
Klinické nálezy u členů české rodiny s retinitis pigmentosa podmíněnou mutací v ORF15 genu RPGR
Clinical Findings in Members of a Czech Family with Retinitis Pigmentosa Caused by the c.2426_2427delAG Mutation in RPGR
Purpose:
To describe the phenotype of members of the first Czech retinitis pigmentosa family with an identified molecular genetic cause (c.2426_2427delAG in RPGR), followed for more than 13 years.
Methods:
Medical records were reviewed and a detailed ophthalmic examination including spectral-domain optical coherence tomography and full-field and multifocal electroretinography (ERG) was performed in two affected males, three female carriers and one unaffected female.
Results:
A 22-year-old male who denied suffering from nyctalopia had a best corrected visual acuity (BCVA) of 0.63 in both eyes. Moderate myopia and myopic astigmatism were present bilaterally. Color vision and contrast sensitivity were normal. There was an eccentric constriction of the visual fields that spared the central 20 degrees in both eyes. Fundus examination revealed bilateral pigmentary changes in the mid-periphery. Full-field ERG documented a 10% rod and 20% cone response. The phenotype of his cousin, also aged 22 years, was more severe. He complained of nyctalopia since 12 years of age. His BCVA was 0.3 in the right eye and 0.5 in the left eye. Myopia and astigmatism were present bilaterally. Contrast sensitivity and color vision were severely impaired. Full field ERG was extinct, but some activity on multifocal ERG was still detectable. The constriction of the visual fields reached 5 degrees in both eyes. Fundus examination showed the typical retinitis pigmentosa appearance. All carriers denied that they suffered from nyctalopia, but two of them had decreased BCVA in at least one eye. None exhibited typical bone spicules or a tapetal-like reflex. Significant refractive errors were present in all eyes of the carriers.
Conclusion:
The finding of moderate or high myopia and astigmatism in males with retinitis pigmentosa as well as refractive errors in female relatives indicates possible X-linked inheritance, which may be especially important in pedigrees where the transmission pattern can not be clearly established. Our study highlights the inter-individual variability in phenotype observed in similar aged patients with identical ORF15 RPGR mutations.
Key words:
retinitis pigmentosa, phenotype, multifocal ERG, mutation, RPGR
Authors:
B. Kousal; P. Skalická 1; P. Diblík 1; P. Kuthan; H. Langrová 1; P. Lišková 2 1,3
Authors‘ workplace:
Oční klinika, 1. lékařská fakulta, Univerzita Karlova v Praze a Všeobecná fakultní nemocnice v Praze, přednosta doc. MUDr. Bohdana Kalvodová, CSc.
1; Oční klinika, Lékařská fakulta v Hradci Králové, Univerzita Karlova v Praze, přednosta prof. MUDr. Pavel Rozsíval, CSc., FEBO
2; Laboratoř biologie a patologie oka, Ústav dědičných metabolických poruch, 1. lékařská fakulta, Univerzita Karlova v Praze a Všeobecná fakultní nemocnice v Praze, přednosta prof. MUDr. Viktor Kožich, CSc.
3
Published in:
Čes. a slov. Oftal., 69, 2013, No. 1, p. 8-15
Category:
Original Article
Overview
Cílem naší práce bylo popsat klinické nálezy u členů první české rodiny s retinitis pigmentosa vázanou na pohlavní chromozom s popsanou molekulárně genetickou příčinou. Konkrétně byla v dané rodině zjištěna patogenní mutace c.2426_2427delAG (p.Glu809fs) v exonu ORF15 genu RPGR.
Analyzovali jsme klinické záznamy za období 13 let a podrobně jsme vyšetřili dva postižené bratrance ve věku 22 let, tři přenašečky a jednu ženu, u které nebyla mutace zjištěna. Kompletní oční vyšetření zahrnovalo také optickou koherenční tomografii se spektrální doménou, zábleskovou a multifokální elektroretinografii (ERG).
U prvního z vyšetřených mužů byla nejlepší korigovaná zraková ostrost (NKZO) 0,63 oboustranně. Kontrastní citlivost a barvocit byly normální. Zaznamenána byla oboustranně střední myopie a myopický astigmatismus. Šeroslepost popíral. Vyšetření fundu odhalilo ve střední periférii přítomnost typických shluků pigmentu ve tvaru kostních buněk. Zorné pole bylo oboustranně excentricky zúženo se zachováním přibližně centrální třetiny. ERG vyšetření prokázalo snížení aktivity tyčinek na 10 % normálních hodnot, funkce čípků dosahovala 20 % normálních hodnot. Druhý muž udával těžkou šeroslepost, kterou pozoroval zhruba od 12 let věku. Jeho NKZO byla 0,3 na oku pravém a 0,5 na oku levém. Zaznamenána byla oboustranně vysoká myopie a myopický astigmatismus, snížená kontrastní citlivost a porucha barvocitu spolu se zúžením zorného pole až k 5 °. Aktivita světločivých buněk byla při klasickém ERG vyšetření již vyhaslá, na multifokální ERG byla však detekovatelná čípková aktivita v centrální oblasti sítnice do 7° excentricity. Vyšetření fundu prokázalo typické známky pokročilé retinitis pigmentosa. Žádná z přenašeček netrpěla subjektivně šeroslepostí, dvě z nich měly sníženou NKZO na alespoň jednom oku. Ani u jedné nebyly zjištěny shluky pigmentu ve tvaru kostních buněk nebo tapetoidní reflex. U všech očí přenašeček byl přítomen astigmatismus větší než 1,5 Dcyl.
Střední nebo těžká myopie kombinovaná s astigmatismem u mužů s retinitis pigmentosa, stejně jako refrakční vady u příbuzných žen, mohou ukazovat na dědičnost vázanou na pohlavní chromozom, což je důležité v rodinách, ve kterých nelze jasně definovat typ přenosu. Naše studie dokumentuje intrafamiliární fenotypovou variabilitu u stejně starých pacientů s mutacemi v genu RPGR.
