
Rasmussenova encefalitída
Rasmussen’s Encephalitis
Rasmussen’s encephalitis is a rare chronic neurologic disease characterized by three main features: refractory epilepsy, progressive unihemispheric atrophy of the brain and progressive neurological deficit. This chronic focal encephalitis typically affects children but adolescents and adults are also involved. Neuropathological and immunological studies indicate that Rasmussen’s encephalitis is probably driven by a T-cell cytotoxic response with potential additional contribution by autoantibodies and activated microglia. Primary cause of the disease remains unsolved. MRI may be a good biomarker in Rasmussen’s encephalitis but we do not have relevant serological markers yet. Immunomodulatory treatments seem to slow disease progressiaon in Rasmussen’s encephalitis but long-term outcomes remain unclear. Functional hemispherectomy remains the only effective treatment for seizures but there are inevitable functional compromises associated with it. The decision and timing of surgery may be problematic in the absence of a dense neurological deficit, and if the patient is older. For patients, their families and treating physicians, choosing the right time to move from medical management to surgery is a real therapeutic dilemma. Due to poor prognosis, patients with susspected Rasmussen’s encephalitis should immediately be referred to an epilepsy centre.
Key words:
chronic encephalitis – refractory epilepsy – diagnostics – immunomodulatory therapy – hemispherectomy
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
:
G. Timárová 1; I. Lisá 1; P. Mikula 2; M. Šimko In Memoriam 3
:
II. Neurologická klinika LF UK a UN Bratislava
1; Rádiologická klinika LF UK, SZU a UN Bratislava
2; I. interná klinika Dionýza Diešku, LF SZU a UN Bratislava
3
:
Cesk Slov Neurol N 2016; 79/112(5): 500-512
:
Minimonography
prolekare.web.journal.doi_sk:
https://doi.org/10.14735/amcsnn2016500
Rasmussenova encefalitída je zriedkavé chronické neurologické ochorenie, ktorého typický klinický obraz predstavujú: farmakorezistentná epilepsia, progredujúca atrofia jednej mozgovej hemisféry a progredujúci neurologický deficit. Postihuje predovšetkým deti, ale nevyhýba sa ani adolescentom a mladým dospelym. Neuropatologické a imunologické štúdie svedčia pre účasť T lymfocytmi sprostredkovaného cytotoxického zápalu s potenciálnou účasťou autoprotilátok a aktivovanej mikroglie v patologickom procese. Primárna príčina ochorenia však zostáva neobjasnená. Magnetická rezonancia mozgu je vhodný biomarker ochorenia, v tejto dobe nemáme spoľahlivé sérologické markery. Súčasné spôsoby imunomodulačnej liečby majú schopnosť zlepšiť a spomaliť priebeh ochorenia, ale dlhodobé východiská imunoterapie sú nejasné. Hemisferektómia predstavuje najúčinnejší spôsob liečby farmakorezistentnej epilepsie, ale nevyhnutné sú nežiaduce funkčné deficity. Rozhodnutie o chirurgickej liečbe môže byť problematické, ak chýba významnejší funkčný deficit a ide o staršieho pacienta. Pre zúčastnených – pacienta, jeho rodinu, včítane ošetrujúcich lekárov – je určenie správneho času, kedy sa posunúť od medikamentóznej liečby k chirurgickej liečbe, náročný rozhodovací proces. Vzhľadom na závažnú prognózu je nevyhnutné pacienta s podozrením na Rasmussenovu encefalitídu neodkladne odoslať do epileptologického centra.
Kľúčové slová:
chronická encefalitída – farmakorezistentná epilepsia – diagnostika – imunomodulačná terapia – hemisferektómia
Úvod
Epilepsia partialis continua (EPC), ktorej príčinou bola lokalizovaná ruská jarne-letná encefalitída, bola po prvýkrát popísaná Koževnikovom a Brunom v roku 1895. V roku 1958 neurochirurg Theodore Rasmussen, neuropatológ Jerzy Olszewski a neurológ Donald Lloyd-Smith popísali chronickú lokalizovanú encefalitídu u troch pacientov, ktorá neskôr dostala meno Rasmussenova encefalitída (RE) [1]. RE je zriedkavé progredujúce zápalové ochorenie mozgu, ktoré typicky postihuje jednu mozgovú hemisféru. Pre RE je charakteristická farmakorezistentná fokálna epilepsia, progredujúca hemiparéza a kognitívny deficit [2].
V posledných rokoch sa objavili nové patogenetické poznatky a narastajú dôkazy o imunitnom patologickom procese ako jej podstate. V uplynulom desaťročí sme zaznamenali výrazný pokrok v zmapovaní klinického obrazu a pochopení etiopatogenézy autoimunitných encefalitíd [3]. Napriek tomu, že RE bola popísaná už pred viac ako polstoročím, jej primárna príčina zostáva stále neznáma a terapeutické pokusy vychádzajú predovšetkým z kazuistík, kazuistických sérií a prospektívnych štúdií na malých súboroch pacientov.
Epidemiológia
RE je zriedkavé sporadické ochorenie, ktoré typicky začína v detskom veku v rozmedzí 1–13 rokov [4] a postihuje deti, ktoré boli predtým zdravé, niekedy po prekonanom infekčnom alebo zápalovom ochorení v čase šiestich mesiacov pred začiatkom RE [5]. Ochorenie má bimodálnu distribúciu (medián veku je 5,3 a 18,9 rokov), pretože asi 10 % prípadov RE začína neskôr, v adolescencii alebo mladom dospelom veku [6]. Priebeh s neskorým nástupom ochorenia je zvyčajne pomalší a neurologický deficit nie je tak ťažký ako u detí [7–10]. RE je zriedkavé ochorenie s odhadovanou prevalenciou 1–2 na milión obyvateľov (0,18 na 100 000 obyvateľov). V štúdii realizovanej v Nemecku bola stanovená incidencia 2–4 prípady na 10 miliónov detí do 18 rokov za rok [11]. Podobne v celonárodnej štúdii realizovanej vo Veľkej Británii bola stanovená incidencia 1–7 na 10 miliónov detí do 16 rokov [12]. Neboli zaznamenané rozdiely v postihnutí mužov a žien ani rozdiely podľa geografickej polohy a etnicity.
Klinický obraz
RE je progredujúce zápalové ochorenie mozgu charakteristické triádou príznakov: atrofiou jednej mozgovej hemisféry, refraktérnymi fokálnymi epileptickými záchvatmi a neurologickým deficitom, ktorý predstavuje progredujúcu hemiparézu s kognitívnym postihnutím. U niektorých pacientov môžu byť prítomné v prodromálnej fáze ochorenia sporadické epileptické záchvaty alebo frustná hemiparéza, čo môže trvať individuálne až niekoľko rokov [13]. Akútna fáza ochorenia je charakteristická početnými epileptickými záchvatmi začínajúcimi v jednej hemisfére. Asi u 50 % pacientov s RE sa fokálne záchvaty počas ochorenia manifestujú ako EPC, u 20 % pacientov samotné ochorenie začne ako fokálny status epilepticus (napr. EPC) [14–16]. V priebehu ochorenia sa v dôsledku progresie patologických zmien v postihnutej hemisfére zvyčajne objavia polymorfné epileptické záchvaty, vč. motorických, somatosenzitívnych, vizuálnych a komplexných parciálnych záchvatov s automatizmami. Záchvaty sú typické rezistenciou na antiepileptiká, ich frekvencia je zvyčajne vysoká, môže byť až desiatky záchvatov za deň [1,13,17]. U detí, ktoré nie sú liečené, sa obvykle do jedného roka vyvinie hemiparéza, hemianopsia a kognitívny deficit a v prípade dominantnej hemisféry afázia [11]. Charakteristické je postihnutie centrálnej motorickej oblasti, ale postihnuté bývajú aj ostatné primárne a asociačné kortikálne oblasti – frontálna, parietálna, laterálna a meziálna temporálna. Hipokampus a amygdala vykazujú rôzny stupeň atrofie a dysfunkcie [18].
Vzťah medzi frekvenciou záchvatov a deterioráciou neurologického nálezu na podklade atrofizácie je komplexný a nelineárny. U niektorých pacientov dominujú epileptické záchvaty, u iných sú menej výrazné [17]. Na základe analýzy 48 pacientov, ktorí boli hospitalizovaní v Montrealskom neurologickom inštitúte (MNI) medzi rokmi 1947 a 1987, autori Oguni et al rozdelili priebeh ochorenia do troch štádií [5,6]. V prvom štádiu, ktoré trvá v priemere tri roky (3 mesiace–10 rokov), sa objavia epileptické záchvaty skôr, ako sa rozvíja hemiparéza. V tomto štádiu postupne frekvencia a intenzita záchvatov narastá. Ide predovšetkým o parciálne simplexné záchvaty z centrálnej oblasti (somatomotorický kortex). Spočiatku môžu mať záchvaty tendenciu k ústupu, ale ich relaps je typický svojou refraktérnosťou na liečbu antiepileptikami. Druhé štádium je typické rozvojom hemiparézy a kognitívneho deficitu, frekvencia a intenzita epileptických záchvatov ďalej narastá, objavujú sa kontinuálne fokálne záchvaty (EPC) a polymorfné záchvaty vychádzajúce z ďalších oblastí hemisféry súbežne s progresiou postihnutia. Druhé štádium trvá priemerne 3,7 roka (2 mesiace–10 rokov). Tretie štádium ochorenia je typické stabilizáciou, neurologický deficit je trvalý a dochádza k poklesu frekvencie záchvatov. Neurologický deficit predstavuje ťažká reziduálna spastická hemiparéza, kognitívne poruchy môžu byť rôzneho stupňa, od ľahkého až po ťažké postihnutie [3,13].
Na základe dlhoročného sledovania 13 pacientov s RE navrhli autori Bien et al podobný trojstupňový model priebehu RE v roku 2002 [7]. Prvé, prodromálne štádium trvá 0–8,1 rokov (medián 7,1 mesiacov), počas neho je nízka frekvencia záchvatov, a ak je vôbec prítomná, tak hemiparéza je ľahká až stredne ťažká. Toto štádium je u detí kratšie ako u adolescentov a dospelých. Druhé, akútne štádium trvá 4–18 mesiacov (medián 8 mesiacov), charakteristický je nárast frekvencie epileptických záchvatov, výskyt kontinuálnych fokálnych záchvatov (EPC) a progresia hemiparézy. Počas tretej, reziduálnej fázy je u pacienta prítomná definitívna, najčastejšie ťažká hemiparéza a pokles frekvencie záchvatov, niekedy pacient nemá ďalej záchvaty.
Asi 10 % prípadov RE začína neskôr, po 12.–13. roku života, s počiatkom ochorenia popísaným až do 37 rokov [19–21]. Vývoj ochorenia je pomalší, hemisferálna atrofia sa rozvíja neskôr, neurologický deficit môže byť veľmi dlho ľahkého stupňa [10,22]. RE s neskorým nástupom ochorenia (Late Onset RE; LORE) s priemerným vekom nástupu 18,9 rokov je typická ľahkým stupňom hemiparézy a neskorým nástupom po dlhšej prodromálnej fáze. Častejšie sú popisované iniciálne atrofie okcipitálne, čo vedie k skorším defektom zorného poľa a neglect syndrómu pri postihnutí nedominantnej hemisféry, niekedy je semiológia viac charakteristická pre epilepsiu temporálneho laloka (TLE) [9,23]. Trvanie tejto fázy až do rozvoja ľahkej hemiparézy môže byť až 8–10 rokov [17,24]. Pomalý priebeh LORE a malý stupeň neurologického deficitu je kľúčový moment v určení terapeutickej stratégie refraktérnej epilepsie, predovšetkým v rozhodovacom procese medzi chirurgickou a medikamentóznou liečbou. U týchto pacientov s malým neurologickým deficitom môže byť uprednostnená imunoterapia a hemisferektómia odložená [3,13].
