrazů u řady pacientů.
Závěr
Tauopatie jsou skupina onemocnění, jejichž klinická diagnostika je relativně obtížná vzhledem k poměrně výrazné heterogenitě klinických projevů. Pro správnou diagnózu je potřeba syntézy výsledků podrobného neurologického vyšetření, neuropsychologického profilu, neuroradiologického zobrazení a analýzy mozkomíšního moku, u genetických forem rovněž molekulárně genetického vyšetření. Jde o onemocnění, která jsou významně poddiagnostikována, i když jsou diagnostická kritéria poměrně dobře definována.
Přesnější klinická diagnostika umožní zahájit účinnější symptomatickou a podpůrnou terapii a prognostickým odhadem dalšího vývoje též zkvalitnit život pacienta.
Postupem navrženým v tomto sdělení bude možné zvýšit záchyt tauopatií a zpřesnit diagnózu těchto onemocnění zejména s ohledem na možné budoucí kauzální terapeutické přístupy.
Seznam použitých zkratek
AGD – demence s argyrofilními zrny
AD – Alzheimerova nemoc
bvFTD – frontotemporální demence (behaviorální varianta)
CBD – kortikobazální degenerace
CBS – kortikobazální syndrom
DLB – demence s Lewyho tělísky
FTLD – frontotemporální lobární degenerace
PART – primary age-related tauopathy
PD – Parkinsonova nemoc
PD-D – Parkinsonova nemoc s demencí
PPA – primární progresivní afázie
PS – parkinsonský syndrom
PSP – progresivní supranukleární obrna
PSP-CBS – PSP s kortikobazálním syndromem
PSP-P – PSP s parkinsonizmem
PSP-PAGF –PSP s akinezí a freezingem chůze
PSP-PNFA – PSP s progresivní nonfluentní afázií
PSP-RS – Richardsonův syndrom
Autoři děkují MUDr. Jiřímu Kellerovi, Ph.D. z Radiodiagnostického oddělení Nemocnice Na Homolce za MR obrázky a neuroradiologické podněty. Mgr. Ondřeji Bezdíčkovi, Ph.D, Mgr. Evě Bolcekové, Mgr. Tomáši Nikolaiovi, Ph.D. z Neurologické kliniky 1. LF UK a VFN v Praze a Mgr. Silvii Johanidesové z Neurologického oddělení Thomayerovy nemocnice za kognitivní a neuropsychologické podněty a zpětnou vazbu.
Podpořeno granty IGA MZ NT 12094-5/2011 a PRVOUK-P26/LF1/4.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Přijato k recenzi: 9. 7. 2015
Přijato do tisku: 21. 9. 2015
doc. MUDr. Robert Rusina, Ph.D.
Neurologická klinika a Centrum klinických neurověd
1. LF UK a VFN v Praze
Kateřinská 30
120 00 Praha 2
e-mail: robert.rusina@lf1.cuni.cz
Sources
1. Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol 2013; 12(6): 609 – 622. doi: 10.1016/ S1474 ‑ 4422(13)70090 ‑ 5.
2. Peden AH, Ironside JW. Molecular pathology in neurodegenerative diseases. Curr Drug Targets 2012; 13(12): 1548 – 1559.
3. Williams DR. Tauopathies: classification and clinical update on neurodegenerative diseases associated with microtubule‑associated protein tau. Intern Med J 2006; 36(10): 652 – 660.
4. Williams DR, Holton JL, Strand C, Pittman A, de Silva R, Lees AJ et al. Pathological tau burden and distribution distinguishes progressive supranuclear palsy ‑ parkinsonism from Richardson‘s syndrome. Brain 2007; 130(6): 1566 – 1576.
5. Rademakers R, Neumann M, Mackenzie IR. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol 2012; 8(8): 423 – 434. doi: 10.1038/ nrneurol.2012.117.
6. Rohan Z, Matej R. Current concepts in the classification and diagnosis of frontotemporal lobar degenerations: a practical approach. Arch Pathol Lab Med 2014; 138(1): 132 – 138. doi: 10.5858/ arpa.2012 ‑ 0510 ‑ RS.
7. Grossman M. The non‑fluent/ agrammatic variant of primary progressive aphasia. Lancet Neurol 2012; 11(6): 545 – 555. doi: 10.1016/ S1474 ‑ 4422(12)70099 ‑ 6.