Klíčová slova:
retinitis pigmentosa, fenotyp, multifokální ERG, RPGR, mutace, pigmentová retinopatie
ÚVOD
Retinitis pigmentosa (RP) je geneticky heterogenní skupina progresivních degenerativních onemocnění sítnice, jejichž společným znakem je primární postižení tyčinek. Šeroslepost je prvním příznakem, poté následuje ztráta periferního vidění. V pozdějších fázích dochází také k odumírání čípků a tím ke snížení centrální zrakové ostrosti. Vyskytovat se může rovněž konkomitantní ztráta tyčinek a čípků, nebo pouze postižení čípků, pak se jedná o tzv. cone-rod dystrofie, které jsou však vyčleňovány jako samostatná klinická jednotka (20, 21). Pro onemocnění je charakteristická velká variabilita klinických nálezů, a to i mezi příslušníky jedné rodiny. Za typický znak je při vyšetření fundu považován nález shluků pigmentu připomínající kostní buňky (20, 19).
RP se vyskytuje buď jako samostatné onemocnění, nebo jako součást řady syndromů. Prevalence nejběžnější nesyndromové RP se udává 1/4000 (20).
Až na výjimky je RP monogenní onemocnění, tj. vykazující Mendelovskou dědičnost. Nejčastěji, přibližně u 20–25 % případů, se zjišťuje autozomálně recesivní forma přenosu. Autozomálně dominantní dědičnost je zastoupena v 15–20 %. Nejméně častá je RP vázaná na pohlavní chromozom, která se zjišťuje u 10–15 % pacientů. Asi 40–55 % všech případů s RP jsou tzv. sporadičtí pacienti, u kterých není přítomna rodinná anamnéza a dědičnost u nich tedy nelze bez znalosti molekulárně genetické příčiny spolehlivě určit (8, 44).
Dosud bylo identifikováno nebo byl podán důkaz o existenci minimálně 50 genů pro toto onemocnění, přičemž se odhaduje, že dalších 40–50 % genů zatím nebylo objeveno (6, 20) (http://www.sph.uth. tmc.edu/Retnet/).
V současné době jsou známy dva geny podmiňující RP vázanou na pohlavní chromozom. Nejčastěji, až u 80 % případů retinitis pigmentosa, se zjišťují mutace v genu pro regulátor GTPázy (Retinitis Pigmentosa GTPase Regulator, RPGR, MIM *312610). Méně často, přibližně ve 20 %, je onemocnění podmíněno mutacemi v genu retinitis pigmentosa 2 (RP2 MIM *300757). Také byly podány důkazy o existenci minimálně tří dalších lokusů, ve kterých se nalézají nové příčinné geny pro tento typ RP, jejich procentuální zastoupení je však minimální (34).
RP vázaná na pohlavní chromozom je jednou z nejzávažnějších forem lidských degenerativních onemocnění sítnice, a to jak vzhledem k věku, ve kterém dochází k prvním projevům, tak k rychlosti progrese. Šeroslepost vzniká u mužů často již v první dekádě života. Ve druhé dekádě se připojuje zúžení zorného pole následované snížením centrální zrakové ostrosti ve třetí a čtvrté dekádě života. Pro onemocnění je charakteristická přítomnost depozit pigmentu, která připomínají vzhledem kostní buňky, zúžení retinálních arteriol, atrofie terče zrakového nervu, zadní subkapsulární opacity čočky a nedetekovatelné nebo snížené amplitudy při elektroretinografickém (ERG) vyšetření (20, 40).
Přenašečky mohou být zcela bez klinických známek onemocnění, většinou se u nich ale pozorují mírné změny na očním pozadí a lehké snížení funkcí sítnice. Často se zjišťuje myopie a astigmatismus, lehký pokles zrakové ostrosti, malé množství pigmentových depozit v periferii sítnice nebo vzhled fundu označovaný jako tapetoidní (kovový) reflex, který je považován za charakteristický nález (13, 14, 33).
U českých pacientů byla molekulárně genetická příčina RP dosud zjištěna pouze v jedné rodině, přičemž byla detekována příčinná mutace c.2426_2427delAG v exonu ORF15 genu RPGR (30). Cílem našeho sdělení je detailně popsat klinické nálezy u příslušníků této rodiny včetně výsledků vyšetření získaných pomocí současných zobrazovacích a elektrofyziologických metod a ukázat některé specifické fenotypové charakteristiky zjišťované v souvislosti s RP vázanou na pohlavní chromozom podmíněnou mutacemi v genu RPGR.
METODY
Vyšetřili jsme 6 členů rodiny s RP vázánou na pohlavní chromozom (obr. 1), ve které jsme již dříve prokázali příčinnou mutaci c.2426_2427delAG v exonu ORF15 genu RPGR. Konkrétně se jednalo o dva postižené muže, dvě obligatorní přenašečky, jednu přenašečku s diagnózou potvrzenou molekulárně genetickým vyšetřením a jednu zdravou ženu, u které bylo přenašečství vyloučeno (30). Studovaná rodina byla jednou z větví rozsáhlého rodokmenu, v němž se již v roce 1979 Klouček a kol. pokusili prokázat vazbu na pohlavní chromozom pomocí tehdy dostupných markerů (29). Variabilita nálezu u některých přenašeček z tohoto rodokmenu byla pak popsána Otradovcem a kol. v roce 1979 (33).