RE typicky postihuje jednu mozgovú hemisféru. Existencia bilaterálnej RE je predmetom diskusie. Bilaterálna RE je extrémne zriedkavá, ale bilaterálne hemisferálne postihnutie bolo pozorované aj u dospelých pacientov, s ľahším priebehom [3]. Niektoré klinické a elektrofyziologické znaky u pacientov s RE svedčia pre obojstranné postihnutie, ako je napr. sekundárne šírenie epileptických záchvatov a interiktálnej epileptiformnej abnormity kontralaterálne, alebo ľahká kontralaterálna atrofia. Volumetrická analýza pomocou magnetickej rezonancie (MR) u pacientov liečených imunoterapiou ukázala, že aj „nepostihnutá“ hemisféra podlieha pomalej progresívnej atrofizácii, ale signifikantne menšieho rozsahu [25]. Je predpoklad, že by sa mohlo jednať o dôsledok Wallerianskej degenerácie komisurálnych vláken a efekt chronickej epilepsie alebo liečby, a nie rozšírenia patologického procesu do druhej hemisféry. Termín bilaterálnej RE by mal byť teda rezervovaný pre prípady so zápalovými zmenami v oboch hemisférach. V súčasnosti boli histopatologicky verifikované tri prípady bilaterálneho postihnutia u detí do 12 rokov z približne 200–300 publikovaných prípadov RE [26–28]. Toto bilaterálne postihnutie u dvoch detí viedlo k fatálnemu koncu. Zatiaľ neboli publikované prípady druho-stranného postihnutia (aj definované klinicky) po chirurgickej liečbe unilaterálnej RE, aj keď sa nejednalo o hemisferektómiu, ale o diskonekčný výkon [13,17].
Postihnutie bazálnych ganglií s jednostrannými extrapyramídovými príznakmi – dystóniou, hemiatetózou a hemichoreou – býva niekedy ťažšie diagnostikovateľné, pretože môže byť prekryté ťažkou epilepsiou a hemiparézou. V dvoch pediatrických kohortách a niekoľkých kazuistikách bolo popísané postihnutie bazálnych ganglií až u 70 % pacientov [29–32].
Iné raritné varianty RE sú formy s neskorým nástupom epilepsie (po 6 mesiacoch–2 rokoch) od rozvoja progresívnej hemisferálnej atrofie a postihnutie kmeňa mozgu [33,34]. RE bez epileptických záchvatov môže byť nepoznanou príčinou progredujúceho neurologického deficitu u detí [35,36].
Diagnostika
Diagnóza RE je založená na klinickom obraze, zobrazovacích vyšetreniach, predovšetkým MR, elektroencefalografickom (EEG) obraze a/alebo histopatologickom náleze.
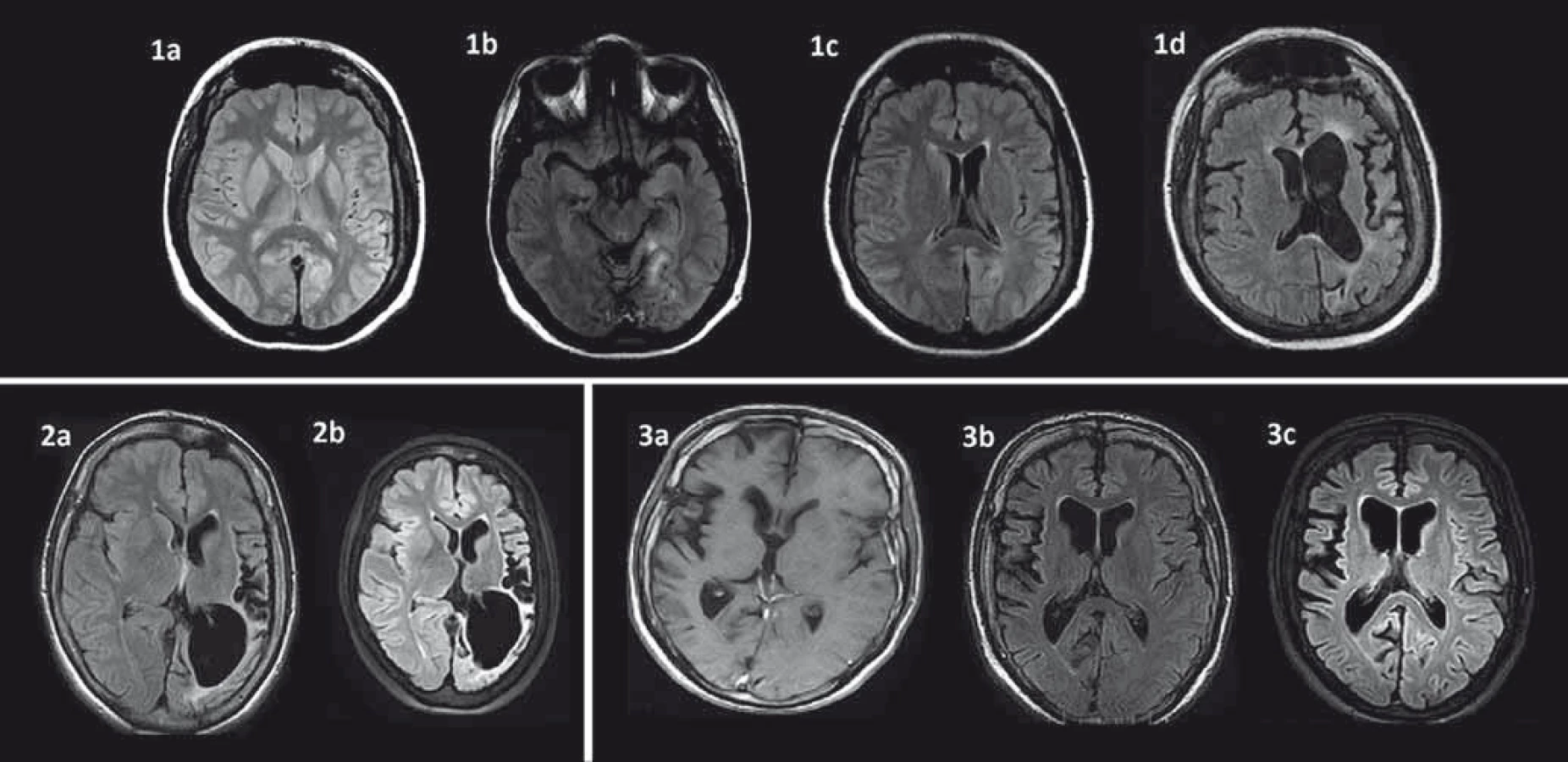
Kľúčovým vyšetrením v diagnostickom procese a sledovaní RE je MR [37,38]. Perisylvická oblasť je predilekčné miesto pre rozvoj atrofie mozgu, zvyčajne sa objavuje počas prvých štyroch mesiacov od začiatku akútnej fázy ochorenia [38,39]. Okrem kortikálnej atrofie je prítomná aj dilatácia komorového systému a atrofia subkortikálnych štruktúr. Ipsilaterálna atrofia caput nuclei caudati je typický príznak hemisferálnej atrofie a môže byť včasným znakom (obr. 1) [38]. Nedávne volumetrické analýzy zistili najrýchlejšiu stratu objemu v prvých ôsmich mesiacoch akútnej fázy a výraznejšiu atrofiu putamen ako caput nuslei caudati [7,25]. V T2/FLAIR váženom MR zobrazení bývajú prítomné hyperintenzitné signály v bielej hmote a kortexe postihnutej hemisféry, ktorých lokalita je heterogénna. U niektorých pacientov bol vo včasnej fáze RE pozorovaný lokálny kortikálny edém. Autori Bien et al korelovali MR obrazy so vzorkami tkanív z regiónov s MR abnormitami. V oblastiach so zvýšeným signálom našli zvýšený počet T buniek, mikrogliálne noduly a GFAP+ astrocyty v porovnaní s oblasťami s atrofiou a bez zvýšeného T2/FLAIR signálu [37]. Niektorí autori T2/FLAIR hyperintenzity vzťahujú ku frekvencii fokálnych epileptických záchvatov [13]. Určitý stupeň atrofie býva viditeľný aj v nepostihnutej hemisfére, pravdepodobne ako následok Walleriánskej degenerácie komisurálnych vláken [29,39,40]. Autori Larionov et al sériou MR vyšetrení u 11 pacientov s RE liečených imunoterapiou potvrdili priemernú stratu tkaniva v postihnutej hemisfére 29,9 cm3 za rok a 6,8 cm3 v nepostihnutej hemisfére [25]. Treba však mať na zreteli, že prvé MR môže byť negatívne napriek prítomnosti EPC alebo fokálnych epileptických záchvatov. Preto sa odporúča opakovať MR (aspoň každých šesť mesiacov), aby bolo možné diagnózu potvrdiť [3]. V rámci diferenciálnej diagnostiky môže byť prínosom T1 MR s použitím gadolínia, MR angiografia alebo angiografia na vylúčenie vaskulitídy s postihnutím len jednej hemisféry. Zvýšenie signálu po podaní gadolínia je u RE veľmi zriedkavé [37,38].
MR spektroskopia (MRS) zameraná na atrofické oblasti zvyčajne zobrazí pokles N-acetyl-aspartátu (NAA), ktorý je markerom neuronálnej integrity. Pri normálnom cholíne (cho) dochádza k poklesu pomeru NAA/cho, čo svedčí pre neuronálnu stratu alebo dysfunkciu [38,41–43]. Prítomnosť laktátu svedčí pre prebiehajúce epileptické záchvaty a EPC [38,44]. Musíme však konštatovať, že súčasné štúdie nezistili pri RE špecifické MRS abnormity [17].
