
Vogt-Koyanagi-Harada syndróm v detskom veku
Vogt-Koyanagi-Harada Syndrome in Children – a Case Report
Vogt-Koyanagi-Harada (VKH) syndrome is a multisystemic disease characterized by granulomatous panuveitis with exsudative retinal detachment and often associated with neurological and skin symptomatology.
In the paper is presented a rare case of probably VHK syndrome in 11-year old caucasian race boy in which was found the bilateral granulomatous panuveitis with exsudative retinal detachment without other systemic symptomatology with typical clinical characteristics and course. Systemic corticosteroid therapy in a patient gradually improved the state, which was then complicated by the occurrence of juxtapapillary subretinal neovascular membrane on both eyes. The following administration of intravitreal injection anti-VEGF (bevacizumab) was modified visual acuity and reduced neovascular membrane.
Key words:
Vogt-Koyanagi-Harada syndrome, children, juxtapapillary choroidal neovascular membrane, anti-VEGF, bevacizumab.
:
B. Bušányová; D. Tomčíková; A. Gerinec
:
Práca bola prednesená na X. sympóziu detskej oftalmológie 27. –28. 5. 2011 v Bratislave.
; Klinika detskej oftalmológie DFNsP-LF UK, Bratislava, prednosta kliniky prof. MUDr. Anton Gerinec, CSc.
:
Čes. a slov. Oftal., 69, 2013, No. 2, p. 81-86
:
Case Report
Práca bola prednesená na X. sympóziu detskej oftalmológie 27.–28. 5. 2011 v Bratislave.
Vogt-Koyanagi-Harada (VKH) syndróm je multisystémové ochorenie charakterizované granulomatóznou panuveitídou s exsudativnou amóciou sietnice a často asociované s neurologickou a kožnou symptomatológiou.
V práci je prezentovaný vzácny prípad pravdepodobného VHK syndrómu u 11-ročného chlapca kaukazskej rasy, u ktorého bola zistená obojstranná granulomatózna panuveitída s exsudatívnou amóciou sietnice bez inej systémovej symptomatológie s typickým klinickým obrazom a priebehom. Pri celkovej kortikosteroidnej liečbe dochádza u pacienta k postupnému zlepšovaniu stavu, ktorý je následne skomplikovaný výskytom juxtapapilárnej choroidálnej neovaskulárnej membrány obojstranne. Po aplikácii anti-VEGF injekcie (bevacizumab) intravitreálne došlo k úprave zrakovej ostrosti a k redukcii neovaskulárnej membrány.
Kľúčové slová:
Vogt-Koyanagi-Harada syndróm, deti, juxtapapilárna choroidálna neovaskulárna membrána, anti-VEGF, Bevacizumab
Úvod
Vogt-Koyanagi-Harada (VKH) syndróm je idiopatické multisystémové T-lymfocytmi sprostredkované autoimúnne ochorenie proti antigénnym komponentom melanocytov, zapríčiňujúce zápal tkanív obsahujúcich melanocyty ako uvea, uši, koža a meningy (14). Charakterizované je granulomatóznou panuveitídou s exsudatívnou amóciou sietnice a často asociované s neurologickou a kožnou manifestáciou (4). Syndróm sa najčastejšie vyskytuje u tmavo pigmentovanej rasy, u aziatov, amerických indiánov, hispáncov a černochov. Vek manifestácie je najčastejšie medzi 20.–50. rokom. Z množstva publikovaných prác bol výskyt u detí pod 16 rokov menej ako 5 %. V literatúre je iba niekoľko zdokumentovaných prípadov VKH syndrómu u detí, v našej práci prezentujeme prípad 11-ročného pacienta s VKH syndrómom, priebeh ktorého bol komplikovaný vznikom choroidálnej neovaskulárnej membrány juxtapapilárne.
Kazuistika
Na naše pracovisko bol odoslaný 11-ročný chlapec kaukazskej rasy s 2-týždňovou anamnézou náhleho poklesu videnia na obe oči. V minulosti sa neliečil na žiadne očné ochorenie, nemal očný úraz ani operáciu. Dva týždne pred začatím problémov bol očkovaný vakcínou PRIORIX. Jediným symptómom bola porucha videnia, iné celkové ťažkosti nemal. Pri prvom vyšetrení u svojho očného lekára bola centrálna zraková ostrosť (CZO) 5/30 obojstranne a pri vyšetrení fundu bola zistená hyperémia terča zrakového nervu (TZN). Následne bol prijatý v spádovej očnej klinike a odoslaný na vyšetrenie mozgu nukleárnou magnetickou rezonanciou (NMR), kde sa zistil nález extraaxiálnej cystickej lézie v pineálnej oblasti. Z dôvodu MRI nálezu bol pacient preložený na Kliniku detskej neurológie DFNsP. Neboli zistené iné neurologické, kožné ani sluchové prejavy a vyšetrenie cerebrospinálneho likvoru (CFS) nezistilo odchýlky.