U všech postižených mužů a přenašeček jsme provedli vyšetření zrakové ostrosti pomocí ETDRS optotypů a vyšetření vidění do blízka pomocí dvou typů Jaegerových tabulek s rozsahem od nejmenšího k největšímu textu 0,4–1,1 a 1–12. Kontrastní citlivost byla zjišťována na Pelli-Robsonových tabulkách (HS Clement Clarke International, Velká Británie) (23) a barvocit Lanthonyho desaturovaným panel D-15 testem. Zorné pole bylo hodnoceno pomocí kinetického (Goldmann kugel perimetr, Carl Zeiss Jena, Německo) i statického automatického perimetru (M-700, Medmont International Pty Ltd., Austrálie). Nález na fundu při biomikroskopickém vyšetření v mydriáze byl fotograficky zdokumentován (FF 450 plus IR Fundus Camera, Carl Zeiss Meditec AG, Německo). Dále bylo provedeno vyšetření optickou koherenční tomografií se spektrální doménou (SD-OCT, Spectral OCT/SLO, OTI Ophthalmic Technologies Inc., Kanada) zobrazující jednotlivé vrstvy sítnice v oblasti makuly a vrstvu nervových vláken sítnice (retinal nerve fiber layer – RNFL) v kruhu o průměru 3,4 mm okolo terče zrakového nervu. Elektrofyziologické hodnocení funkce sítnice bylo provedeno klasickou zábleskovou a multifokální elektroretinografií (ERG; RETI-port plus mfERG system, Roland Consult GmbH, Německo) podle mezinárodních standardů vypracovaných Mezinárodní společností pro klinickou elektrofyziologii zraku (31). Klasickou ERG jsme za skotopických podmínek (noční vidění) sledovali tyčinkovou odpověď, maximální odpověď a oscilační potenciály a za fotopických podmínek (denní vidění) čípkovou odpověď a odpověď na rychle blikající podněty s frekvencí 30 Hz. Multifokální ERG poskytla 61 lokálních odpovědí v centrálních 30º zorného pole.
Hodnotili jsme amplitudy a latence pozitivního vrcholu (P1) odpovědí prvního uspořádání v pěti koncentrických kruzích kolem foveoly. Trojrozměrný obraz hustoty odpovědí znázornil centrální, foveolární odpověď a směrem do periferie klesající aktivitu. Amplitudy 61 místních odpovědí daného pacienta byly porovnány s normálními hodnotami kontrolní skupiny odpovídajícího věku.
VÝSLEDKY
22letý muž (IV:1, obr. 1) popíral jakékoliv subjektivní obtíže včetně šerosleposti, přestože u něj byly již v 9 letech zdokumentovány známky počínající RP s ojedinělými periferními přesuny pigmentu ve tvaru hrudek a zřetelné pigmentové změny charakteru kostních buněk v 15 letech. V 10 letech byla k dosažení NKZO použita následující korekce -2,5 D = -2,0 Dcyl ax 25º vpravo a -0,5 D = -2,0 ax 15º vlevo, v 15 letech -3,0 D = -2,0 Dcyl ax 30º vpravo a -2,5 D = -2,0 Dcyl ax 180º vlevo. Při posledním vyšetření ve 22 letech byla zraková ostrost bilaterálně 0,63 s -3,75 D = -2,0 Dcyl ax 15º vpravo a s -3,5 D = -2,0 Dcyl ax 165º vlevo. Vidění do blízka bylo oboustranně normální.
Biomikroskopické vyšetření očního pozadí v mydriáze odhalilo oboustranně zachovanou makulu včetně vrstvy retinálního pigmentového epitelu (RPE). V periferii sítnice pak byla zjištěna typická chorioretinální atrofie s charakteristickými přesuny pigmentu tvaru kostních buněk lokalizovanými cirkulárně. Přítomno bylo i ztenčení retinálních cév, zvláště arterií (obr. 2a, b). SD-OCT vyšetření neodhalilo ani na jednom oku v makule patologii, v oblasti kolem makuly ale bylo přítomno snížení výšky sítnice a cévnatky (obr. 2c). Terč zrakového nervu se oboustranně jevil růžový, pomocí RNFL byla potvrzena fyziologická tloušťka vrstvy nervových vláken. Nebyla prokázána porucha barvocitu nebo kontrastní citlivosti.
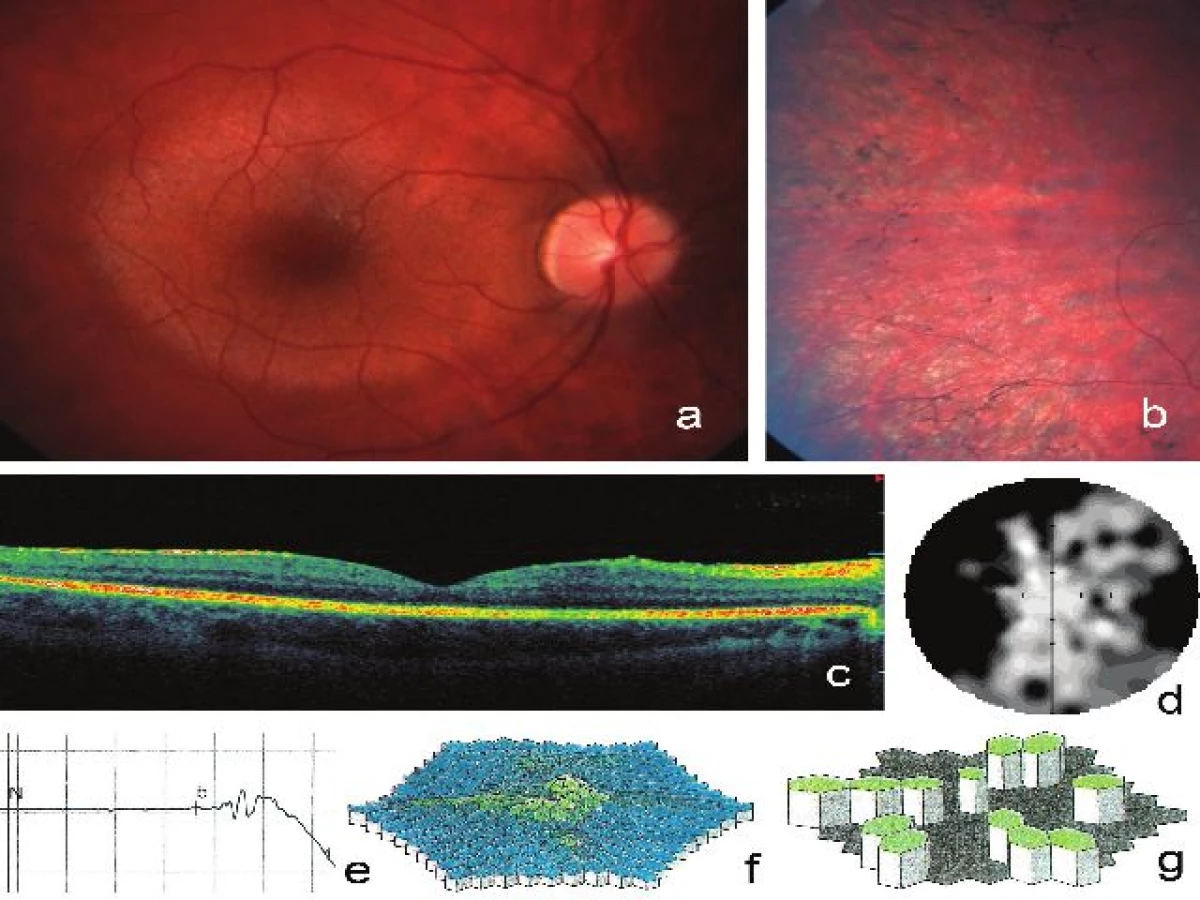
Vyšetření rozsahu zorného pole pomocí počítačové perimetrie bylo prováděno každý rok od 10 let. První hodnotitelné změny ve smyslu ojedinělých relativních skotomů byly zaznamenány v 15 letech věku. Absolutní výpady a charakteristická přítomnost anulárního skotomu mezi 10–40º byla zdokumentována ve 20 a 22 letech (obr. 2d). Vyšetření kinetickým perimetrem ve 22 letech potvrdilo zúžení zorného pole shora ke 20º, v ostatních směrech byl rozsah hodnocen jako normální.