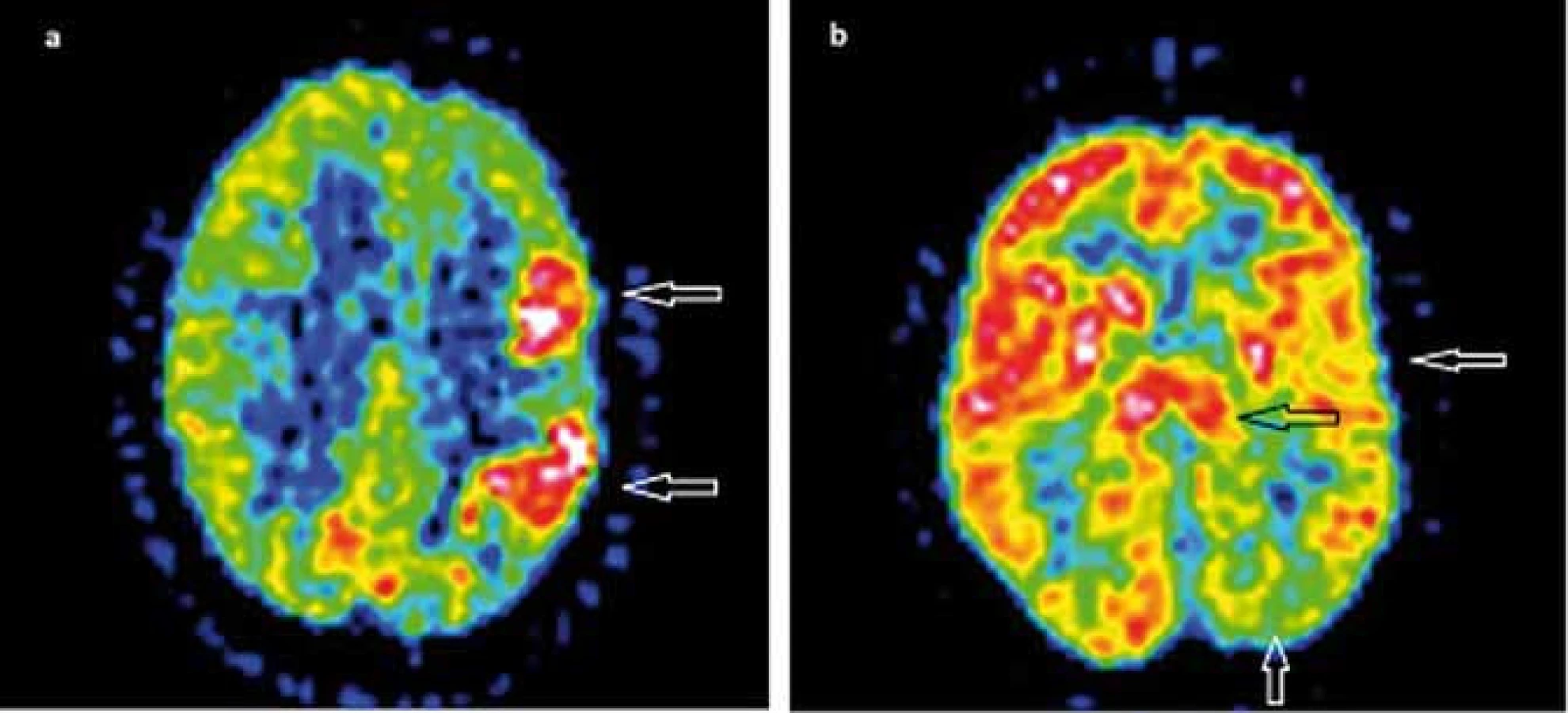
Funkčné zobrazovacie metódy ako 18F-FDG PET a 99mTc-HMPAO SPECT zvyčajne zobrazia poruchy metabolizmu glukózy, event. poruchy prekrvenia v jednej hemisfére, niekedy skôr ako sa vyvinie atrofia. Ako typický korelát v 18F-FDG PET bol pozorovaný rozsiahly interiktálny hypometabolizmus. V niektorých prípadoch bol prítomný pri frekventných záchvatoch a EPC iktálny hypermetabolizmus, často lokalizovaný v okolitom hypometabolizme (obr. 2) [45–48]. Interiktálny a iktálny SPECT priniesol tie isté závery ako PET [38,42,49,50]. PET, SPECT a MRS nie sú vhodné pre definovanie zápalového charakteru ochorenia. Môžu však pomôcť v potvrdení jednostranného postihnutia u suspektnej RE [17].
Vyšetrením likvoru u 50 % pacientov s RE zistíme celularitu a proteíny v normálnom rozsahu, u ostatných sú heterogénne výsledky: môže byť zvýšený počet buniek (16–70/ul, prevažne lymfocytov), zvýšená hladina proteínov (500–1 000 mg/l) a elevácia krivky koloidného zlata v prvej alebo druhej časti. Len u 15 % prípadov boli abnormné všetky tri parametre [51]. Oligoklonálna skladba je nekonštantný nález, býva pozitívna u 0–67 % pacientov [17,52]. Štandardné vyšetrenie likvoru preto nie je vhodné na vylúčenie alebo potvrdenie diagnózy RE. Špecifické testy sa používajú na vylúčenie infekcie centrálneho nervového systému (CNS) známym neurotropným agens [17]. Podobne neexistujú špecifické laboratórne testy, ktoré by vyšetrením séra stanovili diagnózu RE.
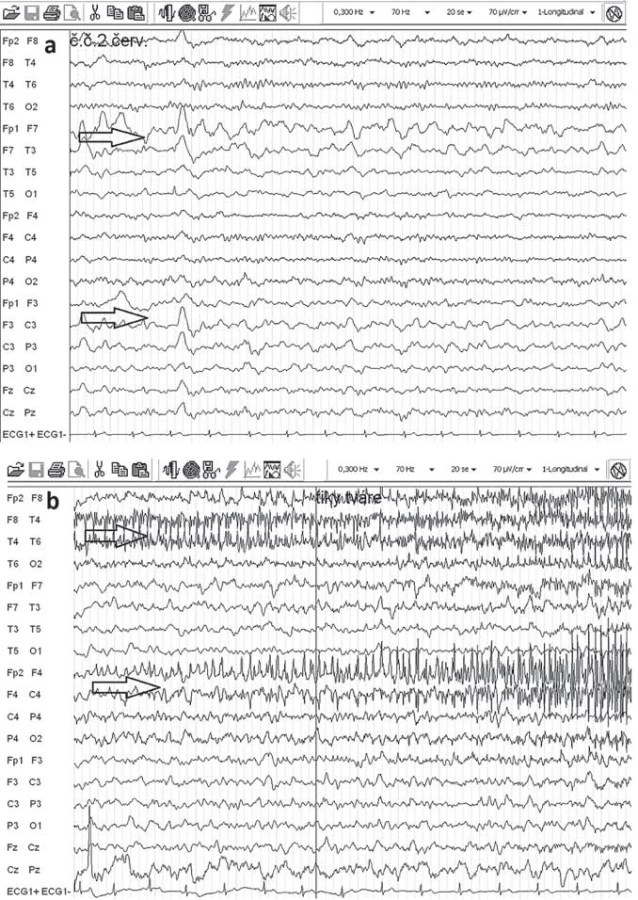
Elektroencefalografia (EEG) nie je špecifická pre RE, hlavný význam je v náleze monohemisferálneho postihnutia. V akútnej fáze ochorenia je v EEG prítomné spomalenie nad postihnutou hemisférou (polymorfná delta), mnohopočetné epileptogénne abnormity a subklinické a klinické iktálne vzorce. EPC nie je vždy asociovaná s rytmickými výbojmi v povrchovej EEG (obr. 3). Neskôr v priebehu ochorenia dochádza k ďalšiemu oplošťovaniu základnej aktivity a ústupu epileptiformných výbojov nad postihnutou hemisférou a zároveň sa stávajú lepšie vyjadrené kontralaterálne, čo niekedy môže viesť k mylnej interpretácii, že nepostihnutá hemisféra sa stala epileptogénnou [17,52].
Biopsia mozgu nie je nutná u všetkých prípadov RE, pretože ostatné vyšetrenia môžu byť postačujúce pre diagnózu. V prípadoch pokročilejšej atrofizácie a malej ihlovej stereotaktickej biopsie môžu byť histopatologické nálezy nekonkluzívne a výrazne zvyšujú riziko falošne negatívnych výsledkov. Preto v prípade, že nie sú kontraindikácie, sa odporúča realizovať otvorenú biopsiu s odobratím vzorky kortexu aj bielej hmoty, niekedy je nevyhnutné odobrať viac vzoriek [53–55]. Podľa odporučenia autorov Biena et al by sa mala bioptická excízia realizovať z neelokventnej oblasti, kde bol v T2/FLAIR MR prítomný zvýšený signál [17,37]. V prípade, že MR je nepriekazná, možno využiť PET alebo SPECT pre určenie miesta biopsie [46]. Histopatologickým vyšetrením je nutné oddiferencovať chronickú vírusovú encefalitídu [56], paraneoplastickú encefalitídu [54,57] a neparaneoplastickú limbickú encefalitídu [58]. Pokiaľ výsledky histopatologického vyšetrenia nie sú jednoznačné, je nutné ďalšie klinické sledovanie a opakovať MR mozgu, pokiaľ sa diagnózu nepodarí stanoviť [17,58].
Histopatologické nálezy u RE rozdelili autori Robitaille et al [53] na základe montrealského materiálu do štyroch skupín podľa trvania ochorenia. Nálezy autorov Robitailla et al potvrdili na základe analýzy 45 chirurgicky riešených prípadov RE v roku 2004 autori Pardo et al [55]. V prvom včasnom štádiu dominuje zápal s početnými nodulmi mikroglie so známkami neuronofágie, perivaskulárnych infiltrátov T lymfocytov [54] a gliovými jazvami. V druhom štádiu sú noduly mikroglie menej výrazné, dominujú perivaskulárne infiltráty T lymfocytov a segmenty kompletnej nekrózy. V treťom štádiu dominuje glióza a strata neurónov, s perzistujúcimi perivaskulárnymi infiltrátmi T lymfocytov a malým počtom nodulov mikroglie. V neskorom štvrtom štádiu dominuje rôzny stupeň gliózy so stratou neurónov, môže byť prítomné malé množstvo nodulov mikroglie a malý rozsah perivaskulárnych infiltrátov. Kvantitatívnou histopatológiou a imunohistochemickým prístupom autori Bien et al našli inverznú koreláciu medzi množstvom nodulov mikroglie, denzity T lymfocytov a aktivovaných astrocytov s trvaním choroby [37]. Rozsiahlejšími imunohistochemickými metódami autori Bien et al potvrdili, že ide o cytotoxickú reakciu T lymfocytov (väčšina sú to CD8+ lymfocyty obsahujúce GrB+granuly) voči neurónom pozitívnym pre MHC triedu I. Z buniek CD68+ HLA – DR+ ma väčšina morfológiu mikroglie, menej ako 5 % má morfológiu makrofágov [59]. B lymfocyty (CD20+) a plazmatické bunky (CD138+) sú zriedkavé, neboli nájdené depozitá imunoglobulínov a ani známky aktivácie komplementu. Inklúzne telieska, ktoré by svedčali pre vírusovú etiológiu neboli nájdené u všetkých pacientov s RE, aj keď v niektorých vzorkách boli nekonštantne prítomné predovšetkým inklúzie herpes vírusov [59]. Morfologické zmeny typu fokálnej kortikálnej degenerácie prítomnej v neskorých štádiách kortikálnej patológie pripomínajú lézie popísané pri excitotoxickom postihnutí kôry [60]. Na základe kazuistík chirurgicky liečených pacientov boli popísané duálne patológie RE, ako napr. kombinácia RE a fokálnej kortikálnej dysplázie, benígnych tumorov, lupus erythematodus, Hashimotovej thyreoitídy, ulceróznej kolitídy alebo tuberóznej sklerózy [22,61]. Nedávno bola popísaná asociácia RE a fokálnej kortikálnej dysplázie typ 2b [62].
V roku 2005 vydali autori Bien et al formálne diagnostické kritéria pre RE ako Konsenzus európskych odborníkov (tab. 1) [17]. V časti A sú neinvazívne kritériá, v časti B je zahrnutá biopsia mozgu ako kritérium pre diagnostiku menej typických prípadov RE.