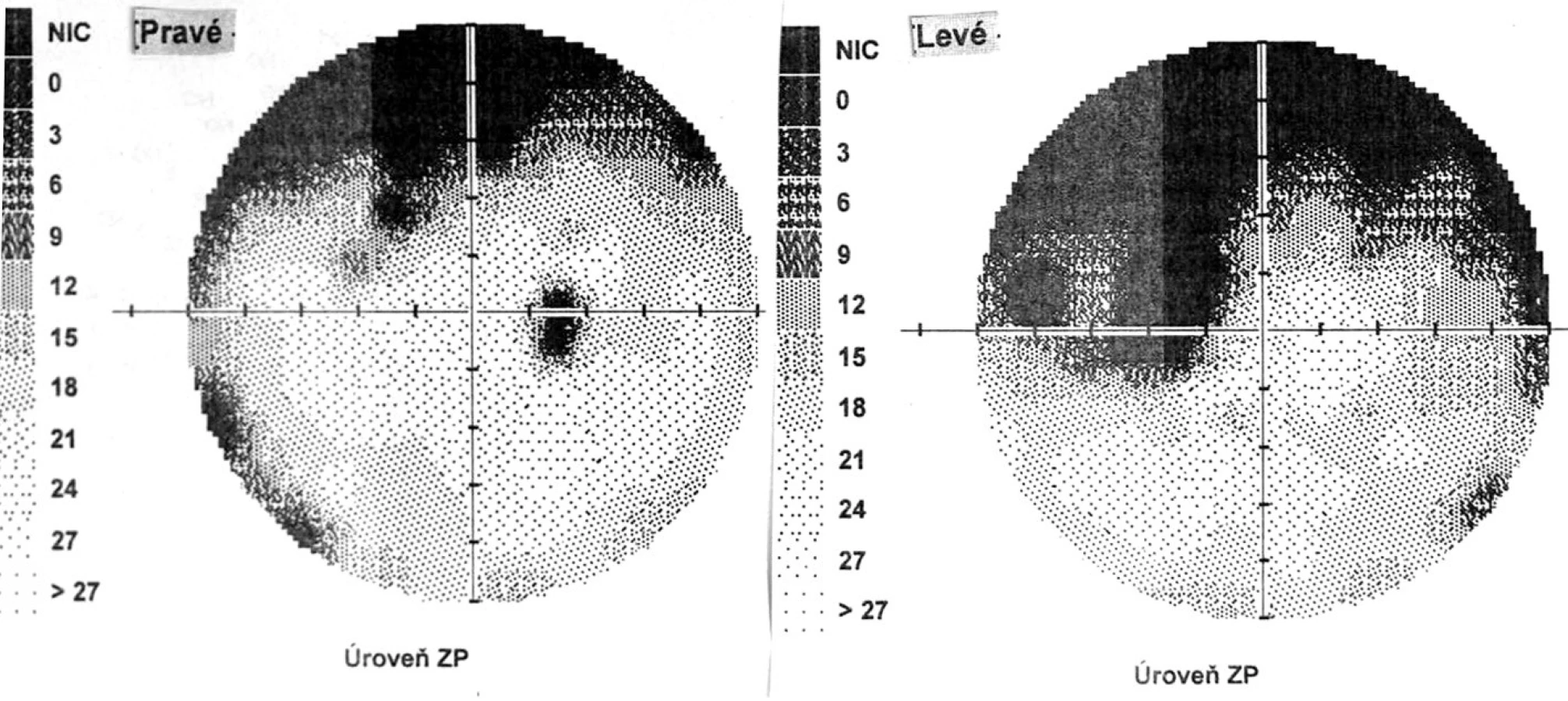
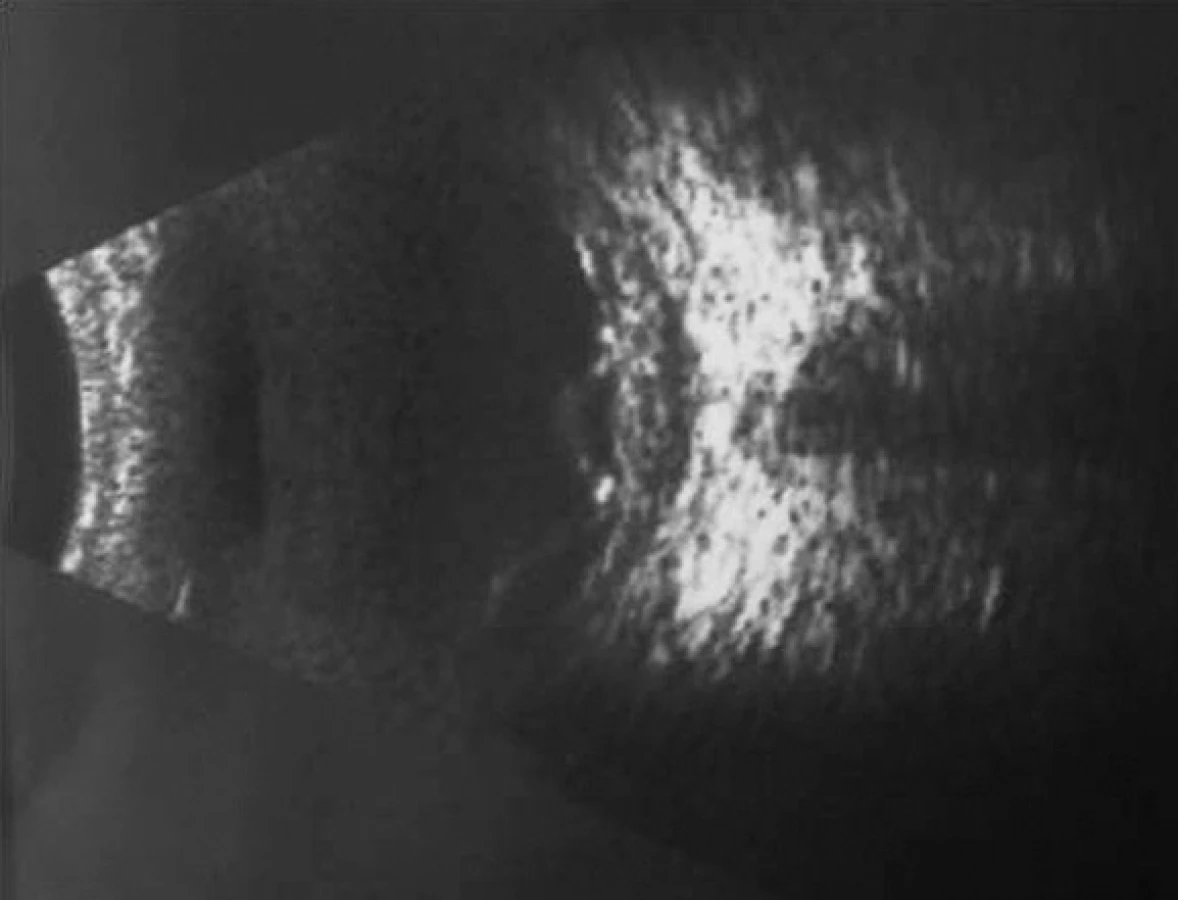
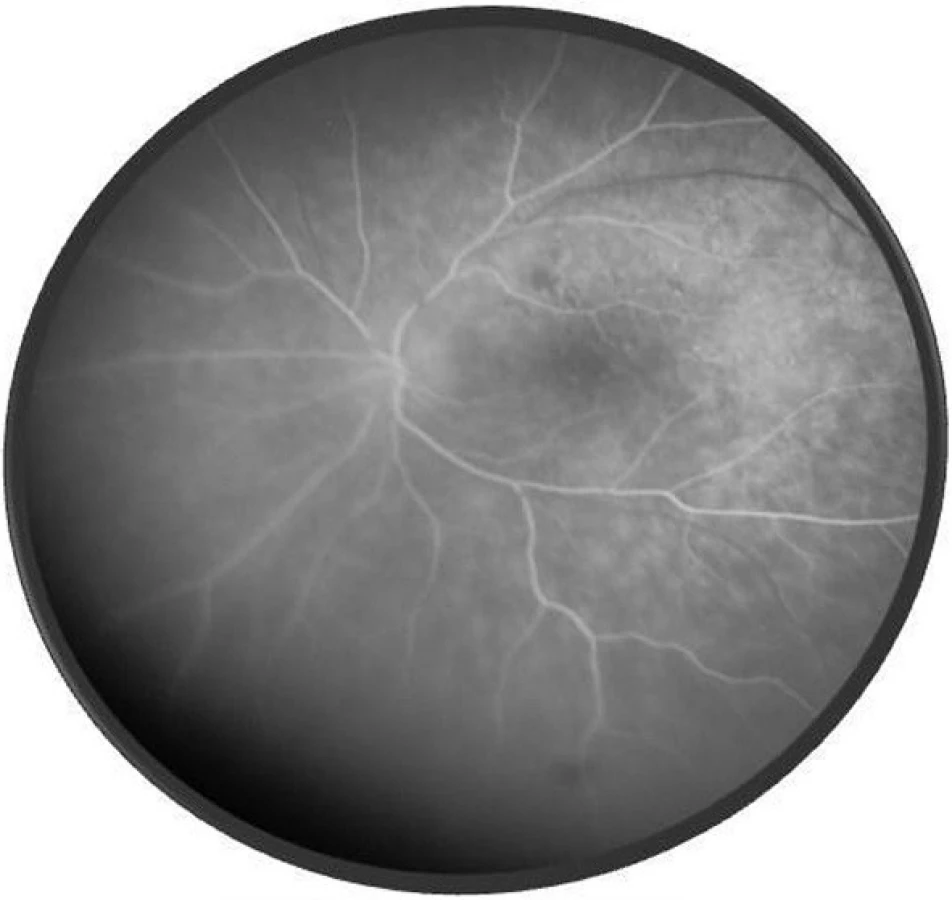
Pri prvom očnom vyšetrení na Klinike detskej oftalmológie DFNsP zistená pretrvávajúca porucha CZO 5/30 obojstranne a obojstranná panuveitída s multifokálnou seróznou amóciou sietnice (obr. 1) a s porušením zorného poľa (obr. 2). Súčasne bolo vyslovené podozrenie na pravdepodobný VKH syndróm. Diagnosticky bola vylúčená iná príčinná súvislosť s iným zápalovým alebo infekčným ochorením. HLA typizáciou zistené HLA I. triedy: HLA A2, A11, B51, B7 a HLA II. triedy: DR01 a DR11. U pacienta ultrasonografický (USG) obraz (obr. 3), nález na optickej koherentnej tomografii (OCT) (obr. 