ERG vyšetřením byla zjištěna reziduální skotopická (cca 10 % normy) i fotopická aktivita (10 % normy na oku pravém a 25 % normy na oku levém) s výrazně prodlouženými latencemi (tabulka 1, obr. 2e). Multifokální ERG prokázala reprodukovatelnou, ale výrazně sníženou foveolární a parafoveolární aktivitu sítnice (cca 20 % normy) s prodlouženými latencemi. Aktivita zevně od 14° excentricity byla jen reziduální (obr. 2f, g).
22letý muž (IV:5, obr. 1) pozoruje šeroslepost od 12 let. První přesuny pigmentu připomínající kostní buňky byly v dokumentaci doloženy ve věku 15 let. Centrální zraková ostrost a myopická korekce se v průběhu let neměnila a při našem posledním vyšetření byla NKZO vpravo 0,3 s korekcí -7,0 D = -1,25 Dcyl ax 10º a vlevo 0,5 s korekcí -6,0 D = -1,0 Dcyl ax 180º. Vidění do blízka bylo oboustranně sníženo na čtení textu s Jaegerovým číslem 1-2. Přední segment vlevo byl bez patologického nálezu, vpravo jsme zjistili počínající zákal v zadním kortexu čočky. Na očním pozadí obou očí byly přítomny některé charakteristické znaky RP: světlejší terče zrakového nervu, jemné zrnění v makulách, cirkulárně ve střední periférii přesuny pigmentu tvaru kostních buněk a velmi úzké cévy až do periférie, dominuje také výrazná chorioretinální atrofie a zviditelnění velkých chorioidálních cév (obr. 3a-d).
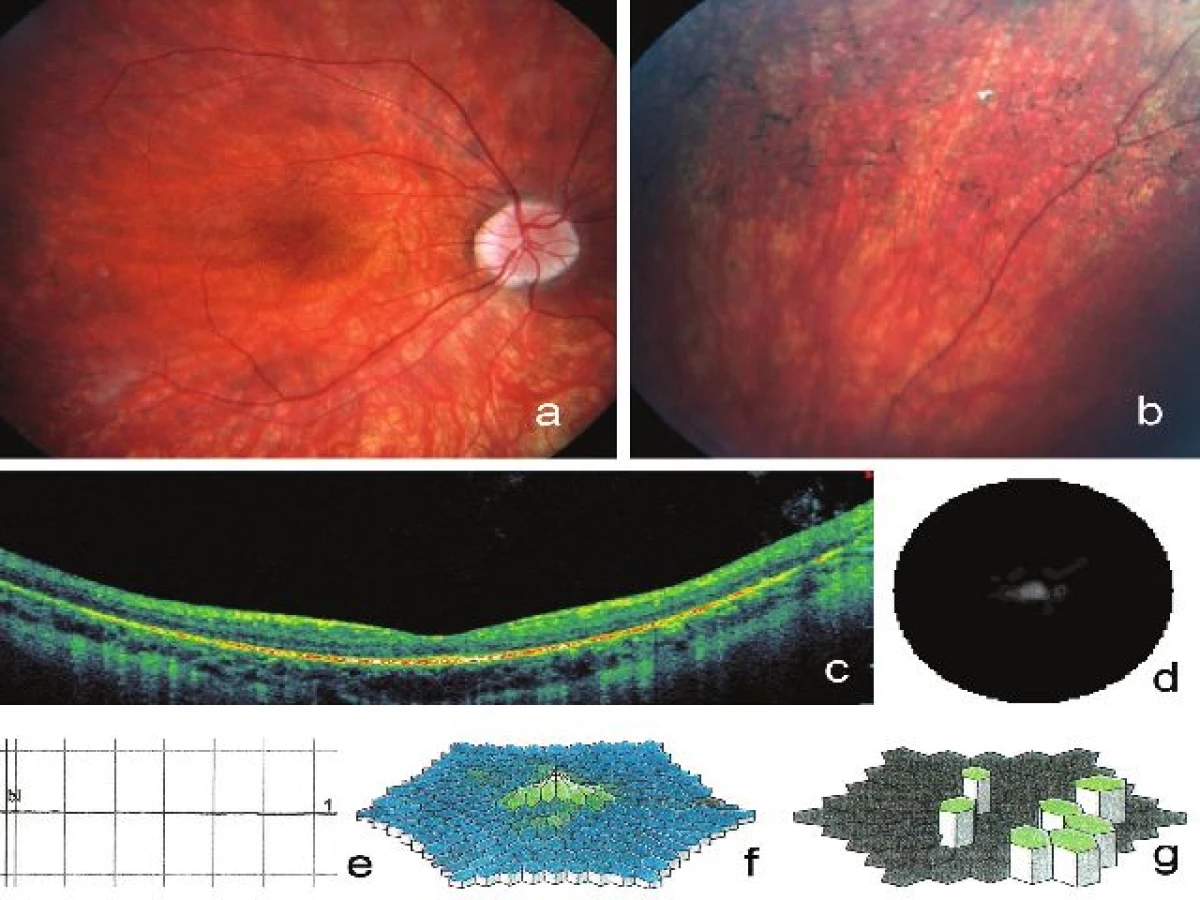
Postupné zužování zorného pole nastalo již v 16 letech s rychlou progresí mezi 19. až 21. rokem. V 16 letech byly zjištěny prakticky cirkulární výpady zorného pole jdoucí z periferie a zasahující na obou očích ke 20–30º. Ve 22 letech již byl zaznamenán pouze centrální zbytek zorného pole s rozsahem menším než 5º (obr. 3d). Také byla zjištěna závažná dyschromatopsie a porucha kontrastní citlivosti (vpravo 0,90, vlevo 1,35). Nález na SD-OCT odhalil snížení výšky sítnice a cévnatky s atrofií vrstvy fotoreceptorů (obr. 3c). Pomocí ERG byla zjištěna pouze reziduální elektrická aktivita sítnice, obtížně odlišitelná od šumu pozadí (obr. 3e-g).
