
Minimální nález u pacienta s Bestovou chorobou podmíněnou mutací c.653G>A v genu BEST1
Minimal Ocular Findings in a Patient with Best Disease Caused by the c.653G>A Mutation in BEST1
Purpose:
To describe the phenotype in an asymptomatic 64-year-old patient with family history of Best disease and to identify the disease causing variant in the BEST1 gene.
Methods:
Detailed ocular examination of the proband including spectral-domain optical coherence tomography (SD-OCT), fluorescein angiography and electrooculography was performed. Direct sequencing approach was used to screen the whole coding sequence of 11 exons of BEST1.
Results:
An early vitelliform stage of Best disease presenting as a small yellowish spot in the macula was observed in the right eye. The fundus appearance in the left eye was normal. SD-OCT of the right macula revealed hypodense space between the retinal pigment epithelium and the neuroretinal layer. Arden ratio was bilaterally mildly reduced; 1.36 in the right and 1.3 in the left eye. Molecular genetic analysis identified a heterozygous change c.653G>A (p.Arg218His) as the disease-causing variant.
Conclusion:
Here we report for the first time a phenotype-genotype correlation in a Czech patient with Best disease. SD-OCT is a fast method that may show the presence of small pathological changes. The screening of BEST1 gene enables identification of disease-causing variants in asymptomatic individuals with normal fundus appearance and thus improves counseling to the affected families.
Key words:
Best disorder, BEST1, mutation, optic coherence tomography, phenotype
:
B. Kousal 1; F. Chakarova 2; G. C. Black 3; S. Ramsden 3; H. Langrová 4; P. Lišková 5
:
Oční klinika, 1. lékařská fakulta, Univerzita Karlova a Všeobecná fakultní nemocnice, Praha, přednosta doc. MUDr. Bohdana Kalvodová, CSc.
1; Institute of Ophthalmology, UCL, London, Velká Británie, ředitel, prof. Philip J. Luther, BSc, MBBS, FRCP, FRCPath, FRCOphth
2; Genetic Medicine Research Group, Manchester Biomedical Research Centre, Manchester Academic Health Sciences Centre, University of Manchester and Central Manchester Foundation Trust, St Mary’s Hospital, Manchester M13 9WL, Velká Británie, ředitel prof. Gra
3; Oční klinika, Lékařská fakulta v Hradci Králové, Univerzita Karlova, Praha, přednosta prof. MUDr. Pavel Rozsíval, CSc.
4; Laboratoř biologie a patologie oka, Ústav dědičných metabolických poruch, 1. lékařská fakulta, Univerzita Karlova a Všeobecná fakultní nemocnice, Praha, přednosta doc. MUDr. Viktor Kožich, CSc.
5
:
Čes. a slov. Oftal., 67, 2011, No. 5-6, p. 170-174
:
Case Reports
Cil:
Popis klinického nálezu a screening genu BEST1 u 64letého asymptomatického pacienta s rodinnou anamnézou Bestovy choroby.
Metody:
U našeho probanda jsme provedli kompletní oční vyšetření včetně optické koherenční tomografie se spektrální doménou (SD-OCT), fluorescenční angiografie a elektrookulografie (EOG). Pro screening 11 kódujících exonů kandidátního genu BEST1 byla použita metoda oboustranného přímého sekvenování.
Výsledky:
Při vyšetření byla zjištěna vpravo počínající viteliformní fáze Bestovy choroby charakterizovaná malou okrskovitou změnou o velikosti 0,3 průměru terče zrakového nervu, která se biomikroskopicky jevila jako výraznější zažloutnutí v oblasti centra makuly. Pomocí vyšetření SD-OCT byl posléze subfoveolárně mezi retinálním pigmentovým epitelem a neuroretinou detekován štěrbinovitý hyporeflexivní prostor. Vlevo byl nález v makule včetně SD-OCT vyšetření normální. Nejlepší zraková ostrost levého oka byla 1,0 a pravého oka 0,4. Toto snížení bylo hodnoceno především jako následek anamnesticky udávané amblyopie, přičemž však nebylo možno vyloučit ani určitý podíl viteliformní léze. Ardenův index byl oboustranně mírně snížen, na oku pravém činila jeho hodnota 1,36; vlevo pak 1,3. Molekulárně genetické vyšetření prokázalo v genu BEST1 příčinnou mutaci c.653G>A vedoucí k záměně argininu za histidin v kodónu 218.
Závěr:
Poprvé bylo provedeno srovnání genotypu s fenotypem u českého pacienta s Bestovou chorobou. SD-OCT vyšetření je rychlou metodu verifikující přítomnost i malých abnormalit a napomáhá k lepší charakterizaci prvotních změn u této klinické jednotky. Screening genu BEST1 umožňuje zjistit příčinnou mutaci i u asymptomatických jedinců bez klinických známek onemocnění, včetně normálních nálezů na EOG vyšetření, a přispívá tak ke zlepšení poradenství postiženým rodinám především z hlediska určení rizika přenosu choroby na potomstvo.
Klíčová slova:
Bestova choroba, BEST1, mutace, optická koherenční tomografie, genotyp
ÚVOD
Bestova choroba (také známá jako viteliformní makulární dystrofie či Bestova viteliformní dystrofie makuly) je oboustranné onemocnění přenášené autozomálně dominantně s řibližnou prevalencí 1 : 10 000 (25).