4) a fluoresceínovej angiografii (FAG) zodpovedali uveitickému štádiu VKH choroby. Započatá pulzná kortikoidná liečba metylprednizolónom 500 mg intravenózne 3 dni s následným perorálnym podaním prednizónu v dávke 1mg/kg/deň.




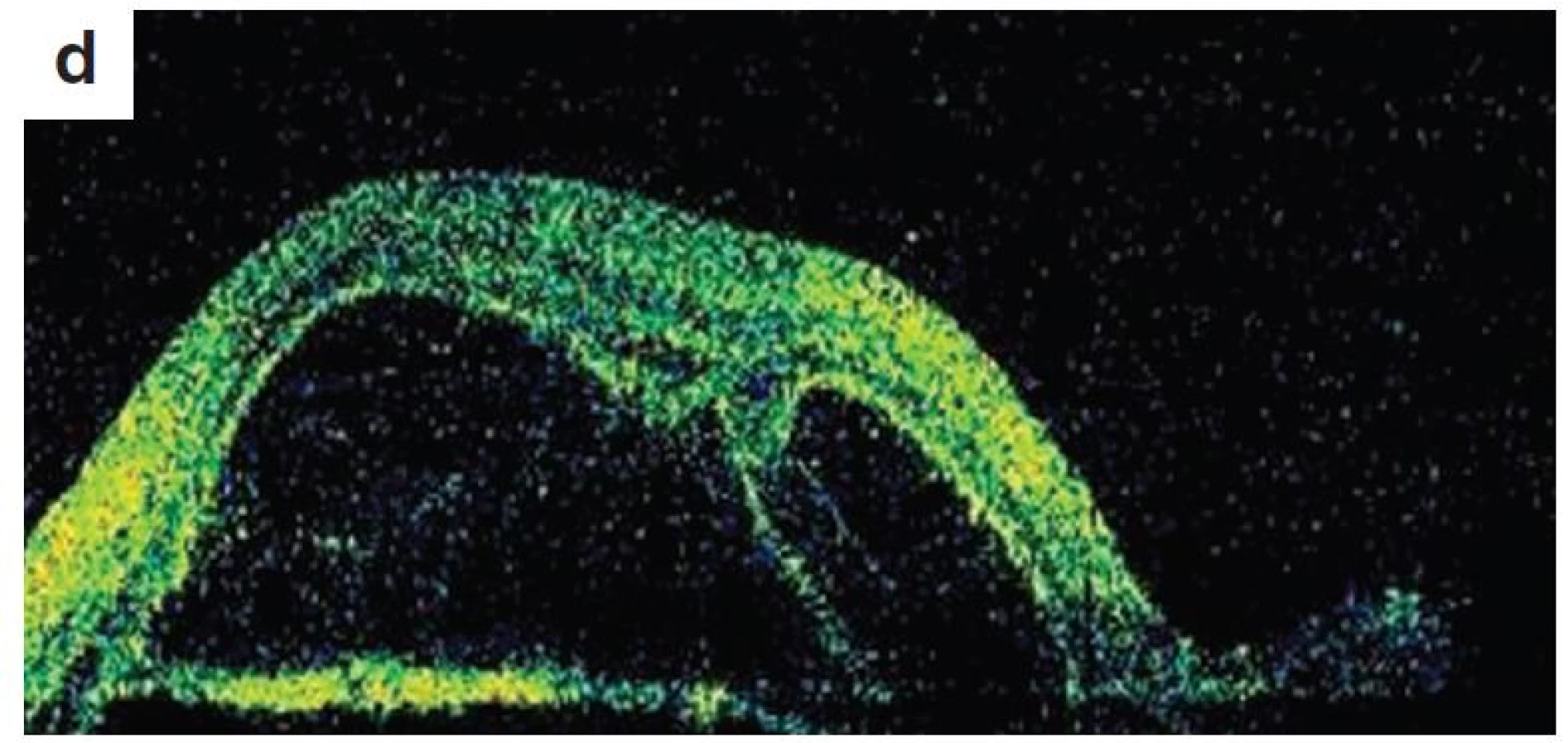
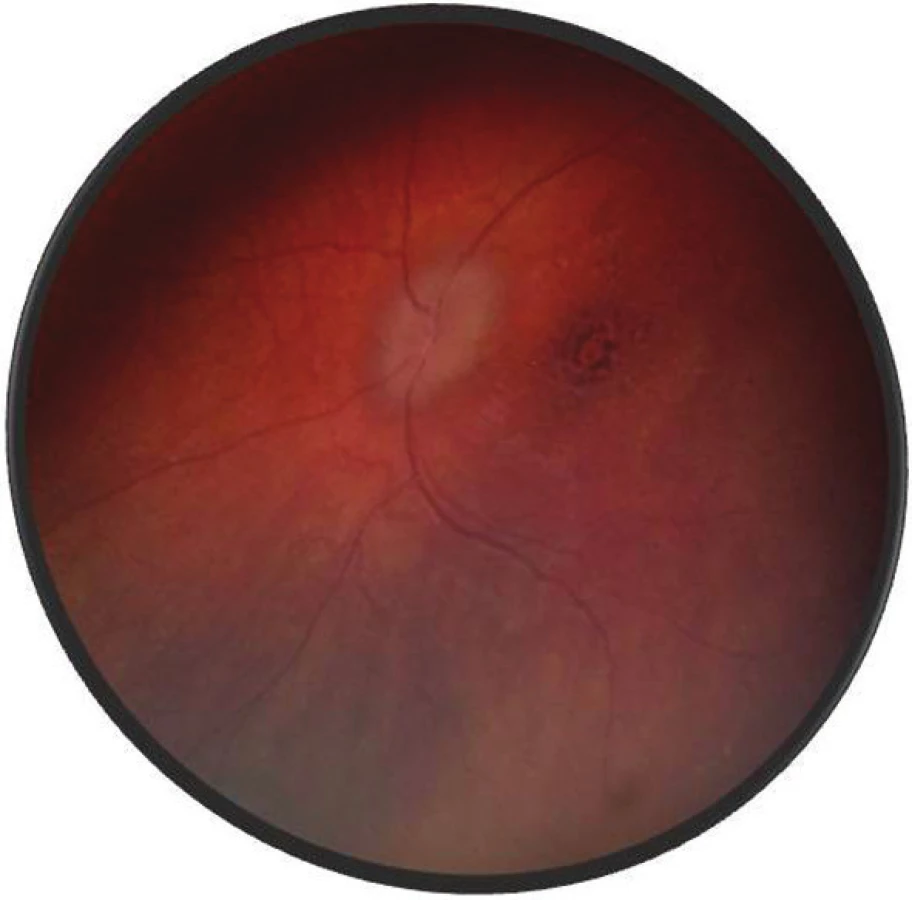
Po 1. mesiaci pri kontrolnom MRI vyšetrení zistený nález cisterna magna permagna a cysta epifýzy. Neurochirurg skonštatoval, že sa jedná o možný bežný nález bez klinického korelátu k zrakovej poruche. Pri celkovej kortikosteroidnej liečbe dochádza u pacienta v období 1,5 mesiaca k postupnému zlepšovaniu CZO na 5/7,5 na pravom oku a 5/10 na ľavom oku, k zlepšeniu zorného poľa, k ústupu