Žena, 44 let (III:1, obr. 1), subjektivně neudávala žádné obtíže. Zjistili jsme nejlepší korigovanou zrakovou ostrost (NKZO) 1,0 vpravo a 0,4 vlevo s korekcí +2,0 D = -2,25 Dcyl ax 15° vpravo a +1,0 D = -3,25 Dcyl ax 175° vlevo. Vidění do blízka bylo vlevo sníženo (Jaegerovo číslo 1,1). Přední segment obou očí byl zcela bez patologického nálezu. Na očním pozadí jsme pozorovali jemné přesuny pigmentu bez podobnosti s kostními buňkami (obr. 4a, b). Vyšetření zorného pole oboustranně neprokázalo žádný abnormální výpad, ale na levém oku bylo zatíženo vysokým procentem ztráty fixace. Na SD-OCT snímku pravého oka se jevily všechny vrstvy sítnice jako normální, na levém oku však byla v porovnání s pravým okem zaznamenána nižší reflexivita komplexu zevních segmentů fotoreceptorů a RPE s výraznějším znázorněním struktur cévnatky. Kontrastní citlivost byla vpravo v normě (1,65), vlevo lehce snížená (1,35). Výsledek vyšetření barvocitu odhalil chyby v modré a v červené ose OL. Při klasickém zábleskovém ERG vyšetření byla zjištěna mírná alterace elektrické aktivity sítnice. Skotopická i fotopická aktivita sítnice byla v mezích normy s výjimkou snížené maximální odpovědi na 90 % normy oboustranně a nízké čípkové odpovědi vpravo (50 % normy) s prodlouženou latencí (tabulka 1). Multifokální ERG odhalila oboustranně sníženou foveolární (85 % normy), parafoveolární (50 % normy) a periferní (80 % normy) aktivitu. Oboustranně bylo zjištěno několik okrskových oblastí redukovaných amplitud (obr. 4 c, d).

39letá sestra (III:5, obr. 1) pozoruje i přes brýlovou korekci snižování zrakové ostrosti od 10 let, v posledních letech připouští, že vidí hůře ve tmě. V dětství nosila okluzor a na levé oko viděla vždy hůře, tomu odpovídala i zjištěná mikroesotropie vlevo. NKZO byla 0,8 vpravo s korekcí -1,0 D = +3,0 Dcyl ax 90º a 0,5 vlevo s korekcí -1,0 D = +3,0 Dcyl ax 90º. Pacientka četla vpravo s korekcí text označený Jaegerovým číslem 1 a vlevo číslem 4. U této obligatorní přenašečky jsme na obou očích zjistili symetricky mírnou chorioretinální atrofii, mírné zúžení retinálních cév a ve střední periférii pak typické změny spočívající v nepravidelném rozložení pigmentu. Zvláště v této oblasti měla sítnice šedavý nádech a zvýšenou viditelnost chorioidálních cév (obr. 4e, f). Při SD-OCT vyšetření byla oboustranně zaznamenána nižší reflexivita rozhraní RPE a neuroretiny s výraznějším znázorněním struktur cévnatky. Automatické perimetrické vyšetření ani vyšetření pomocí kinetického perimetru se značkou IIIe4 nevykazovalo abnormální nález. Vyšetření kontrastní citlivosti a barvocitu bylo v normě. ERG vyšetření ukázalo snížení amplitud skotopických i fotopických odpovědí na 50 % normy s prodloužením latencí. Multifokální ERG odhalila oboustranně snížení amplitud průměrných odpovědí na 50 % normy s mozaikovým rozložením dysfunkčních okrsků (obr. 4g, h).