![Diagnostické kritériá RE – konsenzus európskych odborníkov, autori Bien et al [17].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/da9b43904b649bb50206e2eb8d466682.jpg)
Etiopatogenéza ochorenia
Jednostranná encefalitída, typický nález u RE, ju odlišuje od ostatných zápalových ochorení mozgu. Koncepcia RE ako epileptickej encefalopatie (detská epilepsia s progredujúcou neurologickou deterioráciou) sa opiera o predstavu, že samotná epileptická aktivita vedie k rozvoju neurologického deficitu [17]. Táto predstava sa zakladá na fakte, že neurologický deficit zvyčajne nasleduje po vzniku epilepsie a jeho závažnosť odráža epileptickú aktivitu [17,27]. Efekt steroidov vo včasnej fáze ochorenia zase podporuje názor, že podstatou epilepsie a neurologického deficitu je zápalový proces. Všeobecne sa očakáva, že zápalový proces bude difúzny a bilaterálny, a preto prítomnosť fokálnej epilepsie navodila predstavu, že epilepsia je príčina jednostranného zápalového postihnutia [17]. Na druhej strane existujú závažné argumenty proti tejto koncepcii. Pri Toddovej paréze dochádza k exhauscii, ale nie k trvalému poškodeniu neurónov. Autori Rasmussen et al porovnali histopatologické nálezy u pacientov s častými fokálnymi epileptickými záchvatmi, ale nenašli u nich zápalové zmeny ako u RE [1]. Fokálne epileptické záchvaty sú tak následkom fokálnej encefalitídy [36]. Faktory zodpovedné za asymetrické zápalové postihnutie mozgu zostávajú neznáme.
V súčasnosti narastajú dôkazy, že RE je ochorenie navodené poruchami imunity. Pokusy identifikovať patogénne vírusy ako príčinu RE neviedli k potvrdeniu vírusovej hypotézy [63–70]. Diskutované imunopatogenetické mechanizmy RE možno rozdeliť na tri skupiny: humorálna autoimunita, T lymfocytmi sprostredkovaná cytotoxicita a mikrogliou indukovaná degenerácia [13,17].
Humorálna autoimunita
Tradičný názor, že mozog je chránený proti humorálnej autoimunite, možno dnes považovať za prekonaný. Za posledných 10 rokov bolo popísaných niekoľko autoimunitných encefalitíd v súvislosti s cirkulujúcimi protilátkami proti povrchovým neuronálnym proteínom [2,71,72], kde dominuje poškodenie neurónov protilátkami a/alebo cytokínmi a je menej vyjadrená T bunkami sprostredkovaná cytotoxicita. Autoprotilátka proti tretiemu podtypu glutamátového AMPA receptora (GluR3) bola popísaná ako prvá cirkulujúca protilátka u troch pacientov zo štyroch s RE a zároveň boli referované u niektorých z nich úspechy s plazmaferézou [73]. Okrem GluR3 protilátok bola u pacientov s RE popísaná aj ďalšia protilátka proti NMDA glutamátovému receptoru (NR2B) [74,75]. Tieto protilátky sa okrem RE vyskytujú aj u iných ochorení, ako sú iné typy fokálnych epilepsií (GluR3, GluR1, NR2B), neherpetická akútna limbická encefalitída (NR2B), paraneoplastické encefalopatie (NR2A, NR2B, GluR5, mGluR1), sporadická olivopontocerebelárna atrofia (GluR2), ischemické cievne lézie mozgu a tranzitórne ischemické ataky (NR2A, NR2B) a systémový lupus erythematosus (NR2A, NR2B) [74]. Protilátky proti glutamátovým receptorom (GluR3, NR2B) sa nepotvrdili u všetkých pacientov s RE [76–78], nemožno ich považovať za špecifické pre RE. Ďalšími autormi boli popísané iné typy autoprotilátok (protilátky proti α-7-nikotínovému acetylcholínovému receptoru alebo Munc-18-1 receptoru), ale neskôr neboli potvrdené inými autormi [79–81]. V súčasnosti možno konštatovať, že detekcia glutamátových a iných typov protilátok nemôže prispieť k potvrdeniu diagnózy RE. Pretože každá z protilátok bola objavená len u malej skupiny pacientov s RE a efekt plazmaferézy nie je spoľahlivý u všetkých pacientov s RE, úloha protilátok v patogenéze RE zostáva nejasná [13,17,74]
T bunkami sprostredkovaná cytotoxicita
Cytotoxické T lymfocyty sú považované v súčasnosti za rozhodujúceho hráča v patogenéze RE. Aktívna RE je charakterizovaná Th1 typom imunitnej odpovede, ktorá zahŕňa CD8+ a CD4+ T lymfocyty [82]. Nie je známe, či táto T cytotoxická odpoveď je zameraná proti jednému antigénu. Väčšina zápalových T lymfocytov sú CD8+ lymfocyty a 10 % z nich sú granzým B pozitívne bunky, ktoré sa nachádzajú v apozícii k neurónom (MHC triedy I pozitívnym) a astrocytom (GFAP pozitívnym) s polarizáciou cytotoxických granúl k cieľovej membráne [59]. Akumulácia CD4+ lymfocytov v leptomeningoch a perivaskulárnych priestoroch bola dominujúcou črtou vo vzorkách RE získaných do jedného roka od nástupu záchvatov [82]. Spektrálna typizácia T lymfocytov potvrdila expanziu určitého klonotypu z prekurzorových T buniek odpovedajúcich na antigénny epitop jedného mozgového antigénu [83]. Jeho identita nie je známa, ale predpokladá sa, že ide o antigén spoločný pre neuróny a astrocyty, keďže cytotoxické T lymfocyty atakujú oba typy buniek [84]. Longitudinálne analýzy periférnej krvi potvrdili dominanciu a perzistenciu špecifických CD8+ T bunkových klonov v čase. Tieto údaje silne podporujú hypotézu, že v patogenéze RE zohráva rozhodujúcu úlohu antigénom MHC triedy I spúšťaná, CD8+ T bunkami sprostredkovaná cytotoxická reakcia namierená proti neurónom a astrocytom. Iná možnosť je, že tieto cytotoxické T lymfocyty môžu rozpoznávať cudzorodý antigén z infikovaných neurónov a astrocytov. Keďže bežné vírusy (herpes vírusy a enterovírusy) neboli potvrdené ako kauzálna príčina RE, otvorenou ostáva otázka možného zriedkavého infekčného patogénu [63–70].
Autori Owens et al v roku 2013 publikovali prácu zameranú na porovnanie stupňa zápalovej aktivity kohorty 12 pacientov s RE a 12 pacientov s kortikálnou dyspláziou a fokálnymi záchvatmi, v ktorej sa zamerali na gény kódujúce interferón-γ [82]. Gény CCL5, CCL22, CCL23, CXCL9 a Fas ligand mali u RE vyšší stupeň expresie ako u kortikálnej dysplázie. Tieto gény kódujú špecifické chemokíny a majú prozápalový efekt [13,82]. Otvorenou zostáva otázka využitia terapie zameranej na cytokíny v liečbe RE.
Mikrogliou indukovaná degenerácia
Aktivácia mikroglie je jedna z charakteristických histopatologických čŕt RE. Stupeň aktivácie mikroglie je v rôznych miestach mozgu rôzny, ale koreluje s T lymfocytovou infiltráciou a stupňom kortikálneho poškodenia [55,85]. Mikroglia sa produkciou prozápalových cytokínov podieľa na vzniku epileptických záchvatov a zvýšenia excitability neuronálnych sietí u niektorých epileptických záchvatov [86–89]. U RE okrem aktivácie mikroglie je prítomná aj aktivácia astrocytov, ktorá koreluje s postupom kortikálneho postihnutia [55,84]. V dôsledku cytotoxickej T lymfocytovej reakcie dochádza k úbytku astrocytov [84] a následnému vzostupu albumínu v likvore. V dôsledku downregulácie Kir4.1 káliových kanálikov astrocytov dochádza k nárastu extracelulárneho kália, a tiež NMDA receptorom sprostredkovanej neuronálnej excitabilite, čo môže byť súčasťou procesu iniciácie epileptických záchvatov u RE [13,90].
Autori Ramaswarny et al sa zamerali na štúdium vnútrobunečných mechanizmov aktivovanej mikroglie, tzv. inflamazómu. Vo svojej práci zistili zvýšenú expresiu prozápalových proteínov NLRP1 a NLRP3, ktoré sa v kaskáde s kaspázami 1 a 5 zúčastňujú produkcie interleukínu-1β. V myeloidných a mononukleárnych bunkách bielej hmoty a priľahlého kortexu zistili ďalej zvýšenú expresiu antigénov MHC triedy II a kaspázy 1. Aktivácia v porovnávaných vzorkách pacientov s epilepsiou a sklerózou multiplex bola nižšia [91]. Problémom aktuálnych poznatkov zostáva fakt, že imunohistochemické a imunologické analýzy boli získané počas rozvinutej choroby a nie je jasné, ktoré zo zistených fenoménov sú sekundárne a ktoré môžu byť primárne. V súčasnosti zostáva primárny inzult vedúci k RE neznámy.
Diferenciálna diagnostika
Diferenciálna diagnostika RE zahŕňa ochorenia, ktoré sa môžu prezentovať dysfunkciou jednej hemisféry a refraktérnymi fokálnymi záchvatmi (EPC). Patria sem niektoré vývojové a vrodené poruchy CNS, metabolické, zápalové, infekčné, degeneratívne a nádorové ochorenia (tab. 2).
![Diferenciálna diagnostika Rasmussenovej encefalitídy – upravené, podľa autorov Varadkar et al [13].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/a701b97e605a4c46087154b30c1d95c8.jpg)
Terapia
Závažnosť a rarita ochorenia sú príčinou, prečo ani v súčasnosti nemáme k dispozícii prospektívne kontrolované štúdie, s výnimkou jednej nemeckej randomizovanej štúdie porovnávajúcej efekt intravenóznych imunoglobulínov (IVIG) a takrolimu [11]. Stratégia liečby RE má dva ciele: prvým je dosiahnuť kontrolu epileptických záchvatov a druhým je zamedziť progresiu mozgovej atrofie a zhoršovanie neurologického deficitu [2,17,92].
Antiepileptiká v liečbe RE
Epileptické záchvaty sú u viac ako 80 % pacientov farmakorezistentné, nie je známa žiadna liečba antiepileptikami (AED) v monoterapii alebo v polyterapii, ktorá by bola účinnejšia ako iná [93]. Vo všeobecnosti by sa malo použiť čo najmenej AED a v čo najnižších dávkach. Redukovaná polyterapia (2–3 AED) alebo monoterapia môže byť rovnako efektívna ako mnohopočetná polyterapia. Použitie AED je viazané na predpoklad, že sa dosiahne aspoň čiastočná kontrola sekundárne generalizovaných tonicko-klonických záchvatov a komplexných parciálnych záchvatov, ale EPC a fokálne záchvaty spravidla nereagujú na liečbu antiepileptikami [2,17,92]. V súčasnosti neexistujú žiadne údaje, ktoré by potvrdzovali vyššiu účinnosť nových AED v porovnaní so staršími v liečbe záchvatov pri RE, ale výhodou môže byť nižší potenciál k vedľajším nežiaducim účinkom, lepšia tolerabilita a nižší potenciál ku farmakokinetickým interakciám, čo má význam predovšetkým pri dlhotrvajúcej imunomodulačnej liečbe. AED, ktoré sú enzýmové induktory, znižujú hladiny kortikoidov a cytostatík, na druhej strane enzýmové inhibítory zvyšujú riziko intoxikácie takrolimom a rozvoja encefalopatie. AED, ktoré majú vysokú väzbu na plazmatické bielkoviny, majú nestabilnú hladinu v prípade plazmaferézy [92]. V prípadoch lokalizovanej EPC bol v kazuistických prípadoch použitý s úspechom botulotoxin aplikovaný do cieľových svalov tváre a hornej končatiny, čím sa dosiahla redukcia bolestivých spazmov a zlepšila funkcia postihnutej končatiny [94,95]. Za nefarmakologický pokus ovplyvniť epileptické záchvaty u pacientov s RE možno považovať kazuistiky s použitím stimulátora nervus vagus a transkraniálnej magnetickej stimulácie [96,97].
