
Jsme na dosah cílené terapie Huntingtonovy nemoci? NE
Authors:
doc. MUDr. Marek Baláž, Ph.D.
Authors‘ workplace:
FN u sv. Anny v Brně
; I. neurologická klinika LF MU
Published in:
Cesk Slov Neurol N 2020; 83/116(3): 241
Category:
Controversions
Aktuální farmakologická terapie v léčbě Huntingtonovy nemoci (HN) je symptomatická, zaměřená ze strany neurologa na léčbu motorických příznaků – především chorey (tetrabenazin podávaný v našich podmínkách, v jiných zemích jsou dostupné také deutetrabenazin a valbenazin). Pozornost je v terapii, ve spolupráci s psychiatry, také věnována léčbě nemotorických (velmi často psychiatrických) příznaků, kde lze použít široké spektrum medikace podle jednotlivých symptomů [1].
HN je monogenní onemocnění s dlouhým presymptomatickým obdobím, s buněčnými změnami vyvolanými jediným mutovaným proteinem. Přesto za více než 20 let neuspělo velké množství studii s různými cílovými mechanizmy, jakými jsou např. oxidativní stres, neuroinflamace, dysregulace transkripce, excitotoxicita, mitochondriální dysfunkce a trofická podpora [2]. Testování zacílené na nemoc-modifikující a symptomatickou léčbu za posledních více než 15 let dosáhlo pozitivních výsledků jen v 3,5 % – a ty se týkaly tetrabenazinu a deutetrabenazinu [3].
Dosavadní výzkum se potýkal s problémy, probíhající studie jsou uvedeny v tab. 1.
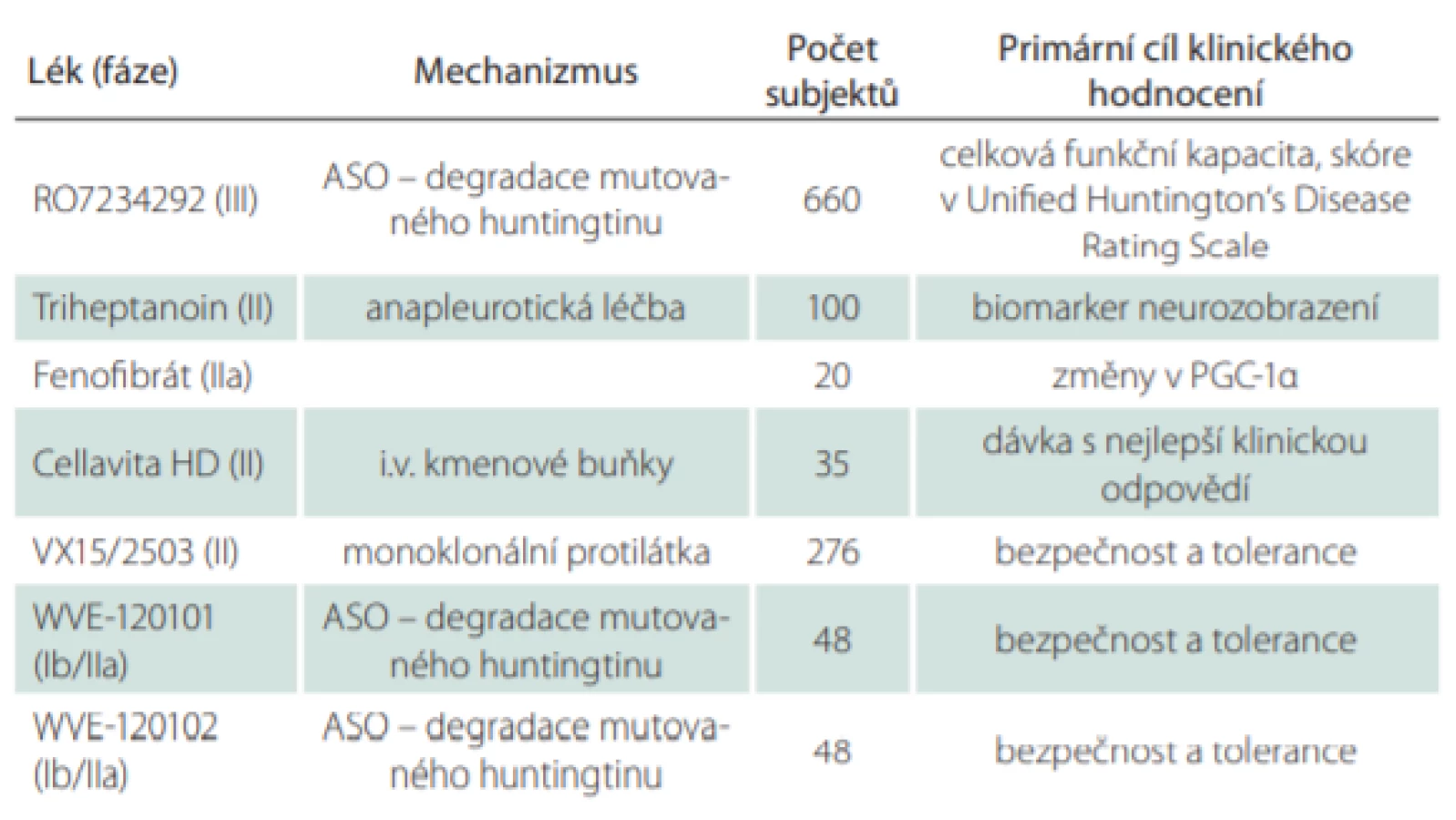
Velká část nadějí k dosažení rozhodujícího průlomu v genetické terapii se upíná k terapii cílené na snížení produkce mutovaného huntingtinu v mozku pomocí léků s aktivitou na úrovni modifikované DNA nazývaných antisense-oligonukleotidy (ASO), které jsou zkoumány v několika studiích. ASO zaznamenaly dramaticky významný efekt u spinální muskulární atrofie a jsou zkoumány u řady dalších nemocí [2]. Hlavní očekávání je spojeno se studií GENERATION-HD1 (intratekálně podávaný preparát s názvem RG6042) s cílem zjistit, zda snížení produkce huntingtinu zpomaluje progresi onemocnění a má pozitivní dopad na příznaky nemoci [3].
Další ze studií zaměřených na ASO a snižování množství (mutovaného) huntigtinu, PRECISION-HD2 (preparát WVE-120102 v tab. 1), prokázala pouze mírnou redukci o 12 %.