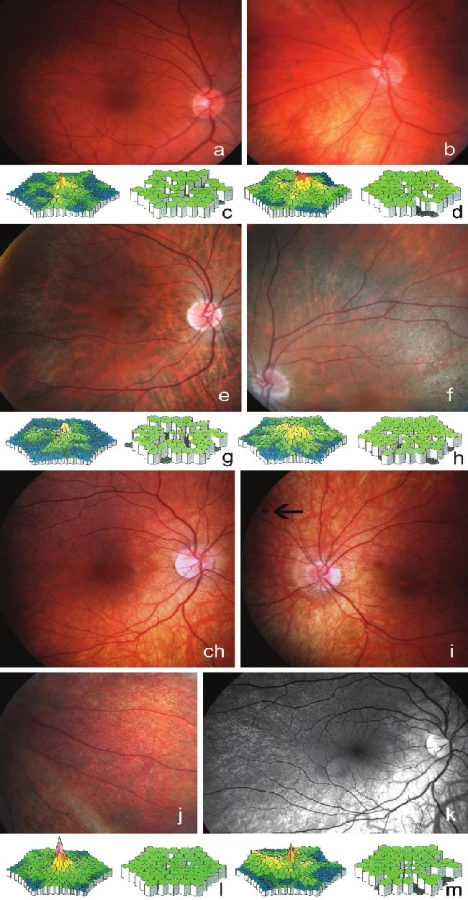
Žena, 20 let (IV:6, obr. 1), šeroslepost popírala. Od dětství má levé oko tupozraké a od 15 let pozoruje snížení NKZO, která byla při našem vyšetření 1,0 s korekcí -4,0 D = -1,5 Dcyl ax 180º vpravo a 0,16 s korekcí -10,0 D = -2,5 Dcyl ax 160º vlevo. Při posouzení vzájemného postavení očí byla zjištěna mikroexotropie levého oka s excentrickou fixací. Nález na fundu odpovídal myopické korekci, nebyly zaznamenány žádné pigmentace tvaru kostních buněk, jen na levém oku v nazálním horním kvadrantu byla přítomna jedna pigmentová hrudka. Temporálně od makuly jsme pak pozorovali oboustranně mírné zrnité zašednutí někdy také popisované jako „fundus sůl a pepř“ (obr. 4ch-k). Na SD-OCT skenech byl patrný myopický charakter retinogramu s nižší reflexivitou rozhraní RPE a neuroretiny s výraznějším znázorněním struktur cévnatky. Vyšetření zorného pole, kontrastní citlivosti, barvocitu nevykazovalo žádné abnormality. ERG vyšetření prokázalo na pravém oku normální amplitudy skotopických i fotopických odpovědí a na levém oku abnormální maximální (80 % normy) a čípkovou (65 % normy) odpověď při normálních až hraničních latencích oboustranně (tabulka 1). Odpovědi pravého oka byly při multifokální ERG hodnoceny jako normální, vlevo bylo zjištěno snížení foveolárních odpovědí na cca 30 % normy, parafoveolární aktivita, stejně jako odpovědi zevně od makuly na 80 % normy (obr. 4l, m).
Žena, 23 let (IV:2, obr. 1), neudávala žádné obtíže. Zjištěná zraková ostrost byla bilaterálně 1,0, vpravo bez korekce, vlevo s korekcí -1,0 D. Vyšetření fundu neprokázalo v souladu s výsledky molekulárně genetického testování patologický nález.
Žádný z vyšetřovaných jedinců nepodstoupil nitrooční chirurgický zákrok. Nitrooční tlak byl u všech ve fyziologickém rozmezí.
DISKUSE
Cílem našeho sdělení je popsat klinické nálezy u příslušníků české rodiny s RP vázanou na pohlavní chromozom, u které byla jako u první publikována molekulárně genetická příčina (30). V předchozí práci jsme prokázali, že onemocnění je ve studovaném rodokmenu podmíněno patogenní mutací c.2426_2427delAG nacházející se v exonu ORF15 genu RPGR, která vede k posunu otevřeného čtecího rámce od aminokyselinového zbytku na pozici 809 (p.Glu809fs) (30) a tím k předčasnému ukončení syntézy proteinu (1). Patogenní změny v ORF15 jsou nejenom nejčastější příčinou RP vázané na pohlavní chromozom, ale vyskytují se odhadem až u 15 % všech případů s tímto onemocněním, tedy s nejvyšší četností ze všech lokusů pro RP (3, 22). Vzhledem k prevalenci v jiných populacích (7, 35) odhadujeme, že v České republice může existovat celkem asi 200–300 jedinců mužského pohlaví s RP vázanou na pohlavní chromozom.
Produktem genu RPGR je protein, který byl lokalizován do přechodu zevního a vnitřního segmentu tyčinek i čípků, podílí se na intraflagelárním transportu, jeho detailní funkce však není zatím známa (25). V souladu s expresí v obou typech světločivých elementů, vyvolávají mutace v RPGR nejenom klinický obraz typické RP, ale i dystrofie čípků, smíšené čípko-tyčinkové dystrofie nebo atrofickou degeneraci makuly (2, 9, 10, 46).
Klinické nálezy u příslušníků námi sledované rodiny byly již v roce 1979 popsané Otradovcem a kol. (33). Studiem literatury jsme dospěli k závěru, že z původního rodokmenu jsme opětovně vyšetřili dvě obligatorní přenašečky (označené na obr. 1 symbolem III:1 a III:5), v té době 2–9leté (33).
Otradovec a kol. (1979) zdokumentovali čtyři postižené muže, u všech vznikly první subjektivní obtíže na začátku 2. decennia a kolem 40. roku života již vykazovali poruchu zraku stupně praktické slepoty (zraková ostrost pod 3/60 nebo binokulární zorné pole menší než 10º (33). Tento průběh jsme pozorovali i my u muže IV:5, který již neměl ve 22 letech známky ERG aktivity a jehož klinický nález tak kontrastoval s výsledky vyšetření u stejně starého bratrance IV:1 s ještě relativně zachovalými zrakovými funkcemi a dosud funkční elektrofyziologickou aktivitou sítnice. ERG vyšetření u tohoto muže prokázalo smíšené poškození tyčinek a čípků, zatím bez snížení zrakové ostrosti do blízka, beze změn barvocitu nebo centrálních výpadů zorného pole.
Rozdíly mezi oběma námi vyšetřenými jedinci jsou příkladem často pozorované variability klinického nálezu, stěžující poradenství pacientům a jejich rodinným příslušníkům při odhadování prognózy vývoje ztráty zrakových funkcí. Příčina tohoto jevu nebyla dosud jednoznačně objasněna. Soudí se, že podkladem mohou být modifikující genetické a epigenetické faktory (11, 35).