Imunomodulačná liečba
V liečbe RE bolo použitých v kazuistikách, malých kazuistických sériach a v nedávnej prospektívnej randomizovanej štúdii z Nemecka [11] niekoľko spôsobov imunosupresívnej a imunomodulačnej liečby s cieľom ovplyvniť progresiu atrofie a refraktérnosť epileptických záchvatov. Z týchto zdrojov sa ukazuje, že kortikosteroidy, intravenózne imunoglobulíny (IVIG), plazmaferéza/imunoadsorpcia, takrolimus a azatioprin môžu byť efektívne v prevencii progresie ochorenia. Imunomodulačná liečba však vykazuje individuálne rozdiely v efektivite a zväčša časovo obmedzený efekt. Diskrepancia medzi spomalením atrofizácie mozgovej hemisféry a vývoja neurologického deficitu môže byť problémom pri indikácii hemisferektómie z dôvodu refraktérnej epilepsie [13,92].
Kortikosteroidy v liečbe RE majú pozitívny efekt predovšetkým v počiatočných štádiách ochorenia. Použité boli vysoké dávky prednizonu/prednizonolonu s postupnou redukciou dávky [27,98,99]. Pre dlhodobú liečbu sa odporúča začať bolusom i.v. metylprednizolonu v dávke 400 mg/m2/deň alebo u detí 20 mg/kg/deň a potom pokračovať p. o. prednizonom/prednizonolonom v dávke 1–2 mg/kg/deň [98,99]. Túto dávku sa odporúča pomaly redukovať. Nežiaduce účinky dlhodobej kortikoterapie sú početné a dobre známe. Krátke bolusy kortikosteroidov možno využiť na prerušenie status epilepticus [17,98,99].
Použitie IVIG v liečbe RE sa ukázalo ako efektívne vo viacerých kazuistických sériach a nedávno aj v kontrolovanej štúdii autorov Biena et al [11,98,100,101]. Pozitívne výsledky s použitím IVIG u RE s neskorým nástupom trvajúce viac ako 12 mesiacov [99–101] viedli k odporučeniu IVIG ako liečby prvej línie predovšetkým u RE s neskorým nástupom. Doporučená je úvodná kúra v dávke 0,4 g/kg/deň počas 3–5 dní a pokračovanie v dávke 0,4–2 g/kg v intervale jeden mesiac, dávku možno rozdeliť na 1–5 nasledujúcich dní [99,100]. Vedľajšie nežiaduce účinky IVIG sú zriedkavé. V prípade nedostatočného efektu samotného IVIG sa ukázala prospešná kombinácia IVIG s kortikosteroidmi [92,98].
Plazmaferéza v liečbe RE bola aplikovaná v sériach 3–6 veľkoobjemových výmen v intervale 2–8 týždňov [99,102]. U RE s neskorým nástupom bola s pozitívnym výsledkom použitá imunoadsorpcia [103]. V nasledujúcich týždňoch po plazmaferéze bolo pozorované u niektorých pacientov zlepšenie neurologickej funkcie a pokles frekvencie záchvatov. Skúsenosti s dlhodobou aplikáciou sú veľmi limitované [17,98].
Cytostatiká takrolimus a azatioprin boli použité v niekoľkých sériach pacientov [11, 104,105] s pozitívnym efektom. Autori Bien et al v prvej kazuistickej sérii porovnali sedem pacientov s 12 historickými prípadmi RE. Zistili, že takrolimus je schopný spomaliť progresiu atrofie, ale ukázal sa neúčinný na epileptické záchvaty [104]. V prospektívnej randomizovanej štúdii autori Bien et al rozdelili pacientov na dve skupiny [11]. Jedna bola liečená takrolimom a druhá IVIG. Konštatovali, že v porovnaní s historickými prípadmi RE u liečených pacientov neprogredovala neurologická dysfunkcia, v skupine liečenej IVIG na rozdiel od takrolimu neboli zaznamenané závažné nežiaduce účinky. Štúdia však nemala silu na porovnanie superiority jedného z týchto dvoch liekov. Cyklofosfamid bol vyskúšaný v sérii štyroch pacientov, ale nebol efektívny [99].
Z ďalších terapeutických pokusov vyšli neúspešne lieky zamerané na potenciálnu vírusovú infekciu – gancyklovir, interferón alfa a zidovudín [106].
Biologická liečba predstavuje nové výzvy v liečbe RE. V nepublikovanej otvorenej kalifornskej pilotnej štúdii autorov Laxera et al s použitím rituximabu (monoklonálna protilátka proti CD20+, t.j. proti B lymfocytom) bolo pozorované zlepšenie epileptických záchvatov u ôsmich z deviatich pacientov, navyše došlo k zlepšeniu neurologického deficitu. Odvtedy sa objavili ďalšie kazuistiky s dobrým efektom rituximabu v liečbe predovšetkým RE s neskorým nástupom [107,108]. Otvorené ostávajú otázky optimálneho protokolu a dlhodobého efektu rituximabu. Nádejnými kandidátmi môžu byť monoklonálne protilátky blokujúce vstup T lymfocytov do CNS. Sľubné výsledky prinieslo použitie natalizumabu v jednej kazuistike RE [109].
Chirurgická liečba
Chirurgická liečba je najúčinnejšia metóda, ktorou sa dá dosiahnuť bezzáchvatovosť u RE, avšak za cenu funkčného deficitu, pretože efekt má iba kompletná diskonekcia postihnutej hemisféry (hemidiskonekcia), nezávisle od toho, či ide o funkčnú hemisferektómiu alebo hemisferotómiu. Bezzáchvatovosť v rôznych sériach pacientov sa pohybuje medzi 62,5 a 85 % [110–113]. Mortalita v týchto sériach sa pohybovala od 0 do 4 % [11,111,112]. Diskonekčné výkony (funkčná hemisferektómia a hemisferotómia) sú spojené s nižším rizikom komplikácií v porovnaní s anatomickou hemisferektómiou [114]. Malé resekcie s cieľom uchovať funkciu neposkytli trvalú kontrolu záchvatov [115]. Rozhodnutie pre hemisferektómiu môže byť komplikované v prípadoch starších detí a dospelých, hlavne ak RE postihuje dominantnú hemisféru alebo neurologický deficit je veľmi ľahký.
Načasovanie diskonekčných výkonov má byť riadené závažnosťou epileptických záchvatov. Podľa niektorých názorov včasná hemisferektómia môže byť prevenciou pred poškodením druhej zdravej hemisféry opakovanými záchvatmi. V sériach detí po hemisferektómii a dosiahnutej kontrole záchvatov bolo pozorované zlepšenie kongitívnych funkcií, s lepšími výsledkami v nedominantnej hemisfére a horšími výsledkami u pacientov v dominantnej hemisfére a perzistujúcimi záchvatmi po operácii [116–118]. Objavenie sa nezávislých epileptiformných výbojov v kontralaterálnej hemisfére sa považuje za nepriaznivý prognostický znak pre kognitívne zhoršenie po operácii [16]. V prípadoch pomalšej progresie neurologického deficitu, ako je to u dospelých, ale niekedy aj u detí, sa odporúča s hemisferektómiou počkať [6,7]. Rozhodnutie pre chirurgickú liečbu teda ovplyvní dominancia hemisféry a očakávaný neurologický deficit, čo musí byť detailne prediskutované s pacientom, v prípade detí s ich rodičmi.
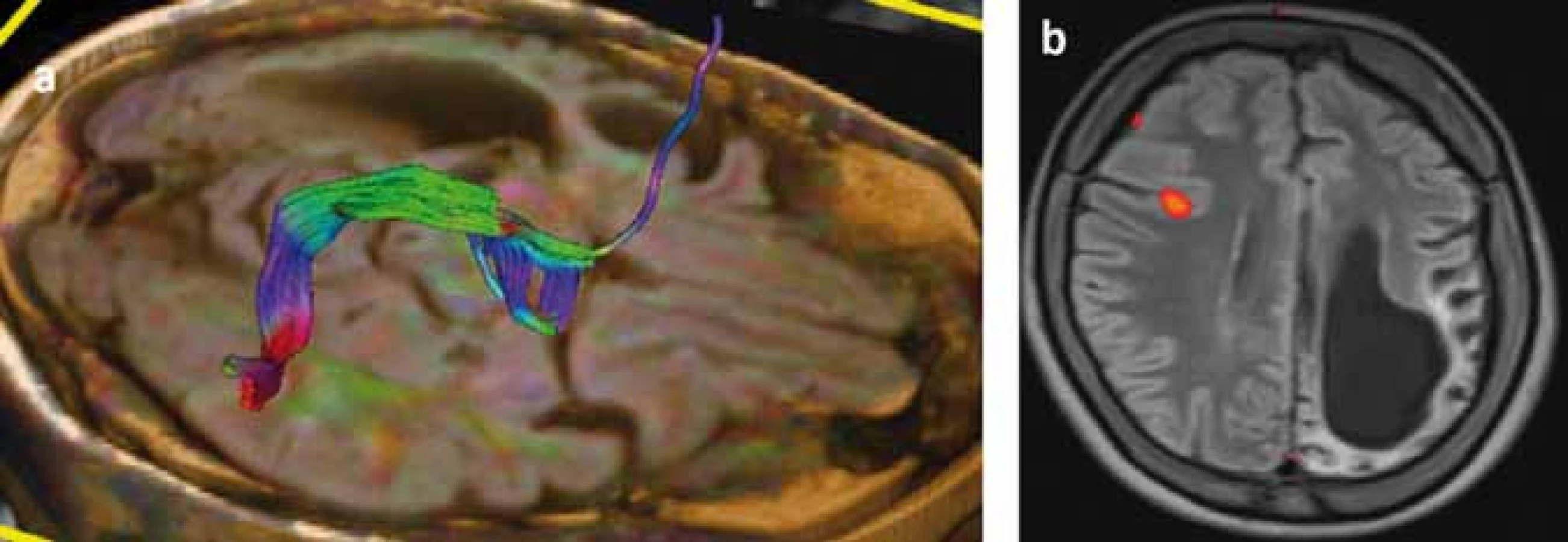
Následky hemisferektómie v motorickej oblasti sa prejavia ako kontralaterálna spastická hemiparéza so stratou jemnej motoriky ruky (funkčne významnou) [119], ale väčšina pacientov je schopná chôdze bez asistencie. Ďalší nežiaduci účinok hemisferektómie je kontralaterálna homonymná hemianopsia, ktorá môže byť prítomná už pred resekciou, a podľa väčšiny lekárov, ktorí pacientov s RE liečia, neinterferuje závažným spôsobom s pacientovým životom [114]. Najzložitejšia je situácia s rečovými schopnosťami pri postihnutí dominantej hemisféry. Pri porovnaní rečových schopností u pacientov s hemisferektómiou v dominantnej hemisfére pred operáciou a po operácii došlo u nich ku signifikantnému zhoršeniu expresívnej funkcie reči, ale nie celkovej inteligencie, perceptívnej rečovej funkcie, vizuálne-motorických praktických schopností, behaviorálneho a adaptívneho správania [113]. Transfer rečových funkcií z dominantnej hemisféry je závislý na veku. Postihnutie rečovo dominantnej hemisféry pred 5.–6. rokom života zvyčajne vedie k transferu rečových funkcií do druhej hemisféry [17]. Šesť detí v jednej sérii vo veku 5–10 rokov rok po hemisferektómii dosiahlo pôvodné perceptívne rečové schopnosti, ale boli schopné len „telegrafickej reči“ [120]. Pre predikciu možného transferu rečových schopností je v súčasnosti prínosom fMR (obr. 4), ale zlatým štandardom zostáva Wadov test. V rozhodovacom procese je nutné zvážiť riziká perzistujúcich záchvatov a stratených funkcií v porovnaní s benefitmi operácie prísne individuálnym prístupom.