8. Dickson DW, Rademakers R, Hutton ML. Progressive supranuclear palsy: pathology and genetics. Brain Pathol 2007; 17(1): 74 – 82.
9. Mathew R, Bak TH, Hodges JR. Diagnostic criteria for corticobasal syndrome: a comparative study. J Neurol Neurosurg Psychiatry 2012; 83(4): 405 – 410. doi: 10.1136/ jnnp ‑ 2011 ‑ 300875.
10. Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF,Abner EL, Alafuzoff I et al. Primary age‑related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 2014; 128(6): 755 – 766. doi: 10.1007/ s00401 ‑ 014 ‑ 1349 ‑ 0.
11. Josephs KA, Petersen RC, Knopman DS, Boeve BF, Whitwell JL, Duffy JR et al. Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology 2006; 66(1): 41 – 48.
12. Litvan I, MacIntyre A, Goetz CG, Wenning GK, Jellinger K, Verny M et al. Accuracy of the clinical diagnoses of Lewy body disease, Parkinson disease, and dementia with Lewy bodies: a clinicopathologic study. Arch Neurol 1998; 55(7): 969 – 978.
13. Onyike CU, Diehl ‑ Schmid J. The epidemiology of frontotemporal dementia. Int Rev Psychiatry 2013; 25(2): 130 – 137. doi: 10.3109/ 09540261.2013.776523.
14. Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J et al. Sensitivity of revised diagnostic criteria for the behavioral variant of frontotemporal dementia. Brain 2011; 134(9): 2456 – 2477. doi: 10.1093/ brain/ awr179.
15. Gorno ‑ Tempini ML, Hillis AE, Weintraub S, Kertesz A,Mendez M, Cappa SF et al. Classification of primary progressive aphasia and its variants. Neurology 2011; 76(11): 1006 – 1014. doi: 10.1212/ WNL.0b013e31821103e6.
16. Josephs KA, Duffy JR, Strand EA, Machulda MM, Senjem ML, Master AV et al. Characterizing a neurodegenerative syndrome: primary progressive apraxia of speech. Brain 2012; 135(5): 1522 – 1536. doi: 10.1093/ brain// aws032.
17. Wadia PM, Lang AE. The many faces of corticobasal degeneration. Parkinsonism Relat Disord 2007; 13 (Suppl 3): S336 – S340. doi: 10.1016/ S1353 ‑ 8020(08)70027 ‑ 0.
18. Hassan A, Whitwell JL, Josephs KA. The corticobasal syndrome – Alzheimer‘s disease conundrum Expert Rev Neurother 2011; 11(11): 1569 – 1578. doi: 10.1586/ ern.11.153.
19. Johanidesová S, Rusina R, Houška P, Keller J, Matěj R.Alzheimerova nemoc probíhající pod obrazem kortikobazální degenerace – kazuistika. Cesk Slov Neurol N 2012; 75/ 108(3): 373 – 377.
20. Ling H, O’Sullivan SS, Holton JL, Revesz T, Massey LA, Williams DR et al. Does corticobasal degeneration exist? A clinicopathological re‑evaluation. Brain 2010; 133(7): 2045 – 2057. doi: 10.1093/ brain/ awq123.
21. Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy. A heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical supranuclear gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol 1964; 10 : 333 – 359.
22. Liscic RM, Srulijes K, Gröger A, Maetzler W, Berg D. Differentiation of progressive supranuclear palsy: clinical, imaging and laboratory tools. Acta Neurol Scand 2013; 127(5): 362 – 370. doi: 10.1111/ ane.12067.
23. Menšíková K, Kaňovský P, Kaiserová M, Nestrašil I, Bareš M. Proměnlivá tvář parkinsonské neurodegenerace. Cesk Slov Neurol N 2013; 76/ 109(1): 26 – 34.
24. Respondek G, Roeber S, Kretzschmar H, Troakes C, Al ‑ Sarraj S, Gelpi E et al. Accuracy of the National Institute for Neurological Disorders and Stroke/ Society for Progressive Supranuclear Palsy and neuroprotection and natural history in Parkinson plus syndromes criteria for the diagnosis of progressive supranuclear palsy. Mov Disord 2013; 28(4): 504 – 509. doi: 10.1002/ mds.25327.