Další specifickou charakteristikou klinického nálezu u jedinců s RP podmíněnou mutacemi v genech RPGR a RP2 lokalizovaných na pohlavním chromozomu je výskyt refrakčních vad s dosud neuspokojivě vysvětleným mechanismem vzniku. Často je zjišťována myopie v kombinaci s myopickým astigmatismem nebo smíšený astigmatismus (16, 28, 34, 35). Tyto refrakční vady jsme pozorovali i u našich pacientů. Při dlouholetém sledování jednotlivých rodinných příslušníků, které trvalo více než 13 let, jsme u jednoho z mužů prokázali postupný nárůst myopie a astigmatismu. Také Otradovec a kol. zjistili u tří ze čtyř postižených vyšetřených mužů střední nebo vysokou myopii od -6,0 D do -15,0 D (33).
Klinické nálezy u všech tří námi vyšetřených přenašeček byly relativně podobné. NKZO byla snížená na alespoň jednom oku, pozorovány byly rovněž závažnější refrakční vady s anizometropií a s ní související amblyopií. Všechny přenašečky měly na očním pozadí změny ve smyslu mírného prořídnutí RPE, užších cév v periférii a ojedinělých přesunů až shluků pigmentu bez typického tvaru kostních buněk, což bývá v mladším a středním věku častým nálezem (35). U přenašečky IV:6 s vysokou myopií však bylo nemožné odlišit, do jaké míry se tato vada spolupodílí na zjištěných změnách fundu. U žádné ze tří přenašeček jsme nezjistili tapetoidní reflex, o kterém se soudí, že vzniká pravděpodobně na podkladě zvýšené reflexivity vnějších segmentů fotoreceptorů (5). V literatuře se uvádí, že tento nález je přítomen u pacientek s mutacemi v RPGR přibližně v polovině případů (15).
Nálezy zjištěné Otradovcem a kol., kteří vyšetřili ze studovaného rodokmenu 10 obligatorních přenašeček a 7 žen, o kterých se domnívali na základě klinického nálezu, že jsou také nositelkami patogenní mutace, byly velmi variabilní: od plně vyvinutého obrazu typické RP, přes sektorovou retinitis punctata albescens, sektorové výpady zorného pole, přítomnost tapetoidního reflexu v oblasti zadního pólu až po pouhé změny na očním pozadí, které byly hodnoceny jako myopický fundus. Stejně jako v našem souboru, všechny přenašečky měly sníženou zrakovou ostrost na alespoň jednom oku. U dvou z nich byl v dětském věku zjištěn konvergentní strabismus a lehká amblyopie šilhajícího oka, jednou z nich byla pravděpodobně námi vyšetřená žena III:5. Z refrakčních vad byl přítomen hypermetropický astigmatismus, střední a vysoká myopie (33).
Výskyt refrakční vad u přenašeček RP je v literatuře dobře zdokumentován (15, 27, 39), již Fishman a kol. v roce 1986 upozorňoval na fakt, že se u nich často zjišťuje astigmatismus větší než 1,5 Dcyl (14).
Klasická záblesková ERG u vyšetřených mužů ukazovala na postižení jak tyčinek, tak i čípků, přičemž u jedince IV:5 byla aktivita již vyhaslá. Přenašečky vykazovaly různé nálezy od mírné alterace elektrické aktivity s amplitudami skotopických i fotopických odpovědí ještě v normálním rozmezí při normálních až hraničních latencích, po snížení amplitud různého stupně s prodloužením latencí. Vysoká myopie nad -6,0 D se může sama o sobě podílet na redukci amplitud při klasické ERG (45).
Zajímavé byly výsledky multifokální ERG, které jsou v souvislosti s RP u českých pacientů publikovány poprvé. Okrsková alterace byla zjištěna i na pravém oku ženy IV:6, přestože celková elektrofyziologická aktivita sítnice byla hodnocena jako normální. Toto mozaikovité rozložení dysfunkce sítnice při vyšetření multifokální ERG je u přenašeček charakteristickým nálezem (43). Multifokální ERG prokázala u obou mužů několik zachovalých okrsků aktivit především ve foveolárních oblastech, klinicky odpovídající dosud relativně dobré NKZO. Při multifokální ERG lze fotopické odpovědi v oblasti makuly často prokázat i u mužů s pokročilou ztrátou zrakových funkcí s již nedetekovatelným klasickým zábleskovým ERG záznamem, jehož hlavní složkou jsou odpovědi z periferie sítnice. Toto jsme pozorovali i my u jednoho z postižených pacientů (IV:5), podobně jako jiní autoři (17, 24, 37).
Variabilita klinických nálezů u přenašeček se dříve vysvětlovala nerovnoměrnou inaktivací chromozomu X, tato teorie však byla experimentálně vyloučena. V současné době se soudí, že stejně jako u postižených mužů má hlavní vliv na fenotypové vyjádření přítomnost modifikujících genetických faktorů buď v samotném příčinném genu, nebo v jiných genech (32, 34).
Vzhledem k tomu, že Otradovec a kol. (33) zjistili u některých přenašeček ze studované rodiny zcela normální nálezy, včetně aktivity při klasickém zábleskovém ERG vyšetření, stejně jako i další autoři (38, 43), je průkaz anebo vyloučení patogenní mutace zvláště přínosný u dcer obligatorních přenašeček, které mají 50% pravděpodobnost, že se také stanou nositelkami této sekvenční varianty. To má dále význam při genetickém poradenství a stanovení rizika přenosu choroby na další generace. V našem rodokmenu (obr. 1) se jednalo o ženy IV:2 a IV:6.
Přestože nelze u RP na základě klinického nálezu většinou usuzovat na konkrétní gen, který by mohl onemocnění podmiňovat, byla u pacientů s mutacemi v RPGR zjištěna rychlejší progrese onemocnění než např. u jedinců s autozomálně dominantní RP způsobenou změnami v genu rodopsinu (RHO). Průměrný věk vzniku těžké slabozrakosti je u pacientů s mutacemi v RPGR 45 let, což je o 32 let dříve, než ve skupině pacientů s mutacemi v genu RHO (36).