amócie sietnice a k vymiznutiu prednej uveitídy. V oftalmoskopickom obraze pretrvával edém TZN a poruchy pigmentového epitelu sietnice (PRE), ktorý bol deštruovaný v makule. Objavili sa depigmentácie v periférii a strednej periférii (obr. 5), čomu zodpovedal aj FAG obraz (obr. 6) a na OCT bola vymiznutá amócia sietnice (obr. 7 a, b). Hodnoty vnútroočného tlaku boli v norme. Stav sme zhodnotili ako chronické štádium VKH syndrómu.

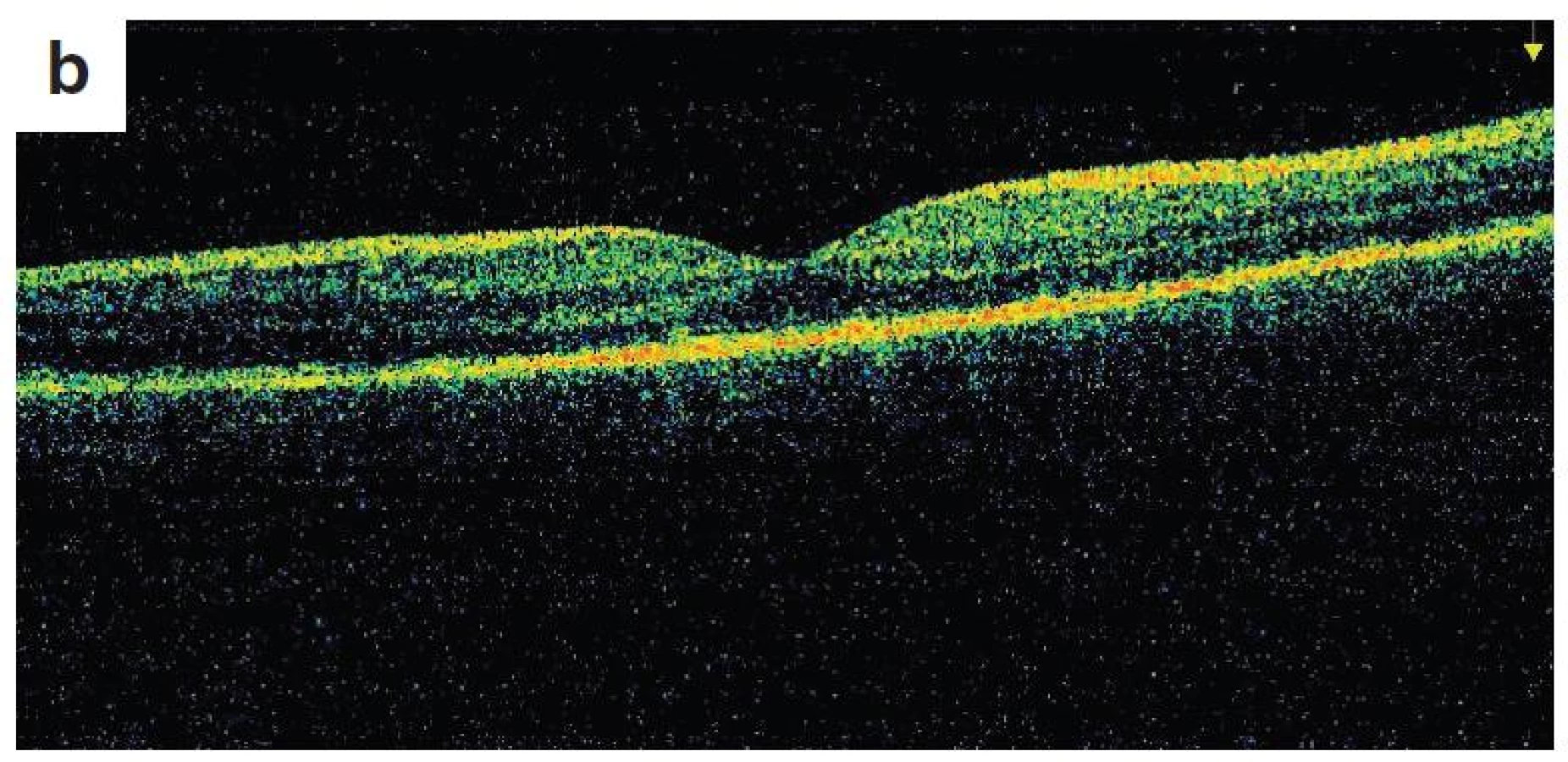
Po 3 mesiacoch od prodrómov ochorenia je CZO 5/5 obojstranne, v klinickom obraze recidíva prednej uveitídy, bez prejavov zadnej uveitídy, pretrváva edém TZN, pacient ponechaný na celkovej kortikoidnej liečbe v dávke 10 mg/deň.