Existuje několik klasifikací fází Bestovy choroby. Dělení průběhu onemocnění vychází nejčastěji z práce Mohlera a Fineho. Ve fázi 0 je vzhled makuly normální, v 1. asymptomatické fázi (previteliformní) jsou přítomny pouze jemné změny retinálního pigmentového epitelu (RPE) (25). Onemocnění se typicky manifestuje v 1. a 2. dekádě života vznikem lehce prominujícího oranžovožlutého ložiska o velikosti 0,5–2,0 průměru terče zrakového nervu, které svým vzhledem připomíná vaječný žloutek (2. viteliformní fáze). V dalším vývoji může dojít k částečné absorpci léze a vzniku tzv. pseudohypopyon (3. fáze). Postupnou dezintegrací začne léze připomínat míchaná vajíčka (4. fáze). V oslední 5. fázi onemocnění, která je provázená často výraznějším poklesem zrakové ostrosti, může dojít ke vzniku atrofické makulopatie, jizvení v makule nebo k tvorbě neovaskularizace nalézající se pod nebo kolem fibrotické makulární jizvy. Ne všichni pacienti procházejí všemi fázemi onemocnění a podle některých autorů může být pořadí 3. a 4. fáze obrácené (22, 25, 33). Většina případů se manifestuje vznikem solitární léze v blasti makuly, popsány však byly také léze multifokální (5, 31).
Bestova choroba je pomalu progredující onemocnění s často stranově asymetrickým nálezem. Obtíže ve smyslu metamorfopsií, rozostřeného vidění a snížení centrální zrakové ostrosti mohou vznikat již v ětství, ale i o několik dekád později. V dlouhodobějším horizontu onemocnění vede u většiny pacientů k oklesu zrakové ostrosti (3, 25).
Klinická diagnóza je potvrzena v aprosté většině případů elektrookulografickým (EOG) vyšetřením, při kterém se pozoruje snížení maximální potenciálové hodnoty naměřené na světle. Její podíl s minimální hodnotou naměřenou potmě x 100 se vyjadřuje ve formě tzv. Ardenova indexu, který je snížen často již v reviteliformní fázi onemocnění. Charakteristická je také blokáda viteliformním materiálem při fluorescenční angiografii a zvýšená autofluorescence žlutých viteliformních lézí (3, 4, 9, 13, 15, 21, 25).
Bestova choroba je podmíněna mutacemi v genu bestrofin 1, označovaném symbolem BEST1, který se nachází na dlouhém raménku 11. chromozomu. Tento gen je také známý pod označením VMD2 a kóduje protein bestrofin 1, který je lokalizovaný v bazolaterální membráně buněk RPE, kde tvoří Cl-kanály. Tyto se aktivují změnami intracelulární koncentrace Ca2+. Množství patogenních mutací vedoucích ke vzniku Bestovy choroby je velké, dosud jich bylo v BEST1 identifikováno více než 200 (http://www-huge.uni-regensburg.de/BEST1_database/home.php).
METODIKA
Klinické vyšetření
Provedli jsme kompletní oční vyšetření 64letého asymptomatického muže, který byl pozván na naše pracoviště vzhledem ke stanovení diagnózy Bestovy choroby u jeho 22letého syna na jiném pracovišti, ostatní rodinná anamnéza byla negativní. Nejlepší korigovaná zraková ostrost byla zjišťována pomocí ETDRS optotypů. Vidění do blízka bylo testováno pomocí Jaegerových tabulek. Provedli jsme biomikroskopické vyšetření fundu v mydriáze, barevnou fotografii očního pozadí a snímek v bezčerveném světle, fluorescenční angiografii (FF 450 plus IR Fundus Camera, Carl Zeiss Meditec AG, Německo), vyšetření zorného pole počítačovým perimetrem (M-700, Medmont International Pty Ltd., Austrálie), vyšetření kontrastní citlivosti na tabuli Pelli–Robson (HS Clement Clarke International, Velká Británie) a barvocitu 15 Hue testem. Pacient byl dále vyšetřen pomocí optické koherenční tomografie se spektrální doménou (SD-OCT, Spectral OCT/SLO, OTI Ophthalmic Technologies Inc., Kanada) s axiálním rozlišením 6 μm a rychlostí 16 snímků/s. Přístroj je kombinací OCT a skenovacího laserového oftalmoskopu a využívá Fourierovu transformaci spojitého signálu k výstupu ve formě řezů sítnicí.
EOG vyšetření (RETI-port plus mfERG system, Roland Consult GmbH, Německo) bylo provedeno podle standardního protokolu Mezinárodní společnosti pro klinickou elektrofyziologii zraku. Výsledky byly hodnoceny jako normální, pokud Ardenův index byl vyšší než 1,85, rozmezí 1,85–1,3 bylo hodnoceno jako mírné snížení a závažné snížení bylo u Ardenova indexu pod 1,3 (21). Studie byla provedena podle zásad Helsinské deklarace.