Na možnost přítomnosti dědičnosti s vazbou na pohlavní chromozom je třeba myslet i u mužů s negativní rodinnou anamnézou nebo u sourozenců mužského pohlaví s časnými známkami onemocnění. Příčinné mutace v genech lokalizovaných na X chromozomu se vyskytují v této skupině až ve 30 % (34). Důležitým vodítkem pro zvolení cíleného genetického screeningu může být i již výše zmíněná přítomnost refrakčních vad.
Znalost molekulárně genetické příčiny RP zlepšuje klinické poradenství pacientům a jejich rodinám. Umožňuje také podstoupit preimplantační diagnostiku, která se používá při in vitro fertilizaci, kdy je ještě před implantací embrya do dělohy provedena kontrola možné přítomnosti vlohy pro dědičnou chorobu. Z očních onemocnění byla tato metoda v literatuře zdokumentována např. pro Stargardtovu chorobu a retinoblastom (12, 18, 41).
Pro onemocnění RP není známa zatím žádná účinná léčba, ale na základě slibných výsledků z klinických studií u pacientů s dystrofií sítnice podmíněnou mutacemi v RPE65 se mnoho nadějí vkládá do genové terapie (26, 42). Zcela nedávno byla publikována studie, která dokumentovala účinnost této léčby u psího modelu pro RP podmíněné mutacemi právě v RPGR (4). Vzhledem k tomu, že patogenní změny v tomto genu jsou jednou z nejčastějších příčin této dystrofie sítnice, očekává se, že výzkum povede ke klinickým zkouškám, a i proto může být pro pacienty důležitá znalost molekulárně genetické příčiny jejich onemocnění.
ZÁVĚR
Provedli jsme porovnání genotypu s fenotypem u první české rodiny s popsanou molekulárně genetickou příčinou RP. Při přítomnosti střední a vysoké myopie a astigmatismu u mužů s tímto onemocněním je třeba pomýšlet na dědičnost s vazbou na pohlavní chromozom, kterou lze pak ověřit cíleným screeningem příčinných genů.
Práce byla vytvořena za podpory programů UNCE 204011, PRVOUK-P24/LF1/3 a krátkodobým cestovním grantem od European Science Foundation programme on Frontiers of Functional Genomics.
Do redakce doručeno dne 3. 12. 2012
Do tisku přijato dne 25. 2. 2013
MUDr. P. Lišková, Ph.D.
Ke Karlovu 2
128 00 Praha 2
petra.liskova@lf1.cuni.cz
Sources
1. Adamian, M., Pawlyk, BS., Hong, DH., et al.: Rod and cone opsin mislocalization in an autopsy eye from a carrier of X-linked retinitis pigmentosa with a Gly436Asp mutation in the RPGR gene. Am J Ophthalmol. 2006; 142 : 515–8.
2. Ayyagari, R., Demirci, FY., Liu, J., et al.: X-linked recessive atrophic macular degeneration from RPGR mutation. Genomics. 2002; 80 : 166–71.
3. Bader, I., Brandau, O., Achatz, H., et al.: X-linked retinitis pigmentosa: RPGR mutations in most families with definite X linkage and clustering of mutations in a short sequence stretch of exon ORF15. Invest Ophthalmol Vis Sci. 2003; 44 : 1458–63.
4. Beltran, WA., Cideciyan, AV., Lewin, AS., et al.: Gene therapy rescues photoreceptor blindness in dogs and paves the way for treating human X-linked retinitis pigmentosa. Proc Natl Acad Sci U S A. 2012; 109 : 2132–7.
5. Berendschot, TT., DeLint, PJ., van Norren, D.: Origin of tapetal-like reflexes in carriers of X-linked retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1996; 37 : 2716–23.
6. Berger, W., Kloeckener-Gruissem, B., Neidhardt, J.: The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res. 2010; 29 : 335–75.
7. Breuer, DK., Yashar, BM., Filippova, E., et al.: A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa. Am J Hum Genet. 2002; 70 : 1545–54.
8. Daiger, SP., Bowne, SJ., Sullivan, LS.: Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol. 2007; 125 : 151–8.
9. Demirci, FY., Rigatti, BW., Wen, G., et al.: X-linked cone-rod dystrophy (locus COD1): identification of mutations in RPGR exon ORF15. Am J Hum Genet. 2002; 70 : 1049–53.
10. Ebenezer, ND., Michaelides, M., Jenkins, SA., et al.: Identification of novel RPGR ORF15 mutations in X-linked progressive cone-rod dystrophy (XLCORD) families. Invest Ophthalmol Vis Sci. 2005; 46 : 1891–8.
11. Fahim, AT., Bowne, SJ., Sullivan, LS., et al.: Allelic heterogeneity and genetic modifier loci contribute to clinical variation in males with X-linked retinitis pigmentosa due to RPGR mutations. PLoS One. 2011; 6: e23021.
12. Fiorentino, F., Magli, MC., Podini, D., et al.: The minisequencing method: an alternative strategy for preimplantation genetic diagnosis of single gene disorders. Mol Hum Reprod. 2003; 9 : 399–410.
13. Fishman, GA., Farber, MD., Derlacki, DJ.: X-linked retinitis pigmentosa. Profile of clinical findings. Arch Ophthalmol. 1988; 106 : 369–75.
14. Fishman, GA., Weinberg, AB., McMahon, TT.: X-linked recessive retinitis pigmentosa. Clinical characteristics of carriers. Arch Ophthalmol. 1986; 104 : 1329–35.
15. Flaxel, CJ., Jay, M., Thiselton, DL., et al.: Difference between RP2 and RP3 phenotypes in X linked retinitis pigmentosa. Br J Ophthalmol. 1999; 83 : 1144–8.
16. Garcia-Hoy
Labels
OphthalmologyArticle was published in
Czech and Slovak Ophthalmology

2013 Issue 1
Most read in this issue
- Klinické nálezy u členů české rodiny s retinitis pigmentosa podmíněnou mutací v ORF15 genu RPGR
- Oboustranná neuroretinitis jako projev nemoci kočičího škrábnutí u devítiletého chlapce
- Benigní maskující syndrom u pacientky s retinitis pigmentosa
- Evaluace parametrů jednoduchého binokulárního vidění na synoptoforu u zdravé dospělé populace