Rozhodovací proces pre liečbu RE je viacstupňový. Ťažká farmakorezistentná epilepsia, ktorá pacienta ohrozuje závažnými komplikáciami, je indikáciou k chirurgickej liečbe, predovšetkým u detí a v prípade, že nie je prítomné vysoké riziko zhoršenia motorických a rečových funkcií (nedominantná hemisféra). Kde nám ide o zachovanie motorických a rečových funkcií, ktoré chorobou ešte nie sú výrazne postihnuté, predovšetkým u starších detí, adolescentov a dospelých, sa uprednostňuje imunoterapia. U malých detí do šiestich rokov so závažnejším motorickým deficitom, kde v rámci mozgovej plasticity možno očakávať transfer postihnutých funkcií do druhej hemisféry, je hemisferektómia indikovaná aj v prípade postihnutia dominantnej hemisféry. Keďže operačná liečba vo všeobecnosti vedie k zlepšeniu celkových kognitívnych schopností, má sa realizovať vo fáze, keď ešte nevyhasli všetky dôležité funkcie, a nie ako metóda poslednej voľby [2,13,17].
Záver
RE je klinicky a histopatologicky dobre definované chronické zápalové ochorenie mozgu, ktorého príčina zostáva napriek veľkému úsiliu neobjasnená. Historicky jediným možným efektívnym riešením farmakorezistentnej epilepsie bola hemisferektómia. Pokroky v odhaľovaní imunopatologických procesov v etiopatogenéze ochorenia priniesli nové imunomodulačné a imunosupresívne metódy liečby RE. Ich výhodou je, že dokážu spomaliť progresiu ochorenia a u mnohých pacientov dostať pod kontrolu epileptické záchvaty. V súčasnosti však nemáme dostatok informácií o dlhodobom efekte imunoterapie. Problémom pri tvorbe dostatočne valídnych štúdií pre hodnotenie efektu liečby je však rarita ochorenia a veľká variabilita klinických obrazov u jednotlivých pacientov, čo sa týka veku, závažnosti epilepsie a progresie neurologického deficitu. Tieto problémy môžu preklenúť iba multicentrické štúdie. Vzhľadom na závažnú prognózu ochorenia je nevyhnutné pacienta s podozrením na RE neodkladne odoslať do epileptologického centra.
Zoznam použitých skratiek
AMPA – kyselina α-amino-3-hydroxy-5-metyl-4-izoxazolpropionová
CAPS – katastrofický antifosfolipidový syndróm
CD – cluster designation (cluster of differentiation) – nomenklatúra hybridómov
CMV – cytomegalovírus
CNS – centrálny nervový systém
DR – vzťahuje sa k D antigénu (D-antigen Related – väzobné miesto T lymfocytov)
EEG – elektroencefalografia
EPC – epilepsia partialis continua
18F-FDG PET – fluorodeoxyglukózová pozitrónová emisná tomografia značená 18F fluórom
fMR – funkčná magnetická rezonancia
GAD – dekarboxyláza kyseliny glutámovej
GFAP – gliálny fibrilárny kyslý proteín
GluR3 – tretí podtyp AMPA glutamátového receptora
Gr – granzým
HIV – vírus ľudskej imunodeficiencie
HLA – systém antigénov ľudských leukocytov (súbor génov kódujúci MHC)
HSV – herpes simplex vírus
IVIG – intravenózny imunoglobulín
Kir4.1 – rodina inward rectifier typu káliových kanálikov (presun kália intracelulárne)
LORE – RE s neskorým nástupom
MELAS – mitochondriálna encefalopatia s laktátovou acidózou a stroke-like epizódami
MHC – veľký komplex histokompatibility (Major Histocompatibility Complex)
MNI – Montrealský neurologický inštitút
MR – magnetická rezonancia
MRS – magneticko-rezonančná spektrometria
Munc-18 – mammalian uncoordinated-18 protein, člen rodiny proteínov Sec1/munc-18 (riadi exocytózu neurónov)
NAA – N-acetyl-aspartát
NMDA – kyselina N-metyl-D-asparágová
NMDAR – NMDA receptor
NR2B – podtyp 2B (podjednotka epsilon 2) glutamátových NMDA receptorov, má význam v dlhodobej potenciácii (LTP)
RE – Rasmussenova encefalitída
SPECT – jednofotónová emisná tomografia
SREAT – na steroidy reagujúca encefalopatia asociovaná s autoimúnnou thyreoiditídou
TLE – epilepsia temporálneho laloka
Autori nezískali v súvislosti s touto prácou žiadnu finančnú podporu ani grantovú, ani súkromnej osoby, ani subjektov z oblasti farmaceutického priemyslu a zdravotníckej techniky.
Touto prácou chceme poďakovať in memoriam za dlhoročnú spoluprácu primárovi Imunologickej ambulancie SZU M. Šimkovi, ktorý nás po tragickej udalosti opustil v roku 2015.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Gabriela Timárová, PhD.
II. neurologická klinika
LF UK a UN Bratislava
Limbová 5
833 05 Bratislava
e-mail: gtimarova@gmail.com
Prijato k recenzii: 11. 4. 2016
Prijato do tlače: 20. 6. 2016
MUDr. Gabriela Timárová, PhD.

MUDr. Gabriela Timárová, PhD. absolvovala štúdium všeobecného lekárstva na Lekárskej fakulte Univerzity Komenskéhos Cenou ministra školstva za mimoriadne študijné výsledky. Od roka 1987 pracuje na II. neurologickej klinike LF UK postupne ako sekundárna lekárka, ordinárka pre klinickú neurofyziológiu a od roku 1995 ako odborná asistentka. V roku 1994 získala atestáciu II. stupňa v odbore neurológia. Po odbornej stránke sa venovala problematike cievnych mozgových príhod, bolestí hlavy, postupne prevážila problematika intraoperačného monitoringu v neurochirurgii, a epileptológie. V roku 2004 obhájila dizertačnú prácu na tému „Emisná tomografia mozgu a parciálna epilepsia“. Aktívne sa zúčastňuje domácich i zahraničných kongresov, kde predniesla viac ako 150 odborných prednášok, je autorkou a spoluautorkou desiatok publikácií v domácich a zahraničných časopisoch, autorkou monografie „Neurológia pre všeobecných praktických lekárov“ (2011). So svojimi spolupracovníkmi v Nemocnici akademika L. Dérera UN v Bratislave založila epileptochirurgický program pre dospelých pa cientov. Je recenzentkou časopisu Clinical and Applied Thrombosis and Haemostasis, recenzovala články pre Českú a slovenskú neurológiu a neurochirurgiu. Je členkou výboru Slovenskej ligy proti epilepsii, členkou výboru Sekcie pre bolesť hlavy Slovenskej neurologickej spoločnosti a členkou výboru Neurofarmakologickej sekcie Slovenskej neurologickej spoločnosti.
Sources
1. Rasmussen T, Olszewski J, Loyd-Smith D. Focal seizures due to chronic localized encephalitis. Neurology 1958; 8 (6): 435–45.
2. Sheybani L, Schaller K, Seeck M. Rasmussen’s encephalitis: an update. Schweizer Archiv Neurol Psych 2011; 162 (6): 225–31.
3. Krýsl D, Elišák M. Autoimunitní encefalitidy. Cesk Slov Neurol N 2015; 78/111 (1): 7–23. doi: 10.14735/amcsnn20157.
4. Mastrangelo M, Mariani R, Menichella A. Eponym Rasmussen syndrome. Eur J Pediatr 2010; 169 (8): 919–24. doi: 10.1007/s00431-010-1148-0.
5. Oguni H, Andermann F, Rasmussen TB. The syndrome of Chronic Encephalitis and Epilepsy. Adv Neurol 1992; 57 : 419–33.
6. Oguni H, Andermann F, Rasmussen TB. The natural history of the syndrome of chronic encephalitis and epilepsy: a study of the MNI series of forty-eight cases. In: Andermann F, ed. Chronic encephalitis and epilepsy – Rasmussen’s syndrome. Boston: Butterworth-Heinemann 1991 : 7–35.
7. Bien CG, Widman G, Urbach H, et al. The natural history of Rasmussen’s encephalitis. Brain 2002; 125 (8): 1751–9. doi: org/10.1093/brain/awf176.
8. Gray F, Serdaru M, Baron H, et al. Chronic localised encephalitis (Rasmussen’s) in an adult with epilepsia partialis continua. J Neurol Neurosurg Psychiatry 1987; 50 (6): 747–75.
9. Hart YM, Andermann F, Fish DR, et al. Chronic encephalitis and epilepsy in adults and adolescents: a variant of Rasmussen’s syndrome? Neurology 1997; 48 (2): 418–24.
10. Villani F, Pincherle A, Antozzi C, et al. Adult-onset Rasmussen’s encephalitis: anatomical-electrographic-clinical features of 7 Italian cases. Epilepsia 2006; 47 (5): 41–6. doi: 10.1111/j.1528-1167.2006.00876.x.
11. Bien CG, Tiemeier H, Sassen R, et al. Rasmussen encephalitis: incidence and course under randomized therapy with tacrolimus or intravenous immunoglobulins. Epilepsia 2013; 54 (3): 543–50. doi: 10.1111/epi.12042.
12. Lamb K, Scott WJ, Mensah A, et al. Prevalence and clinical outcome of Rasmussen encephalitis in children from the United Kingdom. Dev Med Child Neurol 2013; 55 (Suppl 1): 14.
13. Varadkar S, Bien CG, Kruse CA, et al. Rasmussen’s encephalitis: clinical features, pathobiology, and treatment advances. Lancet Neurol 2014; 13 (2): 195–205. doi: 10.1016/S1474-4422 (13) 70260-6.
14. Thomas JE, Reagan TJ, Klass DW. Epilepsia partialis continua. A review of 32 cases. Arch Neurol 1977; 34 (5): 266–75.
15. Obeso JA, Rothwell JC, Marsden CD. The spectrum of cortical myoclonus. From focal reflex jerks to spontaneous motor epilepsy. Brain 1985; 108 (1): 193–224.
16. Longaretti F, Dunkley C, Varadkar S, et al. Evolution of the EEG in children with Rasmussen’s syndrome. Epilepsia 2012; 53 (9): 1539–45. doi: 10.1111/j.1528-1167.2012.03565.x.