Přímé sekvenování genu BEST1
DNA byla extrahována z 5 ml vzorku venózní krve pomocí DNA extrakčního kitu (Nucleon DNA Isolation Kit BACCII, GE Healthcare, Velká Británie). Screening 11 exonů genu BEST1 byl proveden pomocí oboustranného přímého sekvenování exonů a hranic intronů s xony, jak již bylo dříve popsáno (8). Generovaná data byla srovnávána s referenční sekvencí NM_004183.2.
VÝSLEDKY
64letý muž anamnesticky udával tupozrakost pravého oka, oboustrannou hypermetropii a astigmatismus. Ve věku 57 let podstoupil na jiném pracovišti operaci katarakty s implantací nitrooční čočky na pravém oku a ve věku 62 let stejný zákrok na oku levém. Známky Bestovy choroby nebyly při předchozích vyšetřeních fundu popsány.
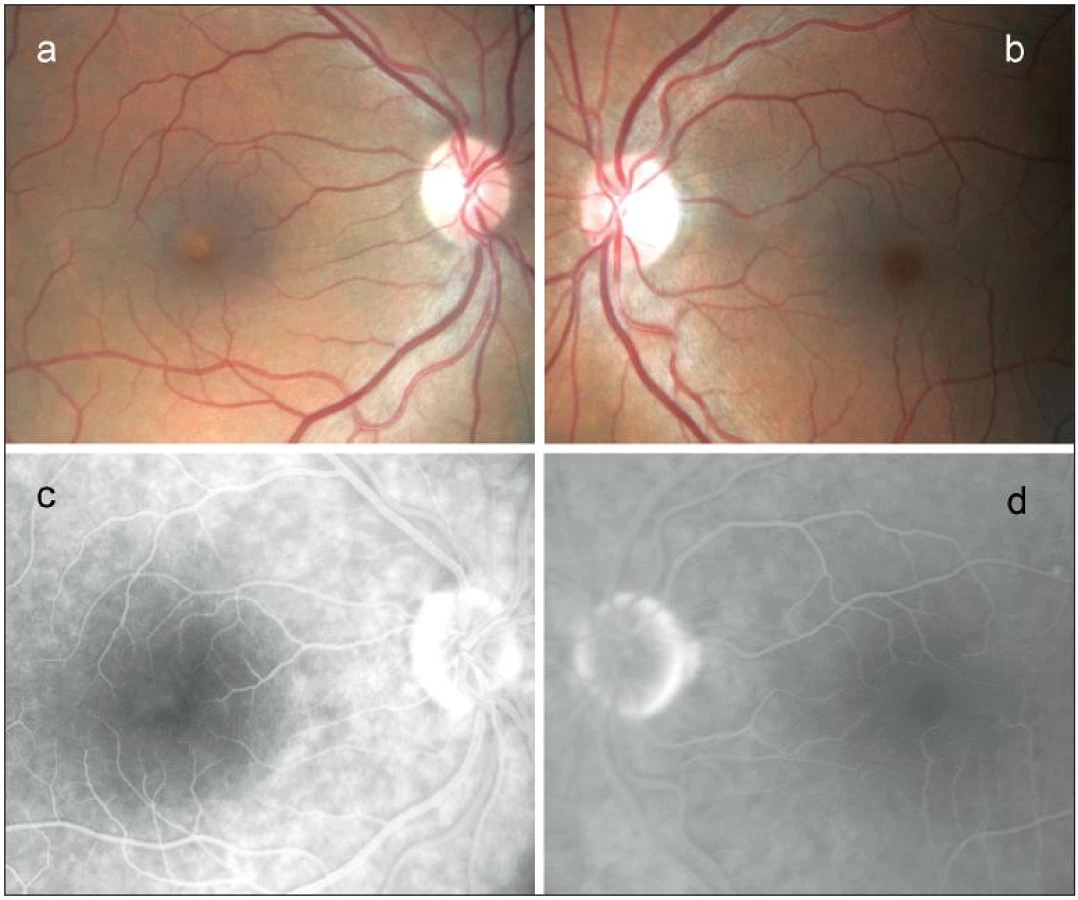
Při našem vyšetření byla nejlepší zraková ostrost na oku pravém 0,4 naturálně a na oku levém 1,0 také naturálně. Z Jaegerových tabulek pacient četl pravým okem číslo 6, levým okem číslo 1. V centru makuly pravého oka bylo přítomno malé, nažloutlé, ostře ohraničené, prominující subretinální ložisko, biomikroskopicky velikosti 0,3 průměru terče zrakového nervu (obr. 1a). Nález v makule levého oka se jevil normální (obr. 1b). Žádná další patologie nebyla při detailním oftamologickém vyšetření zjištěna. V angiografickém obraze pravého oka jsme nalezli v centru léze v časných fázích dole nehomogenně blokovanou fluorescenci a v pozdních fázích nahoře velmi mírnou hyperfluorescenci. Angiografický nález makuly levého oka byl normální (obr. 1c, d).
