
Moderní instrumentální metody studia isoflavonů
Modern instrumentation for the studies of isoflavones
Isoflavones belong to the natural substances exhibiting a number of physiological effects in living organisms. The substances are synthesized in plant tissues as protective agents against biotic stress (i.e. bacterial infection). Isoflavones are also an important dietary constituent in human nutrition. The review discusses modern trends in the studies of isoflavones in plant materials and foodstuffs and procedures for chemical analyses of isoflavones in human body fluids and plant tissues. Highly effective extraction and purification techniques, i.e. solid–phase extraction (SFE), accelerated–solvent extraction (ASE), and Soxhlet extraction, are presented. Latest procedures in chromatographic separation of isoflavones that apply different types of stationary phases are described. Immunochemical analysis, electrochemical sensing of isoflavones, spectrometric and other analytical techniques and their applications are also mentioned. Special attention is focused on a highly selective and sensitive technique of mass spectrometry and its application for identification of isoflavones and their glucosides in plants. Studies of interactions of isoflavones with cell receptors and a number of biologically active substances such as DNA and proteins are described. The reason for the presentation of the review was not to give a full overview of the presented topics but mainly to show modern and the most recent methods in the studies of isoflavones.
Key words:
chromatography – isolation – mass spectrometry – phytoestrogens – human health
Authors:
J. Vacek 1,2; B. Klejdus 2; L. Lojková 2; V. Kubáň 2
Authors‘ workplace:
Akademie věd České republiky, Biofyzikální ústav, Brno
1; Mendelova zemědělská a lesnická univerzita Brno, Ústav chemie a biochemie
2
Published in:
Čes. slov. Farm., 2008; 57, 85-94
Category:
Review Articles
Overview
Isoflavony jsou přírodní látky s řadou fyziologických účinků na živé organismy a představují významný doplněk ve výživě člověka. V předkládaném přehledu jsou popsány moderní instrumentální přístupy ke studiu isoflavonů v rostlinných materiálech a potravinářských produktech, jakož i vhodné techniky chemické analýzy isoflavonů v lidských tělních tekutinách. Článek je zaměřen na chromatografickou analýzu isoflavonů. Jsou rovněž diskutovány efektivní techniky izolace, jako je extrakce pevnou fázi, extrakce kapalinou v nadkritickém stavu nebo Soxhletova extrakce. Jsou zde zmíněny i aplikace některých dalších analytických technik, jako je imunochemická analýza, spektrometrické (především hmotnostní spektrometrie) a elektrochemické metody. Část textu je věnována studiu interakcí isoflavonů s celou řadou biologicky aktivních látek, jako je DNA a proteiny. Cílem práce není podat ucelený přehled dané problematiky, spíše poukázat na moderní přístupy ve studiu isoflavonů.