17. Bien CG, Granata T, Antozzi C, et al. Pathogenesis, diag - nosis and treatment of Rasmussen encephalitis: a European consensus statement. Brain 2005; 128 (3): 454–71. doi: org/10.1093/brain/awh415.
18. Bauer J, Bien CG. Encephalitis and epilepsy. Semin Immunopathol 2009; 31 (4): 537–44. doi: 10.1007/s00281-009-0176-1.
19. Cheong JY, Wong C, Bleasel A, et al. Late onset Rasmussen’s encephalitis with tripple pathology. J Clin Neurosci 2009; 16 (12): 1677–81. doi: 10.1016/j.jocn.2009.02.042.
20. Gambardella A, Andermann F, Shorvon S, et al. Limited chronic focal encephalitis: another variant of Rasmussen syndrome? Neurology 2008; 70 (Suppl 6): 13–8. doi: 10.1111/j.1528-1167.2008.01751.x.
21. McLachlan RS, Girvin JP, Blume WT, et al. Rasmussen’s chronic encephalitis in adults. Arch Neurol 1993; 50 (3): 269–74.
22. Hart YM, Andermann F, Robitaille Y, et al. Double pathology in Rasmussen’s syndrome: a window on the etiology? Neurology 1998; 50 (3): 731–5.
23. Hennessy MJ, Koutroumanidis M, Dean AF, et al. Chronic encephalitis and temporal lobe epilepsy: a variant of Rasmussen’s syndrome? Neurology 2001; 56 (5): 678–81.
24. Kashihara K, Ohno M, Takahashi Y. Twenty-one-year course of adult onset Rasmussen’s encephalitis and bilateral uveitis: case report. J Neurol Sci 2010; 294 (1–2): 127–30. doi: 10.1016/j.jns.2012.10.021.
25. Larionov S, Konig R, Urbach H, et al. MRI brain volumetry in Rasmussen encephalitis: the fate of affected and „unaffected“ hemispheres. Neurology 2005; 64 (5): 885–7.
26. Tobias SM, Robitaille Y, Hickey WF, et al. Bilateral Rasmussen encephalitis: postmortem documentation in a five-year-old. Epilepsia 2003; 44 (1): 127–30.
27. Chinchilla D, Dulac O, Robain O, et al. Reappraisal of Rasmussen’s syndrome with special emphasis on treatment with high doses of steroids. J Neurol Neurosurg Psychiatry 1994; 57 (11): 1325–33.
28. Peariso K, Standridge SM, Hallinan BE, et al. Presentation, diagnosis and treatment of bilateral Rasmussen’s encephalitis in a 12-year-old female. Epileptic Disord 2013; 15 (3): 324–32. doi: 10.1684/epd.2013.0594.
29. Bhatjiwale MG, Polkey C, Cox TC, et al. Rasmussen’s encephalitis: neuroimaging findings in 21 patients with a closer look at the basal ganglia. Pediatr Neurosurg 1998; 29 (3): 142–8.
30. Ben-Zeev B, Nass D, Polack S, et al. Progressive unilateral basal ganglia atrophy and hemidystonia: new form of chronic focal viral encephalitis. Neurology 1999; 51 (Suppl 1): 42.
31. de Leva MF, Varrone A, Filla A, et al. Neuroimaging follow-up in a Case of Rasmussen’s Encephalitis with Dyskinesias. Mov Disord 2007; 22 (14): 2117–21. doi: 10.1016/j.pediatrneurol.2008.03.001.
32. Frucht S. Dystonia, athetosis, and epilepsia partialis continua in a patient with late-onset Rasmussen’s encephalitis. Mov Disord 2002; 17 (3): 609–12.
33. Koen MA, Zupanc ML. Unusual presentation and MRI findings in Rasmussen’s syndrome. Pediatr Neurol 1999; 21 (5): 839–42.
34. McDonald D, Farrell MA, McMenamin J. Rasmussen’s syndrome associated with chronic brain stem encephalitis. Eur J Paediatr Neurol 2001; 5 (5): 203–6.
35. Bien CG, Elger CE, Leitner Y, et al. Slowly progressive hemiparesis in childhood as a consequence of Rasmussen encephalitis without or with delayed-onset seizures. Eur J Neurol 2007; 14 (4): 387–90.
36. Korn-Lubetzki I, Bien CG, Bauer J, et al. Rasmussen encephalitis with active inflammation and delayed seizures onset. Neurology 2004; 62 (6): 984–6.
37. Bien CG, Urbach H, Deckert M, et al. Diagnosis and staging of Rasmussen’s encephalitis by serial MRI and histopathology. Neurology 2002; 58 (2): 250–7.
38. Chiapparini L, Granata T, Farina L, et al. Diagnostic imaging in 13 cases of Rasmussen’s encephalitis: can early MRI suggest the diagnosis? Neuroradiology 2003; 45 (3): 171–83.
39. Wagner J, Schoene-Bake JC, Bien CG, et al. Automated 3D MRI volumetry reveals regional atrophy differences in Rasmussen encephalitis. Epilepsia 2012; 53 (4): 613–21. doi: 10.1111/j.1528-1167.2011.03396.x.
40. Bien CG, Elger CE, Wiendl H. Advances in pathogenic concepts and therapeutic agents in Rasmussen’s encephalitis. Expert Opin Investig Drugs 2002; 11 (2): 981–9.
41. Geller E, Faerber EN, Legido A, et al. Rasmussen encephalitis: complementary role of multitechnique neuroimaging. Am J Neuroradiol 1998; 19 (4): 445–9.
42. Sener RN. Diffusion MRI and spectroscopy in Rasmussen’s encephalitis. Eur Radiol 2003; 13 (9): 2186–91.
43. Cendes F, Andermann F, Silver K. Imaging of axonal damage in vivo in Rasmussen’s syndrome. Brain 1995; 118 (3): 753–8.
44. Sener RN. Rasmussen’s encephalitis: proton MR spectroscopy and diffusion MR findings. J Neuroradiol 2000; 27 (3): 179–84.
45. Fiorella DJ, Provenzale JM, Edward CR,et al. 18F-fluorodeoxyglucose positron emission tomography and MR imaging findings in Rasmussen encephalitis. Am J Neuroradiol 2001; 22 (7): 1291–9.
46. Lee JS, Juhasz C, Kaddurah AK, et al. Patterns of cerebral glucose metabolism in early and late stages od Rasmussen’s syndrome. J Child Neurol 2001; 16 (11): 798–805.
47. Maeda Y, Oguni H, Sato Y, et al. Rasmussen syndrome: multifocal spread of inflammation suggested from MRI and PET findings. Epilepsia 2003; 44 (8): 1118–21.
48. Shah JR, Juhasz C, Kupsky WJ, et al. Rasmussen encephalitis associated with Parry-Romberg syndrome. Neurology 2003; 61 (3): 395–7.
49. Burke GJ, Fifer SA, Yoder J. Early detection of Rasmussen’s syndrome by brain SPECT imaging. Clin Nucl Med 1992; 17 (9): 730–1.
50. Hartley LM, Gordon I, Harkness W, et al. Correlation of SPECT with pathology and seizure outcome in children undergoing epilepsy surgery. Dev Med Child Neurol 2002; 44 (10): 676–80.
51. Rasmussen T, Andermann F. Update on the syndrome of chronic encephalitis and epilepsy. Cleveland Clin J Med 1989; 56 (Suppl 2): 181–4.
52. Granata T, Gobbi G, Spreafico R, et al. Rasmussen’s encephalitis: early characteristics allow diagnosis. Neurology 2003; 60 (3): 422–5.
53. Robitaille Y. Neuropathologic aspects of chronic encephalitis. In: Andermann F, ed. Chronic encephalitis and epilepsy. Rasmussen’s syndrome. Boston: Butterworth - Heinemann 1991 : 79–110.
54. Farrell MA, Droogan O, Secor DL, et al. Chronic encephalitis associated with epilepsy: immunohistochemical and ultrasturctural studies. Acta Neuropathol Berl 1995; 89 (4): 313–21.
55. Pardo CA, Vining EP, Guo L, et al. The pthology of Rasmussen syndrome: stages of cortical involvement and neuropathological sudies in 45 hemispherectomies. Epilepsia 2004; 45 (5): 516–26.
56. Booss J, Esiri MM. Viral encephalitis in humans. Washington DC: ASM Press 2003.
57. Graus F, Ribalta T, Campo E, et al. Immunohisto-chemical analysis of the immune reaction in the nervous system in paraneoplastic encephalomyelitis. Neurology 1990; 40 (2): 219–22.
58. Bien CG, Schulze-Bonhage A, Deckert M, et al. Limbic encephalitis not associated with neoplasm as a cause of temporal lobe epilepsy. Neurology 2000; 55 (12): 1823–8.
59. Bien CG, Bauer J, Deckwerth TL, et al. Destruction of neurons by cytotoxic T cells: a new pathogenetic mechanism in Rasmussen’s encephalitis. Ann Neurol 2002; 51 (3): 311–8.
60. Wang Y, Qin ZH. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosi 2010; 15 (11): 1382–402. doi: 10.1007/s10495-010-0481-0.
61. Takei H, Wilfong A, Malphrus A, et al. Dual pathology in Rasmussen’s encephalitis: a study of seven cases and review of the literature. Neuropathology 2010; 30 (4): 381–91. doi: 10.1111/j.1440-1789.2009.01079.x.
62. Iyer A, Zurolo E, Spliet WG, et al. Evaluation of the innate and adaptive immunity in type I and type II focal cortical dysplasias. Epilepsia 2010; 51 (9): 1763–73. doi: 10.1111/j.1528-1167.2010.02547.x.
63. Friedman H, CH’ien L, Parham D. Virus in brain of child with hemiplegia, hemiconvulsions, and epilepsy. Lancet 1977; 2 (8039): 666.
64. Walter GF, Renella RR. Epstein-Barr virus in brain and Rasmussen’s encephalitis. Lancet 1989; 1 (8632): 279–80.
65. Power C, Poland SD, Blume WT, et al. Cytomegalovirus and Rasmussen’s encephalitis. Lancet 1990; 336 (8726): 1282–4.
66. Farrell MA, Cheng L, Cornford ME, et al. Cytomegalovirus and Rasmussen’s encephalitis. Lancet 1991; 337 (8756): 1551–2.
67. Vinters HV, Wang R, Wiley CA. Herpesviruses in chronic encephalitis associated with intractable childhood epilepsy. Hum Pathol 1993; 24 (8): 871–9.
68. McLachlan RS, Levin S, Blume WT. Treatment of Rasmussen’s syndrome with ganciclovir. Neurology 1996; 47 (4): 925–8.
69. Jay V, Becker LE, Otsubo H, et al. Chronic encephalitis and epilepsy (Rasmussen’s encephalitis: detection of cytomegalovirus and herpes simplex virus 1 by the polymerase chain reaction and in situ hybridization. Neurology 1995; 45 (1): 108–17.
70. Atkins MR, Terrell W, Hulette CM. Rasmussen’s syndrome: a study of potential viral etiology. Clin Neuropathol 1995; 14 (1): 7–12.
71. Vincent A, Bien CG, Irani SR, et al. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol 2011; 10 (8): 759–72. doi: 10.1016/S1474-4422 (11) 70096-5.