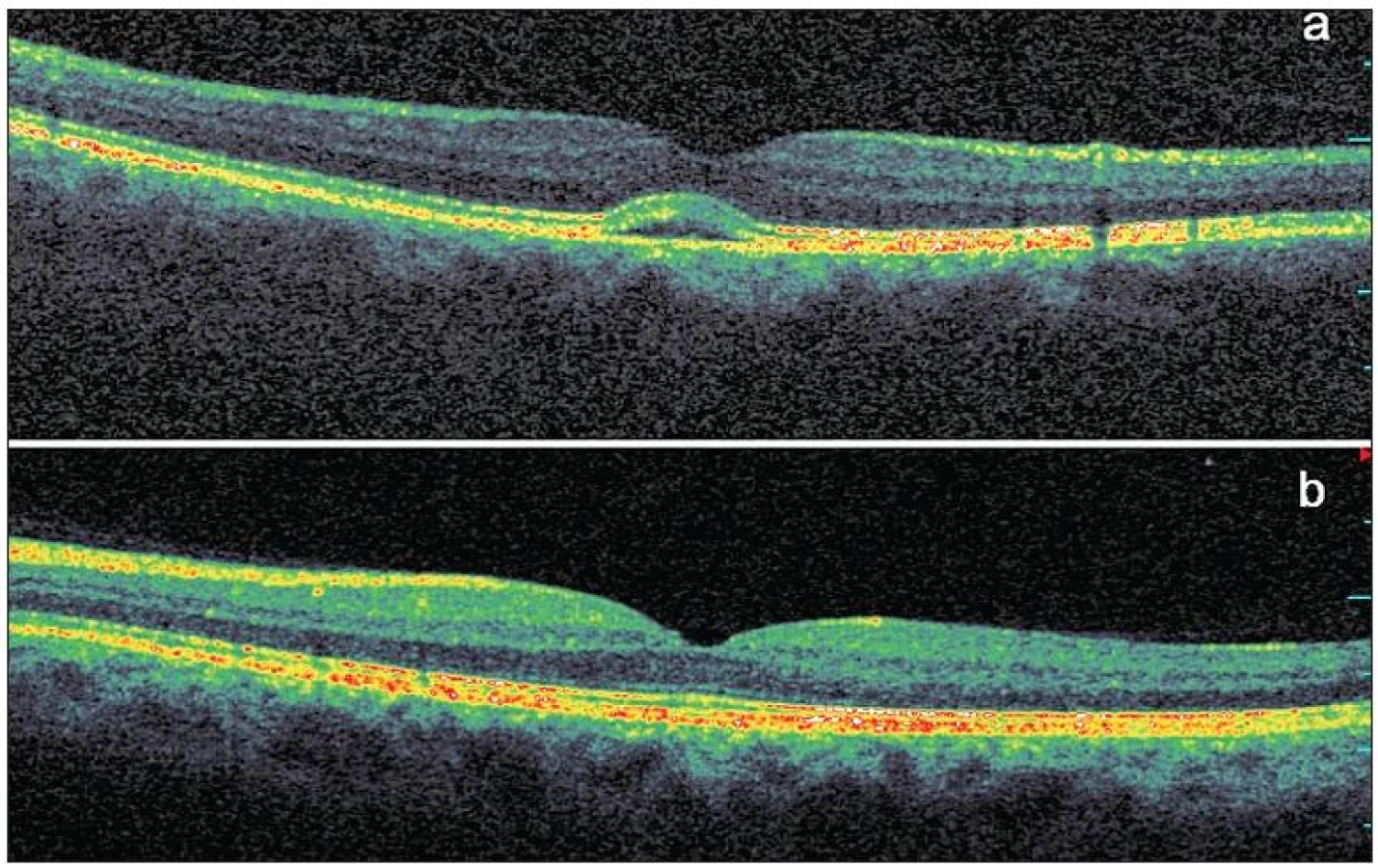
Na SD-OCT skenech pravého oka byla patrná subfoveolárně lokalizovaná ohraničená oblast ztluštění RPE se štěrbinovým prostorem velmi nízké nehomogenní reflexivity mezi RPE a neuroretinou (obr. 2a). Velikost baze léze byla 1,20 x 1,04 mm, výška léze v maximu 0,13 mm. Byla přítomna foveolární deprese se ztenčenou neuroretinou na 0,13 mm. Na OCT skenech levého oka byl normální nález, výška neuroretiny ve fovee činila 0,20 mm (obr. 2b).
Kontrastní citlivost byla na pravém oku snížená na hodnotu 1,35; na levém byla její hodnota fyziologická 1,65. Barvocit byl na obou očích normální. Vyšetření zorného pole se jevilo oboustranně zcela bez známek patologie.
Klinický nález koreloval s EOG vyšetřením, bilaterálně bylo zjištěno mírné snížení Ardenova indexu, jeho hodnota činila vpravo 1,36 a vlevo 1,3.