25. Richardson JC, Steele J, Olszewski J. Supranuclear ophthalmoplegia, pseudobulbar palsy, nuchal dystonia and dementia. A clinical report on eight cases of „heterogenous system degeneration“. Trans Am Neurol Assoc 1963; 88 : 25 – 29.
26. Williams DR, de Silva R, Paviour DC, Pittman A, Watt HC,Kilford L et al. Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson‘s syndrome and PSP ‑ parkinsonism. Brain 2005; 128(6): 1247 – 1258.
27. Imai H, Nakamura T, Kondo T, Narabayashi H. Dopa ‑ unresponsive pure akinesia or freezing. A condition within a wide spectrum of PSP? Adv Neurol 1993; 60 : 622 – 625.
28. Williams DR, Holton JL, Strand K, Revesz T, Lees AJ. Pure akinesia with gait freezing: a third clinical phenotype of progressive supranuclear palsy. Mov Disord 2007; 22(15): 2235 – 2241.
29. Josephs KA, Boeve BF, Duffy JR, Smith GE, Knopman DS,Parisi JE et al. Atypical progressive supranuclear palsy underlying progressive apraxia of speech and nonfluent aphasia. Neurocase 2005; 11(4): 283 – 296.
30. Thal DR, Schultz C, Botez G, Del Tredici K, Mrak RE, Griffin WS et al. The impact of argyrophilic grain disease on the development of dementia and its relationship to concurrent Alzheimer‘s disease‑related pathology. Neuropathol Appl Neurobiol 2005; 31(3): 270 – 279.
31. Matěj R, Koukolík F. Nemoc s argyrofilními zrny: kazuistické sdělení prvních dvou případů diagnostikovaných v ČR a přehled literatury. Cesk Patol 2006; 42(2): 66 – 70.
32. Rosen HJ, Gorno ‑ Tempini ML, Goldman W, Perry RJ, Schuff N, Weiner M et al. Pattern of brain atrophy in frontotemporal dementia and semantic dementia. Neurology 2002; 58(2): 198 – 208.
33. Rusz J, Bonnet C, Klempíř J, Tykalová T, Baborová E,Novotný M et al. Speech disorders reflect differing pathophysiology in Parkinson’s disease, progressive supranuclear palsy and multiple system atrophy. J Neurol 2015; 262(4): 992 – 1001. doi: 10.1007/ s00415 ‑ 015 ‑ 7671 ‑ 1.
34. Dugger BN, Adler CH, Shill HA, Caviness J, Jacobson S, Driver ‑ Dunckley E et al. Concomitant pathologies among a spectrum of parkinsonian disorders. Parkinsonism Relat Disord 2014; 20(5): 525 – 529. doi: 10.1016/ j.parkreldis.2014.02.012.
35. Rusina R, Pazdera L, Kulišťák P, Vyšata O, Matěj R. Pick and Alzheimer diseases: a rare comorbidity presenting as corticobasal syndrome. Cogn Behav Neurol 2013; 26(4): 189 – 194. doi: 10.1097/ WNn.0000000000000011.
36. Menšíková K, Matěj R, Tučková L, Rusina R, Ehrmann J, Kaňovský P. Progressive supranuclear palsy phenotype mimicking synucleinopathies. J Neurol Sci 2013; 329(1 – 2): 34 – 37. doi: 10.1016/ j.jns.2013.03.008.
37. Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele ‑ Richardson ‑ Olszewski syndrome): report of the NINDS ‑ SPSP international workshop. Neurology 1996; 47(1): 1 – 9.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2015 Issue 5
- Memantine Eases Daily Life for Patients and Caregivers
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Metamizole at a Glance and in Practice – Effective Non-Opioid Analgesic for All Ages
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Advances in the Treatment of Myasthenia Gravis on the Horizon
Most read in this issue
- Diagnostika pacienta s akutní závratí
- Recidivující tranzitorní globální amnézie – čtyři kazuistiky
- Normativní studie testu Reyovy‑ Osterriethovy komplexní figury v populaci českých seniorů
- Přínos vyšetření čichu pro diagnostiku neurodegenerativních onemocnění