Z historie výzkumu HN vyplývá, že v klinických studiích selhaly desítky preparátů, které neuspěly v dosažení cílů jednotlivých studií a především ve snaze o ovlivnění rychlosti a stupně progrese neurodegenerace [4]. Agregace huntingtinu je zjevná u myších modelů už v embryonálních stádiích a existují důkazy o tom, že mutace u HN ovlivňuje vývoj mozku u pacientů, u kterých se příznaky projeví až v dospělosti. Podle dostupných důkazů se metabolické abnormity, neurodegenerace a subklinické fenotypové projevy mohou objevit celá desetiletí před propuknutím nemoci. Existuje riziko, že úspěšné snížení hladin mutovaného huntingtinu nemusí mít dopad na klinické projevy nebo progresi onemocnění [3].
Naděje se tedy upínají nyní k jedné studii ve fázi III, s přibližně 800 pacienty ve třech ramenech studie. V případě, že by výsledky studie nenaplnily očekávání, pravděpodobně nebude možné počítat s tím, že by byla terapie ASO dostupná v nejbližších 10 letech.
Léky ze skupiny ASO v léčbě spinální svalové atrofie patří mezi nejdražší terapie, v případě dosažení pozitivního efektu v klinických studiích obdobných medikamentů u HN bude nutné vyřešit otázku dostupnosti léčby pro pacienty.
Na tomto místě považuji za významné připomenout, že i v době, kdy je očekáván průlom se zavedením cílené léčby HN, je stále k dispozici technologie reprodukční medicíny – preimplantační genetická diagnostika – s potenciálem zásadně redukovat výskyt nemoci [3].
doc. MUDr. Marek Baláž, Ph.D.
I. neurologická klinika LF MU, FN u sv. Anny v Brně
Sources
1. Ustohal L, Obdržálková M, Víchová M. Huntingtonova choroba jako neuropsychiatrický problém. Psychiatr praxi 2018; 19 (2): 64–66.
2. Wild EJ, Tabrizi SJ. One decade ago, one decade ahead in huntington‘s disease. Mov Disord 2019; 34 (10): 1434–1439. doi: 10.1002/mds.27849.
3. Testa CM, Jankovic J. Huntington disease: a quarter century of progress since the gene discovery. J Neurol Sci 2019; 396 : 52–68. doi: 10.1016/j.jns.2018.09.022.
4. Travessa AM, Rodrigues FB, Mestre TA et al. Fifteen years of clinical trials in Huntington‘s disease: a very low clinical drug development success rate. J Huntingtons Dis 2017; 6 (2): 157–163. doi: 10.3233/JHD-170245.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2020 Issue 3
- Memantine Eases Daily Life for Patients and Caregivers
- Possibilities of Using Metamizole in the Treatment of Acute Primary Headaches
- Memantine in Dementia Therapy – Current Findings and Possible Future Applications
- Advances in the Treatment of Myasthenia Gravis on the Horizon
Most read in this issue
- Glioblastom grade IV – dlouhodobé přežití
- Primární progresivní afázie
- Bolesti hlavy v graviditě
- Kognitivní poruchy u dětí s epilepsií