Klíčová slova:
chromatografie – hmotnostní spektrometrie – izolace – fytoestrogeny – zdraví člověka
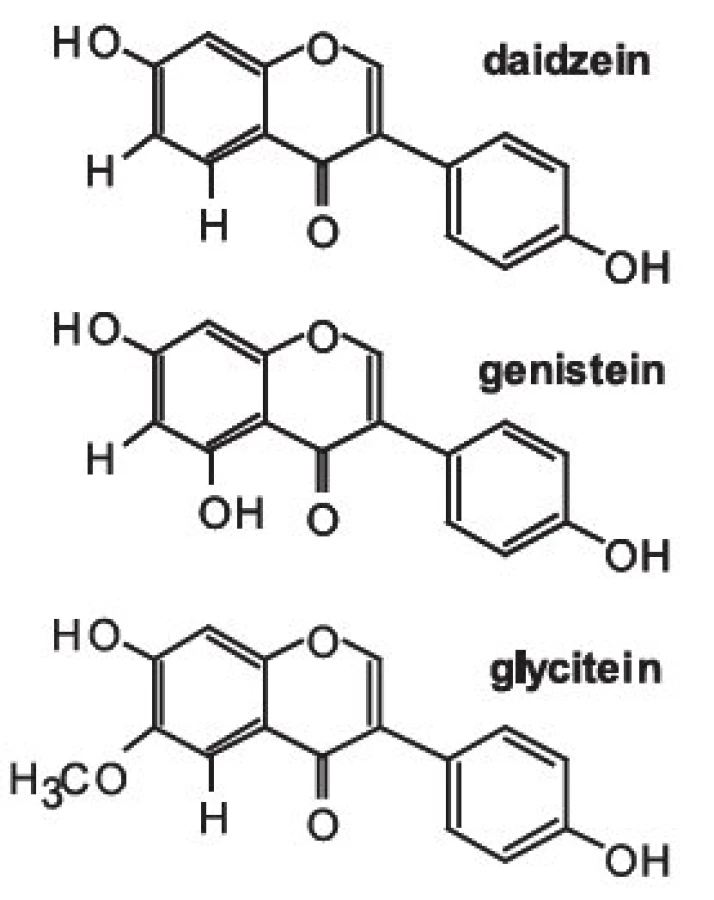
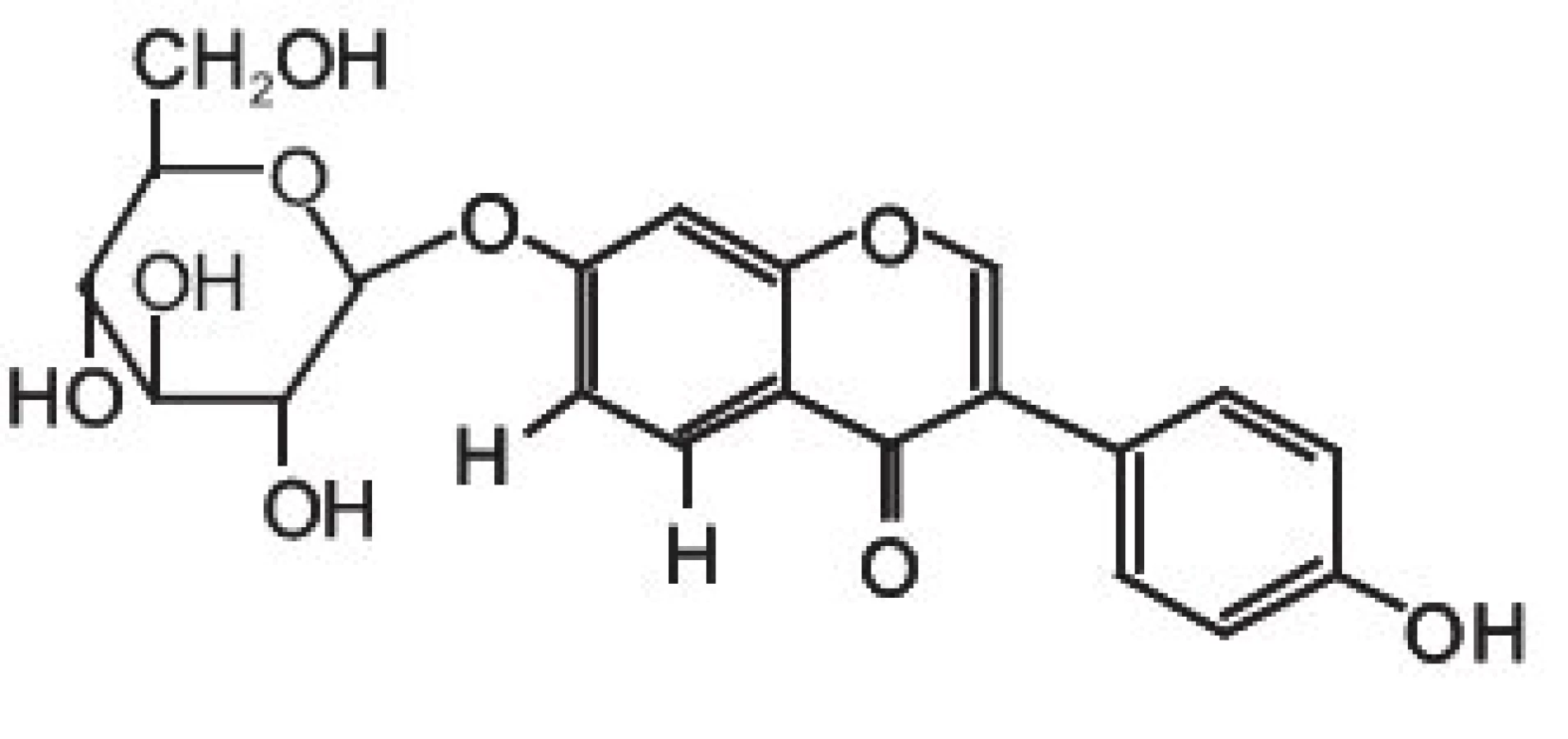
Isoflavony patří společně s flavanony, flavonoly, flavony, katechiny a anthokyanidiny do rozsáhlé skupiny přírodních látek označovaných jako flavonoidy. Vzorce tří vybraných isoflavonů (ve formě aglykonů) jsou zobrazeny na obrázku 1. Rostliny syntetizují isoflavony, ale i celou řadu dalších polyfenolických látek jako „obranné sloučeniny“ při napadení rostlinných pletiv bakteriálním, nebo jiným infekčním agens. V rostlinných pletivech se isoflavony často vyskytují ve formě ß-glykosidů, popřípadě jejich derivátů (strukturní vzorec daidzinu je na obrázku 2).
Jedná se o sekundární rostlinné metabolity (polyfenoly) vykazující celou řadu fyziologických účinků v těle člověka a dalších savců. Isoflavony našly své uplatnění při léčbě rakoviny a kardiovaskulárních poruch 1, 2). Kromě pozitivního vlivu isoflavonů na lidské zdraví byly zveřejněny publikace o jejich toxických účincích, které souvisí s podanou dávkou a celou řadou dalších faktorů 3). Některé isoflavony vykazují estrogenní aktivitu (podobné účinky jako estradioly), čehož je využíváno při potlačení některých symptomů menopauzy a v léčbě dalších hormonálně podmíněných poruch. Kromě isoflavonů řadíme do této skupiny i některé další flavonoidy, jako jsou lignany 4).