Molekulárně genetické vyšetření
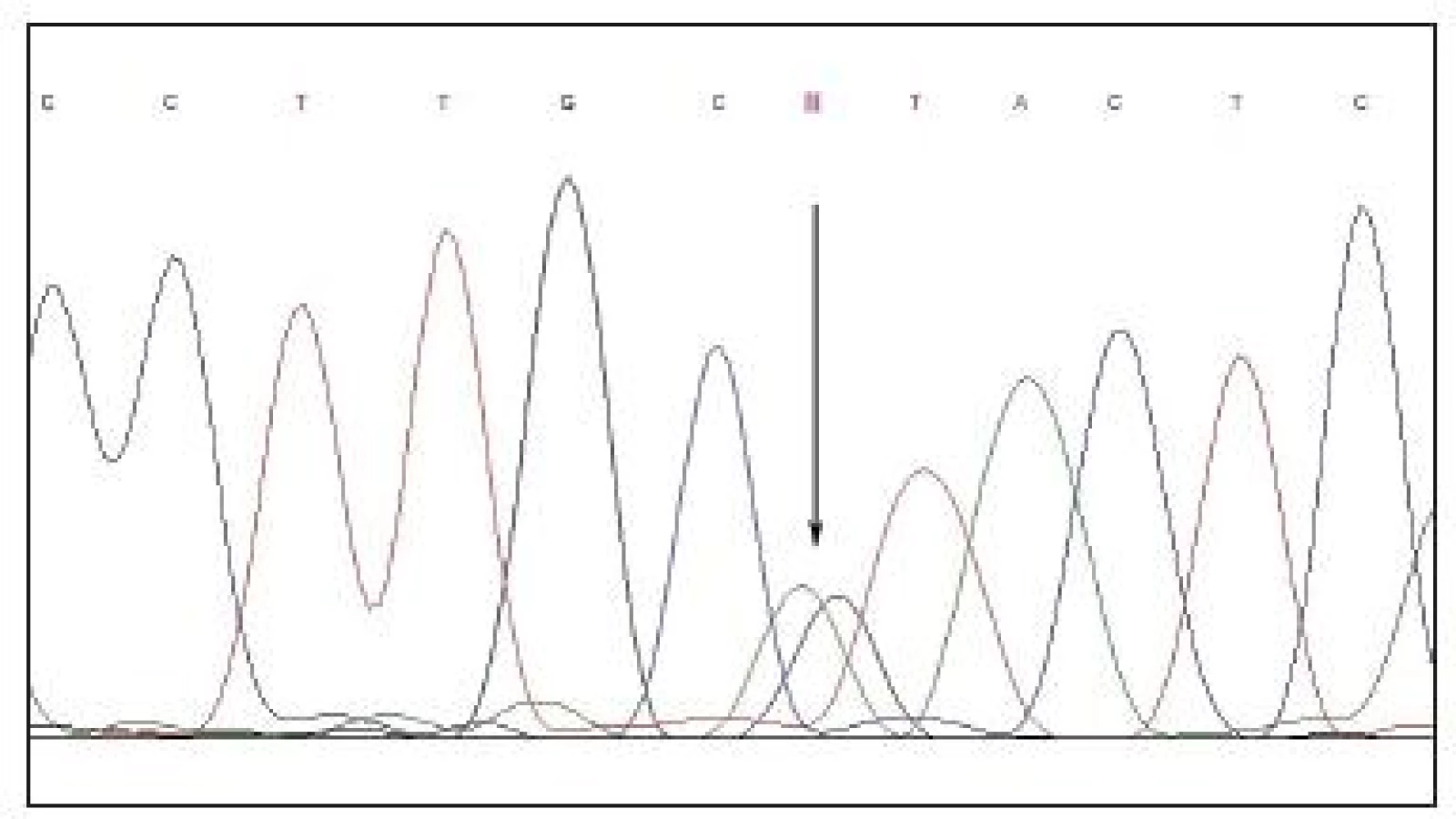
Přímé sekvenování kódujících sekvencí genu BEST1 prokázalo přítomnost heterozygotní mutace c.653G>A vedoucí na proteinové úrovni k záměně argininového aminokyselinového zbytku za histidin v kodónu 218 (obr. 3).
DISKUSE
Bestova choroba je po Stargardtově chorobě druhou nejčastější makulární degenerací s monogenním typem dědičnosti (22). Screening genu BEST1 vedl k určení molekulárně genetické příčiny tohoto onemocnění u námi vyšetřovaného pacienta. Zjistili jsme substituci nukleotidu G za nukleotid A na pozici 653 v exonu 6, který je po exonu 8 druhým nejčastějším kódujícím úsekem genu BEST1, ve kterém jsou zjišťovány patogenní varianty podmiňující vznik Bestovy choroby (http://www-huge.uni-regensburg.de/BEST1_database/home.php). Detekovaná mutace má za následek záměnu argininového zbytku za histidinový na pozici 218 aminokyselinového řetězce. Arginin na pozici 218 je evolučně (mezidruhově) velmi konzervovaný, což mimo jiné podporuje fakt, že jeho přítomnost má významný vliv na správnou funkci proteinu bestrofinu-1 a jeho záměna vede ke škodlivým následkům (19, 23). U pacientů s Bestovou chorobou se jedná o nejčastější aminokyselinový zbytek, kde se vyskytují mutace podmiňující toto onemocnění. Stejná mutace, která je přítomná u našeho pacienta, byla již dříve popsána u několika dalších probandů původem především z Francie (18, 19, 20).
U pacientů českého původu byla patogenní změna v BEST1 genu detekována doposud pouze v jedné rodině s Bestovou chorobou. Konkrétně byla zjištěna mutace c.76G>C (p.Arg25Gln) nalézající se v exonu 2 genu BEST1. Klinický nález ani jednoho ze tří postižených příslušníků vyšetřované rodiny nebyl detailně popsán (23).