72. Lancaster E, Dalmau J. Neuronal autoantigens – pathogenesis, associated disorders and antibody testing. Nat Rev Neurol 2012; 8 (7): 380–90. doi: 10.1038/nrneurol.2012.99.
73. Rogers SW, Andrews PI, Gahring LC, et al. Autoantibodies to glutamate receptor GluR3 in Rasmussen’s encephalitis. Science 1994; 265 (5172): 648–51.
74. Pleasure D. Diagnostic and Pathogenetic Significance of Glutamate Receptor Autoantibodies. Arch Neurol 2008; 65 (5): 589–92. doi: 10.1001/archneur.65.5.589.
75. Greiner H, Leach JL, Lee KH, et al. Anti-NMDA receptor encephalitis presenting with imaging findings and clinical features mimicking Rasmussen syndrome. Seizure 2011; 20 (3): 266–70. doi: 10.1016/j.seizure.2010.11.013.
76. Wiendl HM, Bien CG, Bernasconi PP, et al. GluR3 antibodies: prevalence in focal epilepsy but no specificity for Rasmussen’s encephalitis. Neurology 2001; 57 (8): 1511–14.
77. Mantegazza R, Bernasconi P, Baggi F, et al. Antibodies against GluR3 peptides are not specific for Rasmussen’s encephalitis but are also present in epilepsy patients with severe, early onset disease and intractable seizures. J Neuroimmunol 2002; 131 (1–2): 179–85.
78. Watson R, Jiang Y, Bermudez I, et al. Absence of antibodies to glutamate receptor type 3 (GluR3) in Rasmussen encephalitis. Neurology 2004; 63 (1): 43–50.
79. Yang R, Puranam RS, Butler LS, et al. Autoimmunity to munc-18 in Rasmussen’s encephalitis. Neuron 2000; 28 (2): 375–83.
80. Watson R, Jepson JE, Bermudez I, et al. Alpha7 - acetylcholine receptor antibodies in two patients with Rasmussen encephalitis. Neurology 2005; 65 (11): 1802–4. doi: 10.1212/01.wnl.0000191566.86977.04.
81. Alvarez-Baron E, Bien CG, Schramm J, et al. Autoantibodies to Munc18, cerebral plasma cells and B-lymphocytes in Rasmussen encephalitis. Epilepsy Res 2008; 80 (1): 93–7. doi: 10.1016/j.eplepsyres.2008.03.007.
82. Owens GC, Huynh M, Chang JW, et al. Differential expression of Interferon-alfa and chemokines genes distinguishes Rasmussen encephalitis from cortical dysplasia and provides evidence for an early Th1 immune response. J Neuroinflammation 2013; 10 : 56–69. doi: 10.1186/1742-2094-10-56.
83. Schwab N, Bien CG, Waschbisch A, et al. CD8+ T cell clones dominate brain infiltrates in Rasmussen encephalitis and persist in the periphery. Brain 2009; 132 (5): 1236–46. doi: 10.1093/brain/awp003.
84. Bauer J, Elger CE, Hans VH, et al. Astrocytes are a specific immunological target in Rasmussen’s encephalitis. Ann Neurol 2007; 62 (1): 67–80. doi: 10.1002/ana.21148.
85. Wirenfeldt M, Clare R, Tung S, et al. Increased activation of Iba1+ microglia in pediatric epilepsy patients with Rasmussen’s encephalitis compared with cortical dysplasia and tuberous sclerosis complex. Neurobiol Dis 2009; 34 (3): 432–40. doi: 10.1016/j.nbd.2009.02.015.
86. Balosso S, Maroso M, Sanchez-Alavez M, et al. A novel non-transcriptional pathway mediates the proconvulsive effects of interleukin-1beta. Brain 2008; 131 (12): 3256–65. doi: 10.1093/brain/awn271.
87. Schafer DP, Lehrman EK, Kautzman AG, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012; 74 (4): 691–705. doi: 10.1016/j.neuron.2012.03.026.
88. Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci 2012; 35 : 369–89. doi: 10.1146/annurev-neuro-061010-113810.
89. Ivens S, Kaufer D, Flores L, et al. TGF-b receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain 2007; 130 (2): 535–47.
90. Takahashi Y, Mine J, Kubota Y, et al. A substantial number of Rasmussen syndrome patients have increased IgG, CD4+ T cells, TNFa, and Granzyme B in CSF. Epilepsia 2009; 50 (6): 1419–31. doi: 10.1111/j.1528-1167.2008.01977.x.
91. Ramaswarny V, Walsh JG, Sinclair DB, et al. Inflammasome induction in Rasmussen’s encephalitis: cortical and associated white matter pathogenesis. J Neuroinflammation 2013; 10 : 152. doi: 10.1186/1742-2094-10-152.
92. Bien CG, Schramm J. Treatment of Rasmussen encephalitis half a century after its initial description: promising prospects and a dilemma. Epilepsy Res 2009; 86 (2): 101–12. doi: 10.1016/j.eplepsyres.2009.06.001.
93. Dubeau F, Sherwin AL. Pharmacologic principles in the management of chronic focal encephalitis. In: Ander - mann F, ed. Chronic encephalitis and epilepsy Rasmussen’s syndrome. Boston: Butterworth-Heinemann 1991 : 179–92.
94. Lozsadi DA, Hart IK, Moore AP. Botulinum toxin A improves involuntary limb movements in Rasmussen syndrome. Neurology 2004; 62 (7): 1233–34.
95. Browner N, Azher SN, Jankovic J. Botulinum toxin treatment of facial myoclonus in suspected Rasmussen encephalitis. Mov Disord 2006; 21 (9): 1500–2.
96. Grujic J, Bien CG, Pollo C, et al. Vagus nerve stimulator treatment in adult-onset Rasmussen’s encephalitis. Epilepsy Behav 2011; 20 (1): 23–5. doi: 10.1016/j.yebeh.2010.10.024.
97. San-Juan D, Calcaneo JD, Gonzalez-Aragon MF, et al. Transcranial direct current stimulation in adolescent and adult Rasmussen’s encephalitis. Epilepsy Behav 2011; 20 (1): 126–31. doi: 10.1016/j.yebeh.2010.10.031.
98. Hart YM, Cortez M, Andermann F, et al. Medical treatment of Rasmussen’s syndrome (chronic encephalitis and epilepsy): effect of high-dose steroids or immunoglobulins in 19 patients. Neurology 1994; 44 (6): 1030–6.
99. Granata T, Fusco L, Gobbi G, et al. Experience with immunomodulatory treatments in Rasmussen’s encephalitis. Neurology 2003; 61 (12): 1807–10.
100. Leach JP, Chadwick DW, Miles JB, et al. Improvement in adult-onset Rasmussen’s encephalitis with long-term immunomodulatory therapy. Neurology 1999; 52 (4): 738–42.
101. Timarova G, Lisa I, Dudasova K, et al. Immunomodulation therapy in three cases of late-onset Rasmussen encephalitis. Abstract. AES Washington 2013. [online]. Available from URL: https://www.aesnet.org/meetings_events/annual_meeting_abstracts/find/Timarova/2/2/0.
102. Andrews PI, Dichter MA, Berkovic SF, et al. Plasmapheresis in Rasmussen’s encephalitis. Neurology 1996; 46 (1): 242–6.
103. Antozzi C, Granata T, Aurisano N, et al. Long-term selective IgG immuno-adsorption improves Rasmussen’s encephalitis. Neurology 1998; 51 (1): 302–5.
104. Bien CG, Gleissner U, Sassen R, et al. An open study of tacrolimus therapy in Rasmussen’s encephalitis. Neurology 2004; 62 (11): 2106–9.
105. Varadkar S, Chong WK, Robinson R, et al. Azathioprine therapy in Rasmussen Syndrome. Epilepsy Currents 2012; 12 (Suppl 1): 417.
106. McLachlan RS, Levin S, Blume WT. Treatment of Rasmussen’s syndrome with gancyclovir. Neurology 1996; 47 (4): 925–8.
107. Thilo B, Stingele R, Knudsen K, et al. A case of Rasmussen encephalitis treated with rituximab. Nat Rev Neurol 2009; 5 (8): 458–62. doi: 10.1038/nrneurol.2009.98.
108. Lockman J, Le S, Fisher RS, et al. Case Report: Rasmussen’s Encephalitis Treated Successfully with Rituximab. Cureus 2013; 5 (8): e136. doi: 10.7759/cureus.136.
109. Bittner S, Simon OJ, Gobel K, et al. Rasmussen encephalitis treated with natalizumab. Neurology 2013; 81 (4): 395–7. doi: 10.1212/WNL.0b013e31829c5ceb.
110. Vining E, Freeman J, Pillas D, et al. Why Would You Remove Half a Brain? The Outcome of 58 Children After Hemispherectomy – the Johns Hopkins Experience: 1968 to 1996. Pediatrics 1997; 100 (2): 163–71.
111. Kossoff EH, Vining EP, Pillas DJ, et al. Hemispherectomy for intractable unihemispheric epilepsy, etiology vs outcome. Neurology 2003; 61 (7): 887–90. doi: org.10.1212/01.
112. Jonas R, Nguyen S, Hu B, et al. Cerebral hemispherectomy: hospital course, seizure, developmental, language, and motor outcomes. Neurology 2004; 62 (10): 1712–21.
113. Pulsifer MB, Brandt J, Salorio CF, et al. The cognitive outcome of hemispherectomy in 71 children. Epilepsia 2004; 45 (4): 243–54. doi: 10.1111/j.1528-1167.2009.02335.x.
114. Villemure JG. Hemispherectomy: Techniques and Complications. In: Wyllie E, ed. The treatment of epilepsy and practices. Philadelphia: Lea and Febiger 1997 : 1081–6.
115. Honavar M, Janota I, Polkey CE. Rasmussen’s encephalitis in surgery for epilepsy. Dev Med Child Neurol 1992; 34 (1): 3–14.
116. Thomas SG, Chacko AG, Thomas MM, et al. Outcomes of disconnective surgery in intractable pediatric hemispheric and subhemispheric epilepsy. Int J Pediatr 2012; 2012 : 527891. doi: 10.1155/2012/527891.
117. Follett P, Vora N, Cross JH. Paediatric Intractable Epilepsy Syndromes: changing concepts in diagnosis and management. Adv Tech Stand Neurosurg 2012; 39 : 45–60. doi: 10.1007/978-3-7091-1360-8_2.
118. Althausen A, Gleissner U, Hoppe C, et al. Long-term outcome of hemispheric surgery at different ages in 61 epilepsy patients. J Neurol Neurosurg Psychiatry 2013; 84 (5): 529–36. doi: 10.1136/jnnp-2012-303811.
119. Van Empelen R, Jennekens-Schinkel A, Buskens E, et al. Functional consequences of hemispherectomy. Brain 2004; 127 (9): 2071–9.
120. Boatman D, Freeman J, Vining E, et al. Language recovery after left hemispherectomy in children with late-onset seizures. Ann Neurol 1999; 46 (13): 579–86.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2016 Issue 5
- Advances in the Treatment of Myasthenia Gravis on the Horizon
- Hope Awakens with Early Diagnosis of Parkinson's Disease Based on Skin Odor
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
Most read in this issue
- Current Corticosteroid Treatment in Brain Tumours
- Rasmussen’s Encephalitis
- Traumatic Brachial Plexus Injuries Represents Serious Peripheral Nerve Palsies
- Detection of Spirochetal DNA from Patients with Neuroborreliosis