Potřebný příjem isoflavonů lidským organismem lze zajistit konzumací potravinářských produktů a případně i léčiv připravených z luštěnin a dalších zdrojů bohatých na tyto látky. Mezi nejrozšířenější zdroje isoflavonů patří sója, hrách, vojtěška nebo jetel a další zástupci čeledi Fabaceae.
Vzhledem ke svému širokému uplatnění v humánní i veterinární medicíně je studium isoflavonů a vývoj nových analytických postupů pro jejich identifikaci a stanovení v rostlinách předmětem zájmu mnoha vědecko-výzkumných týmů. O tom svědčí i skutečnost, že v roce 2006 bylo publikováno kolem 500 prací obsahujících klíčové slovo „isoflavones“ a přes 1700 prací zaměřených na problematiku flavonoidních látek (zdroj: www.wos.cz). V současné době je pozorován výrazný pokrok v instrumentaci pro izolaci, separaci a identifikaci isoflavonů. Tato instrumentace nalézá uplatnění pro analýzu polyfenolických látek nejen v luštěninách, ale především v jiných rostlinných čeledích, u kterých se isoflavony vyskytují ve výrazně nižších koncentracích. Problematice stanovení flavonoidů, resp. fytoestrogenů, nebo konkrétně isoflavonů, bylo věnováno v nedávné době hned několik souhrnných prací 5–8).