Fenotyp u jedinců s Bestovou chorobou může být značně variabilní, a to i v rámci jedné rodiny. Různí se věk vzniku prvních symptomů a závažnost nálezu mezi očima jednoho pacienta. Popsána byla též inkompletní penetrance, kdy pacient je nositelem patologické mutace, ale nevykazuje žádné známky onemocnění, a to ani při EOG vyšetření, přičemž snížení hodnoty Ardenova indexu bylo donedávna považováno za základní diagnostické kriterium. V literatuře lze najít také klinické charakteristiky pacientů s normálním Ardenovým indexem a přítomností viteliformní léze (16, 24, 29, 32, 33, 35, 38, 39). Mechanismy, které jsou příčinou této fenotypové variability, nebyly dosud zcela objasněny. Předpokládá se vliv modifikujících genetických faktorů anebo vlivů okolního prostředí (12, 35).
Přestože typická manifestace Bestovy choroby bývá v . nebo 2. dekádě života (3, 32, 34) a v . a 7. dekádě života se očekává u minimálně poloviny pacientů snížení nejlepší korigované zrakové ostrosti pod 0,5, která však nebývá na lepším oku horší než 0,1–0,05 (3, 25, 32), u námi vyšetřovaného 64letého pacienta byla zjištěna pouze počínající viteliformní fáze onemocnění na pravém oku a normální nález v akule levého oka. Vzhledem k omu, že u očí s iteliformními lézemi je nejlepší korigovaná zraková ostrost většinou normální nebo jen lehce snížená (32, 34, 35), je pravděpodobné, že její pokles na pravém oku vyšetřovaného byl způsoben především anamnesticky udávanou amblyopií.
Kvůli značné variabilitě projevů Bestovy choroby, a to i u jedinců z ednotlivých rodin, je těžké vysledovat těsnější vztah mezi klinickým vyjádřením a patogenními mutacemi. Booij a kol. (3) nedávno poukázal na statisticky větší riziko poklesu zrakových funkcí u pacientů s utací p.Val10Ala ve srovnání s ositeli mutací p.Thr6Pro a p.Tyr227Asn. Popis více jedinců s říčinnou mutaci p.Arg218His by napomohl zjistit, zda tuto lze dávat do souvislosti s írnějším průběhem choroby, jak by mohl naznačovat nález u námi vyšetřeného pacienta. V iteratuře se vyskytuje v omto ohledu jen drobná zmínka: u 72letého otce pacientky s estovou chorobou německého původu bylo zjištěno, že jeho zraková ostrost umožňuje vykonávat všechny běžné denní úkony (19).
Pouze několik prací popisuje OCT nálezy u pacientů s detekovanými mutacemi v BEST1 a zároveň s ormálním či minimálním nálezem při oftalmoskopickém vyšetření fundu (27, 32, 34, 35). První známkou Bestovy choroby před vznikem viteliformní léze je dle pozorování Querquese a kol. (30) ztluštění reflexivní vrstvy mezi normálně vypadajícím RPE a linií junkce zevních a vnitřních segmentů fotoreceptorů, v alším průběhu onemocnění vzniká akumulace tekutiny a materiálu mezi RPE a neuroretinou, kterou jsme pozorovali i my na pravém oku pacienta (31, 37).
U Bestovy choroby bylo prokázáno ukládání toxických prekurzorů a derivátů lipofuscinu, které vznikají jako odpad Waldova cyklu. Akumulovaný materiál se zobrazuje dobře při autofluorescenci fundu a v počátečních fázích choroby se nachází nejenom v PE, ale také ve fotoreceptorech a jejich vnějších segmentech. Soudí se, že onemocnění by mohlo vznikat na podkladě poruchy regulace mikroprostředí buňkami RPE. Následkem hromadění tekutiny v ubretinálním prostoru a separaci vrstev nedochází k fagocytóze vnějších segmentů fotoreceptorů, které obsahují toxické prekurzory lipofuscinu a tím dochází k ejich oxidativnímu poškozování a následně k odumírání. Vnější segmenty fotoreceptorů jsou také vylučovány do subretinálního prostoru a toxické prekurzory lipofuscinu se dále hromadí v RPE, který je fagocytuje po době delší než za fyziologického stavu, kdy se nacházejí v ěsném kontaktu. Lipofuscin pak zřejmě poškozuje buňky RPE a vede k eho atrofii (22, 36).
Řada pacientů s estovou chorobou se může při vyšetření jevit jako sporadické případy bez rodinné anamnézy. Naše práce, v souladu s inými publikovanými studiemi, poukazuje na fakt, že onemocnění lze často prokázat i u dalších příbuzných prvního stupně (3, 35). Molekulárně genetickým testováním lze zjistit přenašeče bez známek onemocnění a potvrdit dominantní dědičnost, což vede ke zlepšení poradenství postiženým pacientům a jejich rodinám.
Kromě Bestovy choroby jsou mutace v enu BEST1 spojovány se vznikem čtyř dalších degenerativních onemocnění sítnice: viteliformní makulární dystrofie s anifestací v dospělosti, autozomálně dominantní vitreoretinální choroidopatie, autozomálně recesivní bestrofinopatie a retinitis pigmentosa, která může být přenášena jak dominantně, tak recesivně (6, 7, 8, 10, 22).