Po 6 mesiacoch od začiatku ochorenia pacient v klinickom obraze bez prejavov prednej uveitídy, pri fundoskopickom vyšetrení zistený vznik juxtapapilárnej choroidálnej neovaskulárnej membrány (CNVM) pri temporálnom okraji TZN obojstranne. Jej prítomnosť bola potvrdená FAG a OCT vyšetrením. CZO bola 5/5 obojstranne, na perimetri rozšírená slepá škvrna (obr. 8).

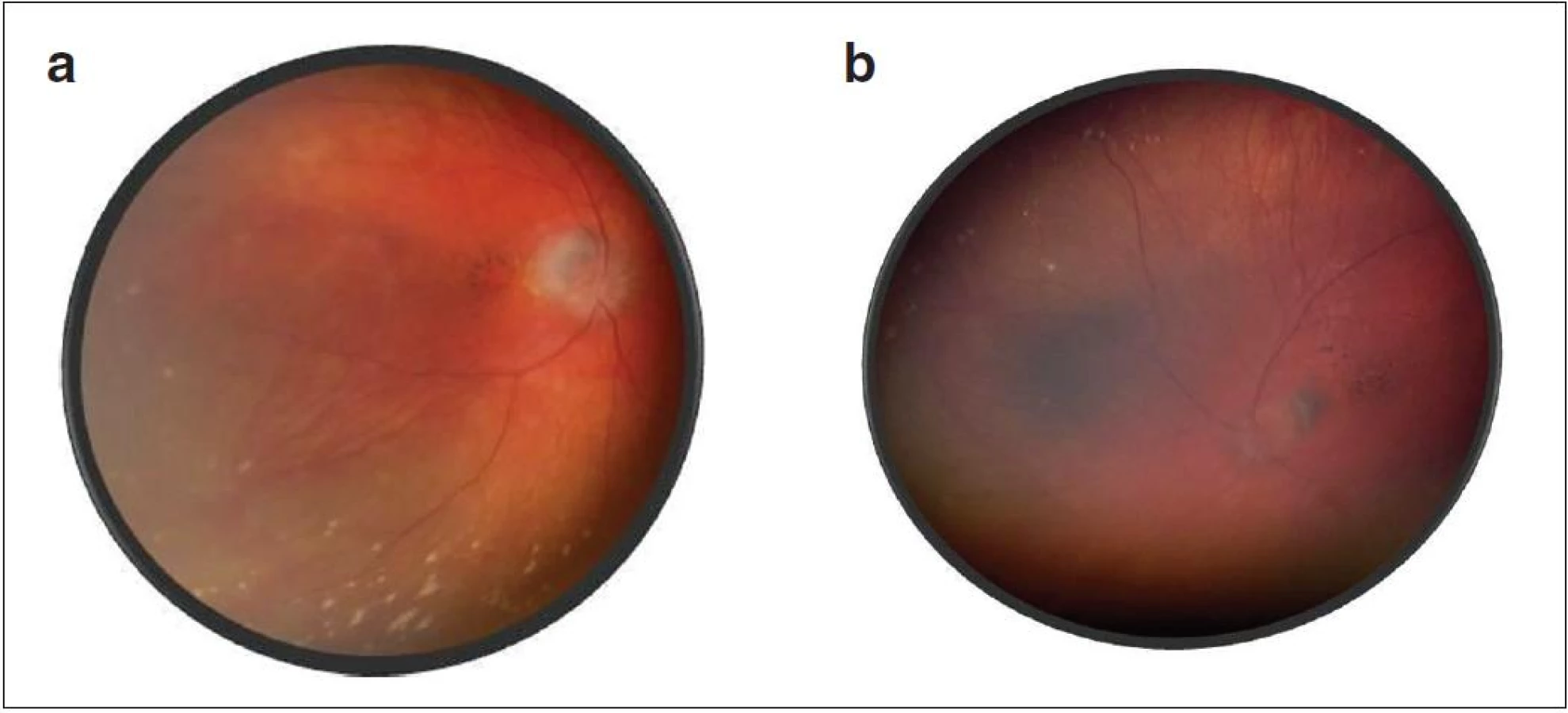
Po 10 mesiacoch od začiatku ochorenia bol pacient bez prejavov prednej uveitídy. Pri fundoskopickom vyšetrení bola zistená progresia juxtapapilárnej CNVM pri temporálnom okraji TZN obojstranne, ktorá zasahovala do makuly (obr. 9a, b). Vo FAG obraze bola zistená na oboch očiach peripapilárne temporálne od terča sa šíriaca hyperfluorescencia zasahujúca oblasť makuly s oneskoreným presakovaním (obr. 10). V OCT obraze bola zistená juxtapapilárna CNVM so submakulárnou tekutinou s centrálnou hrúbkou makuly (CHM) 381 µm na pravom oku a 364 µm na ľavom oku. (obr. 11a, b). CZO bola 5/10 obojstranne, v perimetrickom náleze bol paracentrálny skotómom s rozšírenou slepou škvrnou. Preto sme po udelení informovaného súhlasu zákonného zástupcu pacientovi aplikovali 1 injekciu anti-VEGF-bevacizumab intravitreálne v dávke 0,75 mg/0,1 ml do pravého oka a 1,25 mg/0,1ml do ľavého oka. Následne do 1 mesiaca došlo k úprave CZO na 5/5 na pravom oku a 5/7,5 na ľavom oku s regresiou juxtapapilárnej CNVM viditeľnej na FAG aj OCT vyšetrení (CHM 247/264 µm), avšak pretrváva juxtapapilárna subretinálna fibróza.