Kauzální léčba Bestovy choroby neexistuje. U ohraničených choroideálních neovaskulárních membrán může být provedena termální fotokoagulace nebo fotodynamická terapie (1, 33). Popsána byla také aplikace anti-VEGF léků (17, 26, 33). Všechny tyto terapeutické přístupy vykazovaly dobré výsledky. Indikace léčby je však sporná, neboť prognóza neléčené choroideální neovaskularizace komplikující Bestovu chorobu je rovněž relativně dobrá (14).
ZÁVĚR
V české rodině s Bestovou chorobou jsme identifikovali patogenní mutaci v příčinném genu BEST1 a poprvé provedli srovnání s linickým vyjádřením. Minimální nález svědčící pro přítomnost Bestovy choroby může být zjištěn i u zcela asymptomatických jedinců, jako např. u námi vyšetřeného pacienta, kteří však mohou onemocnění přenášet na potomstvo. Stanovení příčinné mutace umožňuje potvrzení diagnózy, což může být důležité zvláště u pacientů s minimálními známkami onemocnění na fundu a normálním EOG nálezem (28, 38).
Práce byla vytvořena za podpory následujících institucí: Ministerstvo školství, mládeže a tělovýchovy MSM0021620806 a NIHR Manchester Biomedical Research Centre.
Do redakce doručeno dne 3. 8. 2011
Do tisku přijato dne 1. 12. 2011
MUDr. P. Lišková, Ph.D.
Laboratoř biologie a patologie oka, Ústav dědičných metabolických poruch,
1. lékařská fakulta, Univerzita Karlova v Praze a Všeobecná fakultní nemocnice v Praze
Ke Karlovu 2
128 08 Praha 2
e-mail: petra.liskova@lf1.cuni.cz
Sources
1. Andrade, RE., Farah, ME., Costa, RA.: Photodynamic therapy with verteporfin for subfoveal choroidal neovascularization in best disease. Am J Ophthalmol. 2003; 136 : 1179–1181.
2. Antonarakis, SE.: Recommendations for a nomenclature system for human gene mutations. Nomenclature Working Group. Hum Mutat, 1998; 11 : 1–3.
3. Booij, JC., Boon, CJ., van Schooneveld, MJ., et al.: Course of visual decline in relation to the Best1 genotype in vitelliform macular dystrophy. Ophthalmology, 2010; 117 : 1415–1422.
4. Boon, CJ., Klevering, BJ., Keunen, JE., et al.: Fundus autofluorescence imaging of retinal dystrophies. Vision Res. 2008; 48 : 2569–2577.
5. Boon, CJ., Klevering, BJ., den Hollander, AI., et al.: Clinical and genetic heterogeneity in multifocal vitelliform dystrophy. Arch Ophthalmol, 2007; 125 : 1100–1106.
6. Boon, CJ., Klevering, BJ., Leroy, BP., et al.: The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Prog Retin Eye Res. 2009; 28 : 187–205.
7. Burgess, R., MacLaren, RE., Davidson, AE., et al.: ADVIRC is caused by distinct mutations in BEST1 that alter pre-mRNA splicing. J Med Genet. 2009; 46 : 620–625.
8. Burgess, R., Millar, ID., Leroy, BP., et al.: Biallelic mutation of BEST1 causes a distinct retinopathy in humans. Am J Hum Genet, 2008, 82 : 19–31.
9. Cross, HE., Bard, L.: Electro-oculography in Best’s macular dystrophy. Am J Ophthalmol. 1974; 77 : 46–50.
10. Davidson, AE., Millar, ID., Urquhart, JE., et al.: Missense mutations in a retinal pigment epithelium protein, bestrophin-1, cause retinitis pigmentosa. Am J Hum Genet. 2009; 85 : 581–592.
11. den Dunnen, JT., Antonarakis, SE.: Nomenclature for the description of human sequence variations. Hum Genet. 2001; 109 : 121–124.
12. Esumi, N., Kachi, S., Hackler, L., et al.: BEST1 expression in the retinal pigment epithelium is modulated by OTX family members. Hum Mol Genet. 2009; 18 : 128–141.
13. Godel, V., Chaine, G., Regenbogen, L., et al.: Best’s vitelliform macular dystrophy. Acta Ophthalmol Suppl. 1986; 175 : 1–31.
14. Chung, MM., Oh, KT., Streb, LM., et al.: Visual outcome following subretinal hemorrhage in Best disease. Retina. 2001; 21 : 575–580.
15. Jarc-Vidmar, M., Kraut, A., Hawlina, M.: Fundus autofluorescence imaging in Best’s vitelliform dystrophy. Klin Monbl Augenheilkd. 2003; 220 : 861–867.
16. Lacassagne, E., Dhuez, A., Rigaudiere, F., et al.: Phenotypic variability in a French family with a novel mutation in the BEST1 gene causing multifocal best vitelliform macular dystrophy. Mol Vis. 2011; 17 : 309–322.
17. Leu, J., Schrage, NF., Degenring. RF.: Choroidal neovascularisation secondary to Best’s disease in a 13-year-old boy treated by intravitreal bevacizumab. Graefes Arch Clin Exp Ophthalmol. 2007; 245 : 1723–1725.
18. Lotery, AJ., Munier, FL., Fishman, GA., et al.: Allelic variation in the VMD2 gene in Best disease and age-related macular degeneration. Invest Ophthalmol Vis Sci. 2000; 41 : 1291–1296.
19. Marchant, D., Gogat, K., Boutboul, S., et al.: Identification of novel VMD2 gene mutations in patients with Best vitelliform macular dystrophy. Hum Mutat. 2001; 17 : 235.
20. Marchant, D., Yu, K., Bigot, K., et al.: New VMD2 gene mutations identified in patients affected by Best vitelliform macular dystrophy. J Med Genet. 2007; 44: e70.
21. Marmor, MF., Brigell, MG., McCulloch, DL., et al.: ISCEV standard for clinical electro-oculography (2010 update). Doc Ophthalmol. 2011; 122 : 1–7.
22. Marmorstein, AD., Cross, HE., Peachey, NS.: Functional roles of bestrophins in ocular epithelia. Prog Retin Eye Res. 2009; 28 : 206–226.
23. Marquardt, A., Stohr, H., Passmore, LA., et al.: Mutations in a novel gene, VMD2, encoding a protein of unknown properties cause juvenile-onset vitelliform macular dystrophy (Best’s disease). Hum Mol Genet. 1998; 7 : 1517–1525.
24. Miller, SA.: Multifocal Best’s vitelliform dystrophy. Arch Ophthalmol. 1977; 95 : 984–990.
25. Mohler, CW., Fine, SL.: Long-term evaluation of patients with Best’s vitelliform dystrophy. Ophthalmology. 1981; 88 : 688–692.
26. Montero, JA., Ruiz-Moreno, JM., De La Vega, C.: Intravitreal bevacizumab for adult-onset vitelliform dystrophy: a case report. Eur J Ophthalmol. 2007; 17 : 983–986.
27. Pianta, MJ., Aleman, TS., Cideciyan, AV., et al.: In vivo micropathology of Best macular dystrophy with optical coherence tomography. Exp Eye Res. 2003; 76 : 203-211.
28. Pollack, K., Kreuz, FR., Pillunat, LE.: [Best’s disease with normal EOG. Case report of familial macular dystrophy]. Ophthalmologe, 2005; 102 : 891–894.
29. Ponjavic, V., Eksandh, L., Andreasson, S., et al.: Clinical expression of Best’s vitelliform macular dystrophy in Swedish families with mutations in the bestrophin gene. Ophthalmic Genet. 1999; 20 : 251–257.
30. Querques, G., Regenbogen, M., Quijano, C., et al.: High-definition optical coherence tomography features in vitelliform macular dystrophy. Am J Ophthalmol. 2008; 146 : 501–507.
31. Querques, G., Regenbogen, M., Soubrane, G., et al.: High-resolution spectral domain optical coherence tomography findings in multifocal vitelliform macular dystrophy. Surv Ophthalmol. 2009; 54 : 311–316.
32. Querques, G., Zerbib, J., Santacroce, R., et al.: The spectrum of subclinical Best Vitelliform Macular Dystrophy in subjects with mutations in BEST1 gene. Invest Ophthalmol Vis Sci. 201; 52 : 4678–4684.
33. Querques, G., Zerbib, J., Santacroce, R., et al.: Functional and clinical data of Best vitelliform macular dystrophy patients with mutations in the BEST1 gene. Mol Vis. 2009; 15 : 2960–2972.
34. Schatz, P., Bitner, H., Sander, B., et al.: Evaluation of macular structure and function by OCT and electrophysiology in patients with vitelliform macular dystrophy due to mutations in BEST1. Invest Ophthalmol Vis Sci. 2010; 51 : 4754–4765.
35. Sohn, EH., Francis, PJ., Duncan, JL., et al.: Phenotypic variability due to a novel Glu292Lys variation in exon 8 of the BEST1 gene causing best macular dystrophy. Arch Ophthalmol. 2009; 127 : 913–920.
36. Spaide, R.: Autofluorescence from the outer retina and subretinal space: hypothesis and review. Retina, 2008; 28 : 5–35.
37. Spaide, RF., Noble, K., Morgan, A., et al.: Vitelliform macular dystrophy. Ophthalmology, 2006; 113 : 1392–1400.
38. Testa, F., Rossi, S., Passerini, I., et al.: A normal electro-oculography in a family affected by Best disease with a novel spontaneous mutation of the BEST1 gene. Br J Ophthalmol. 2008; 92 : 1467–1470.
39. Wabbels, B., Preising, MN., Kretschmann, U., et al.: Genotype-phenotype correlation and longitudinal course in ten families with Best vitelliform macular dystrophy. Graefes Arch Clin Exp Ophthalmol. 2006; 244 : 1453–1466.
Labels
OphthalmologyArticle was published in
Czech and Slovak Ophthalmology

2011 Issue 5-6
Most read in this issue
- Comparison of Keratometric Values and Corneal Eccentricity of Myopia, Hyperopia and Emmetropia
- Minimal Ocular Findings in a Patient with Best Disease Caused by the c.653G>A Mutation in BEST1
- AquaLase Method – Influence to the Secondary Cataract Appearance and its Safety
- Suprachoroid Hemorrhage without the Connection to the Surgical Procedure