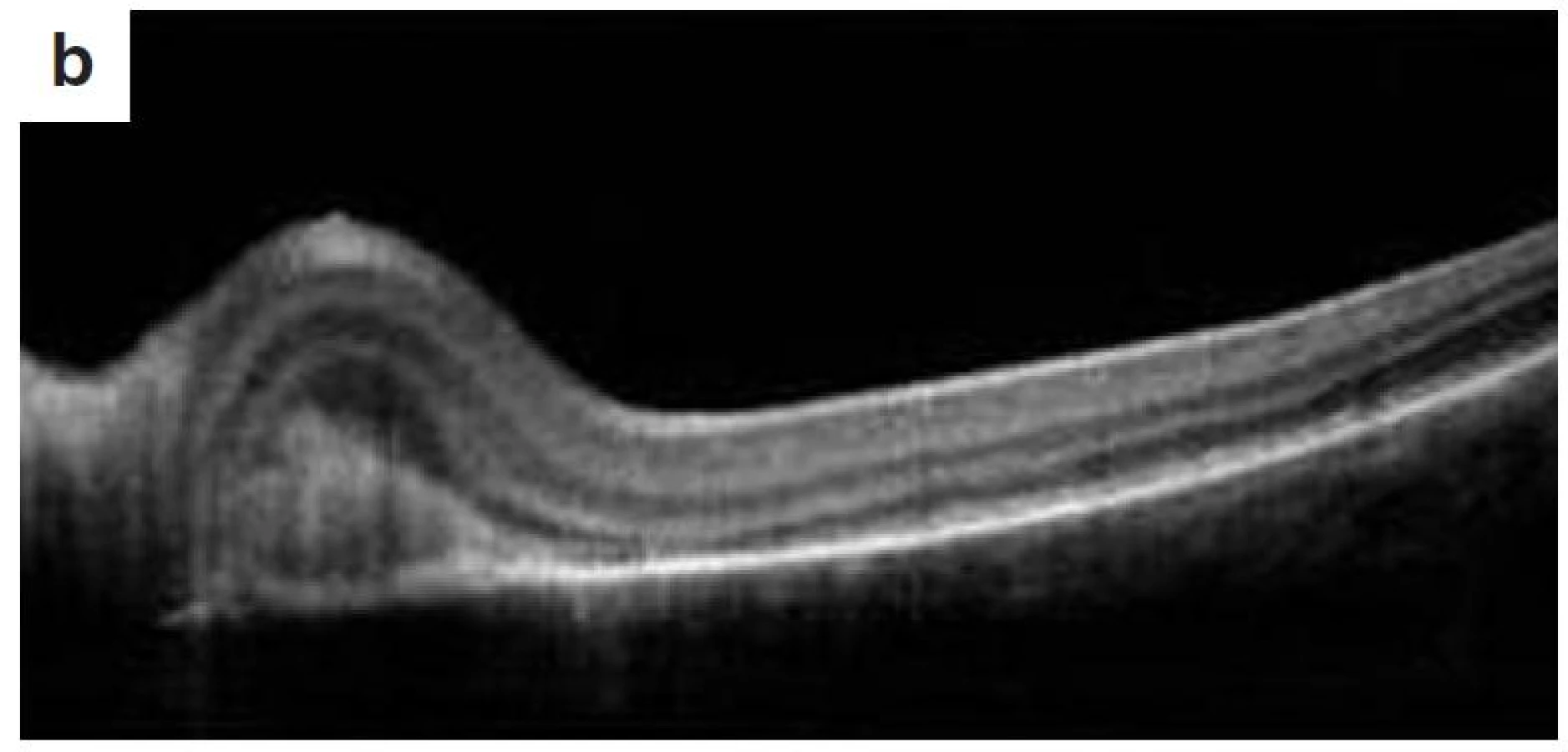
Po 11 mesiacoch od začiatku ochorenia bola ukončená celková liečba prednizónom.
V 17. a 23. mesiaci od začiatku ochorenia sa objavili dve recidívy prednej uveitídy obojstranne, so vznikom zadných synechií (obr. 12), čo zodpovedá recidivujúcemu štádiu ochorenia. Neboli zistené prejavy recidívy zadnej uveitídy. Zápal bol pri prvej recidíve zvládnutý lokálnou kortikoidnou liečbou a pri druhej recidíve perorálnou kortikoidnou liečbou prednizónom v dávke 1 mg/kg s postupným znižovaním po dobu 2 mesiacov. V súčasnosti po 32 mesiacoch od vzniku ochorenia je CZO 5/5 na pravom oku a 5/7,5 na ľavom oku. Pacient je bez známok prednej a zadnej uveitídy obojstranne. U pacienta nebol zistený glaukóm ani katarakta. Avšak u pacienta sú chronické zmeny na prednom a zadnom segmente a pretrváva zneostrenie TZN pre nález juxtapapilárnej subretinálnej fibrózy, v zornom poli pretrváva paracentrálny skotóm v mieste slepej škvrny. Centrálna hrúbka makuly je 249 µm na pravom oku a 255 µm na ľavom oku. Pacient naďalej ostáva v našom sledovaní.

Diskusia
Vogt (1906), Koyanagi ( 9), Harada ( 6) – nezávisle popísali niekoľko pacientov s obojstrannou uveitídou, exsudatívnou amóciou sietnice, s neurologickými abnormalitami a s ochoreniami kože. Napriek rozdielnostiam jednotlivých prípadov a predpokladu, že sa jedná o rôzne ochorenia, následne iní autori dospeli k záveru, že ochorenie by sa malo nazvať Vogt-Koyanagi-Harada syndróm. Medzinárodná komisia pre nomenklatúru stanovila kritéria pre diagnózu VKH choroby. Revidované kritéria definujú 3 kategórie ochorenia: kompletná VHK, nekompletná VKH a pravdepodobná VKH choroba (4, 20) (tab. 1). Náš pacient spĺňal kritériá pre pravdepodobnú VKH chorobu.

Patogenéza ochorenia je neznáma, zápal a strata melanocytov boli zistené vo viacerých orgánoch – v koži, vnútornom uchu, meningoch a uvei. Na základe histopatologických nálezov sa predpokladá infekčný alebo autoimúnny základ ochorenia. VKH choroba sa najčastejšie vyskytuje u pacientov s genetickou predispozíciou k ochoreniu. Bola zistená asociácia s HLA - DRB1, HLA-DR4, HLA-DR5 a HLA-DQ4 (13).
Frekvencia výskytu VKH choroby je nízka, najčastejšie je postihnutá tmavo pigmentovaná rasa. Pigmentácia kože sama nie je predisponujúcim faktorom v patogenéze ochorenia. Ženy sú postihnuté častejšie ako muži. Ochorenie postihuje ľudí medzi 20.–50. rokom, najčastejšie v tretej dekáde, výskyt u detí je raritný. V literatúre je zaznamenaných niekoľko prípadov výskytu u detí (1, 3, 7, 10, 15, 18, 21, 23).
Neurologické symptómy môžu perzistovať týždne, obvykle po kortikoidoch odoznievajú, kožné prejavy napriek liečbe pretrvávajú, sluchové ťažkosti odoznievajú po kortikoidnej liečbe po týždňoch až mesiacoch.
Klinická manifestácia – rozoznávame 4 štádia ochorenia (tab. 2).

Diagnostika ochorenia sa zakladá na klinickom obraze a symptómoch, neexistujú laboratórne testy špecifické pre toto ochorenie. Nápomocné v diagnostike sú FAG, ICG, OCT, USG, MRI, vyšetrenie likvoru (CSF), elektrofyziologické testy, audiometria. HLA typizácia nie je pre diagnózu významná a preto sa rutinne nedoporučuje.
Účelom liečby je zvládnutie uveitídy a prevencia komplikácií. Liečba VKH ochorenia systémovými kortikoidmi má byť včas naštartovaná a agresívna – metylprednizolón do 1 g/d intravenózne niekoľko dní, u detí 10 mg/kg/d, následne prednizón 1 mg/kg/d perorálne, dĺžka a znižovanie dávky individuálne, niekedy 6–12 mesiacov, avšak nie kratšie ako 3 mesiace pre riziko recidívy (2, 12, 19). Toto doporučenie sme rešpektovali aj my. U pacientov nereagujúcich na kortikoidy, alebo u ktorých liečba vyvolala nežiadúce účinky je doporučená imunomodulačná liečba – metotrexát, cyklosporín, infliximab, takrolimus, azathioprin, cyklofosfamid (10, 24).
Liečba u detí detí so syndrómom VKH je náročná. V literatúre boli popísané rôzne modely liečby s rôznym efektom, stále nie je určený definitívny liečebný model a liečba je u detských VKH prípadoch zvyčajne individuálna (3). V predchádzajúcich publikáciách boli u detí kortikosteroidy najúčinnejšie látky v liečbe syndrómu VKH, zatiaľ čo kombinácie terapie s cyklosporínom, metotrexátom alebo azathioprínom boli použité s priaznivými výsledkami v refraktérnych prípadoch (7). V našom prípade bolo ochorenie zvládnuté systémovou kortikoidnou liečbou, na ktorej bol pacient ponechaný 11. mesiacov.
V liečbe komplikácií je najčastejšie nutné riešiť sekundárny glaukóm, kataraktu, ktoré sa u nášho pacienta nevyskytli a choroidálne neovaskulárne membrány (CNVM). Choroideálne neovaskularizácie sa vyskytujú v 15 % u pacientov s VKH a sú asociované so zlou zrakovou prognózou (25). Bevacizumab je humánna monoklonálna protilátka, ktorá sa viaže ku všetkým podtypom cievneho endoteliálneho rastového faktora (VEGF). Bol úspešne použitý pri liečbe sekundárnej CNVM pri rôznych ochoreniach. Intravitreálne podaný bevacizumab je účinný liek na liečbu CNVM na očiach so syndrómom VKH (8,17,18,22). Podobný efekt v liečbe CNVM na očiach so syndrómom VKH bol dosiahnutý aj pri intravitreálnom podaní ranibizumabu (11). V našom prípade bol pacient liečený pre komplikovanú juxtapapilárnu CNVM na oboch očiach bevacizumabom. Jedno intravitreálne podanie bevacizumabu bolo postačujúce a stabilizovalo CZO. Avšak pretrvávajúca subretinálna fibróza je príčinou paracentrálneho skotómu v zornom poli na oboch očiach.
Kortikosteroidy sú používané pre angiostatické a antipermeabilné vlastnosti. Imunosupresiou je nutné tlmiť chronický zápalový proces. Kombinácia intravitreálne aplikovaného anti-VEGF a kortikosteroidu spolu so systémovou imunosupresívnou liečbou bola efektívna v liečbe zápalovej CNVM bez závažnejších systémových alebo očných vedľajších účinkov (22).
Laserová fotokoagulácia, fotodynamická liečba, chirurgická excízia a kortikosteroidná liečba boli použité na liečbu sekundárne vzniknutej CNVM pri VKH s rôznym stupňom úspechu (5, 6, 9, 16).
Vizuálna prognóza VKH u dospelých tak aj u detí je všeobecne priaznivá. Klinický nález pri manifestácii ochorenia, interval medzi objavením sa prvých príznakov a začiatkom liečby, opakovanie zápalu, vývoj komplikácií, používanie intravenóznych kortikosteroidov a spôsob znižovania systémových kortikosteroidov sú významné prognostické faktory.
Záver
VKH syndróm je závažné ochorenie, ktoré raritne postihuje i deti. Pokiaľ nie sú prítomné extraokulárne príznaky, diagnostika môže byť komplikovaná a liečba v akútnej fáze vyžaduje neodkladnú bolusovú kortikoidnú liečbu. VKH syndróm vyžaduje monitorovanie po dlhé roky pre svoj chronický charakter. Prognóza quo ad visum je u detských pacietov priaznivá. Pokles zrakovej ostrosti u pacientov s VKH syndrómom je často zapríčinený komplikáciami ako katarakta, glaukóm a choroidálna neovaskularizácia. Choroidálna neovaskularizácia je hlavná príčina neskoršej straty zraku.
Do redakce doručeno dne 15. 2. 2013
Do tisku přijato dne 27. 5. 2013
MUDr. Beáta Bušányová
Klinika detskej oftalmológie DFNP-LF UK
Limbová 1
833 40 Bratislava
e-mail: b.busany@pobox.sk
Sources
1. Al Hemidan A.I., Tabbara K.F., Althomali T.: Vogt-Koyanagi-Harada associated with diabetes mellitus and celiac disease in a 3-year-old girl. Eur J Ophthalmol, 16; 2006, 1 : 173-7.
2. Benfdil N., Baha Ali T., Jellab B. et al.: Vogt Koyanagi Harada syndrome in children: diagnosis and management. Bull Soc Belge Ophtalmol, 2010; 314 : 15-8.
3. Berker N., Ozdamar Y., Soykan E. et al.: Vogt-Koyanagi-Harada syndrome in children: report of a case and review of the literature. Ocul Immunol Inflamm, 15; 2007, 4 : 351-7.
4. Da Silva, F.T., Damico, F.M., Marin, M.L. et al.: Revised diagnostic criteria for Vogt-Koyanagi Harada disease: considerations on the different disease categories. Am J Ophthalmol, 147; 2009, 2 : 339–345.
5. Farah M.E., Costa R.A., Muccioli C. et al.: Photodynamic therapy with verteporfin for subfoveal choroidal neovascularization in Vogt-Koyanagi-Harada syndrome. Am J Ophthalmol, 2002; 134 : 137–139.
6. Foster R.E., Knight C.D., Lowder C.Y.: Subfoveal choroidal neovascular membrane excision in Vogt-Koyanagi-Harada syndrome. Retina, 2000; 20 : 547–549.
7. García L.A., Carroll M.O., Garza León M.A.: Vogt-Koyanagi-Harada syndrome in childhood. Int Ophthalmol Clin, 48; 2008, 3 : 107-17.
8. Julián K., Terrada C., Fardeau C. et al.: Intravitreal bevacizumab as first local treatment for uveitis-related choroidal neovascularization: long-term results. Acta Ophthalmol, 89; 2011, 2 : 179-84.
9. Ketata, A., Ben Zina, Z., Hajji, D. et al.: Two cases of subretinal neovascular membrane in Vogt-Koyanagi-Harada syndrome. J Fr Ophtalmol, 2006, 29 : 302–306.
10. Khalifa Y.M., Bailony M.R., Acharya N.R.: Treatment of pediatric Vogt-Koyanagi-Harada syndrome with infliximab. Ocul Immunol Inflamm, 18; 2010, 3 : 218-22.
11. Kolomeyer, A.M., Roy, M.S., Chu, D.S.: Case Report The Use of Intravitreal Ranibizumab for Choroidal Neovascularization Associated with Vogt-Koyanagi-Harada Syndrome. Case Reports in Medicine, Volume 2011, Article ID 747648, 3 pages, doi:10.1155/2011/747648
12. Lai T.Y., Chan R.P., Chan C.K. et al.: Effects of the duration of initial oral corticosteroid treatment on the recurrence of inflammation in Vogt-Koyanagi-Harada disease. Eye, 23; 2009, 3 : 543-8.
13. Lucena D.R., Paula J.S., Silva G.C. et al.: Incomplete Vogt-Koyanagi-Harada syndrome associated with HLA DRB1*01 in a 4-year-old child: case report. Arq Bras Oftalmol, 70; 2007, 2 : 340-2.
14. Maezawa, N., Yano, A., Taniguchi, M. et al.: The role of cytotoxic T lymphocytes in the pathogenesis of Vogt - Koyanagi-Harada disease. Ophthalmologica, 185; 1982, 3 : 179–186.
15. Martin T.D., Rathinam S.R., Cunningham E.T. Jr.: Prevalence, clinical characteristics, and causes of vision loss in children with Vogt-Koyanagi-Harada disease in South India. Retina, 30; 2010, 7 : 1113-21.
16. Nowilaty, S.R., Bouhaimed, M.: Photodynamic therapy for subfoveal choroidal neovascularisation in Vogt-Koyanagi-Harada disease. Br J Ophthalmol, 2006; 90 : 982–986.
17. Park H.S., Nam K.Y., Kim J.Y.: Intravitreal bevacizumab injection for persistent serous retinal detachment associated with Vogt-Koyanagi-Harada disease. Graefes Arch Clin Exp Ophthalmol, 249; 2011, 1 : 133-6.
18. Raffa L., Bawazeer A.: Intravitreal bevacizumab injection in a 14-year-old Vogt-Koyanagi-Harada patient with choroidal neovascular membrane. Can J Ophthalmol, 44; 2009, 5 : 615-6.
19. Read, R.W., Yu F., Accorinti, M. et al.: Evaluation of the effect on outcomes oftheroute of administration of corticosteroids in acute Vogt-Koyanagi-Harada disease. Am J Ophthalmol, 142, 2006, 1 : 119 – 24.
20. Reed, R.W., Holland, G.N., Rao N.A. et al.: Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international committee on nomenclature. Am J Ophthalmol, 131; 2001, 5 : 647 – 52.
21. Setiabudiawan, B., Karfiati, F., Ghrahani, R. et al.: Vogt-Koyanagi-Harada disease in an 8-year-old boy. 1; 2011, 2 : 98–103.
22. Sivakami, A.P., Sudhira, P.H.: Management of recurrent inflammatory choroidal neovascular membrane secondary to Vogt-Koyanagi-Harada syndrome, using combined intravitreal injection of bevacizumab and triamcinolone acetate, Indian J Ophthalmol, 60; 2012, 6 : 551–552.
23. Soheilian, M., Aletaha, M., Yazdani, S. et al.: Management of pediatric Vogt-Koyanagi-Harada (VKH)-associated panuveitis. Ocul Immunol Inflamm, 2006; 14 : 91–98.
24. Wang Y., Gaudio P.A.: Infliximab therapy for 2 patients with Vogt-Koyanagi-Harada syndrome. OculImmunol Inflamm, 16; 2008, 4 : 167–71.
25. Wu L., Evans T., Saravia M. et al.: Intravitreal bevacizumab for choroidal neovascularization secondary to Vogt-Koyanagi-Harada syndrome. Jpn J Ophthalmol, 53; 2009, 1 : 57-60.
Labels
OphthalmologyArticle was published in
Czech and Slovak Ophthalmology

2013 Issue 2
Most read in this issue
- Myopia or Hyperopia?
- Vogt-Koyanagi-Harada Syndrome in Children – a Case Report
- The Importance of Angle Kappa for Centration of Multifocal Intraocular Lenses
- Contraception and Ocular Thromboembolic Episodes – A Case Report