V této práci je popsána problematika izolace isoflavonů z rostlinných materiálů a potravinářských produktů pomocí moderních extrakčních metod a jejich separace, kvantifikace a identifikace pomocí vybraných separačních chromatografických a elektromigračních technik. Část textu je věnována nejnovějším pokrokům v detekci isoflavonů a jejich identifikaci UV-Vis spektrofotometrií, hmotnostní spektrometrií, imunoanalytickými a elektrochemickými metodami a dalšími technikami. Cílem práce není podat ucelený přehled dané problematiky, spíše poukázat na moderní metody, které je možné využít pro studium isoflavonů.
Výskyt isoflavonů v rostlinných zdrojích
Základní chemickou strukturou isoflavonů je různě substituovaný 3-fenyl-chromen-4-on. Mezi nejjednodušší isoflavony patří aglykony jako daidzein (7,4--dihydroxyisoflavon), genistein (5,7,4-trihydroxyisoflavon) a glycitein (7,4-dihydroxy-6-methoxyisoflavon). V rostlinných pletivech však převládají jejich cukerné konjugáty (obr 2.), zatímco aglykony se v nich vyskytují v poměrně nízkých koncentracích 9). Například v sóji převládají konjugáty genisteinu a daidzeinu, jako je genistin (genistein-7-O-ß-D-glukosid), 6-O-malonyl--genistin, daidzin (daidzein-7-O-ß-D-glukosid) a 6-O--malonyl-daidzin a konjugáty glyciteinu. Další významný zdroj isoflavonů – jetel luční – obsahuje převážně formononetin a biochanin A (methylderiváty daidzeinu a genisteinu), jejichž výskyt je významně ovlivňován kultivačními a klimatickými podmínkami, za kterých jsou rostliny pěstovány.
Isoflavony byly také identifikovány v čeledích Convolvulaceae, Cyperaceae, Brassicaceae a v mnoha dalších 10). S rozvojem vysoce efektivních izolačních postupů a velmi citlivých detekčních technik jsou v současnosti identifikovány další isoflavony v různých taxonomických skupinách. Mezi nejnovější objevy patří například identifikace glaziovianinu A v pletivech Ateleia glazioviana, u kterého byla prokázána cytotoxická aktivita k lidským HL-60 leukemickým buňkám 11). Nové isoflavony byly také nalezeny v oddencích Belamcanda chinensis 12) a nové deriváty byly objeveny v kořenech Hedysarum scoparium 13). Detailnější informace o výskytu isoflavonů napříč rostlinnou říší a jejich využití v chemotaxonomii lze najít v publikacích 10, 14).
Stabilita isoflavonů
Stabilita rostlinných přírodních látek je limitována celou řadou fyzikálních (světlo, teplota) a chemických faktorů (pH, iontová síla atd.), což musí být zohledněno při přípravě a manipulaci se vzorky pro analýzu. Poznatky o stabilitě isoflavonů mají neoddiskutovatelný význam pro přípravu a skladování potravin a stabilizaci léčiv, které je obsahují.
Flavonoidní látky podléhají fotodegradaci. U roztoků daidzeinu a formononetinu v různých organických rozpouštědlech (acetonitril, methanol, etanol, hexanol) byla z poklesu absorbance v absopčním maximu kolem 250 nm po jejich rozpuštění pozorována na světle jejich degradace 15). Z těchto důvodů je vhodné skladovat standardy, vzorky i preparáty obsahující isoflavony ve tmě. Podobně bylo prokázáno, že také teplota má vliv na stabilitu isoflavonů. Mathias et al. 16) studovali stabilitu malonyl - a acetyl - cukerných konjugátů daidzinu a genisteinu při 25, 80 a 100 °C a při různých hodnotách pH. Z výsledků vyplývá, že uvedené ß-glukosidy a jejich deriváty podléhají degradaci (alkalické hydrolýze) až při vysokých hodnotách pH (konkrétně pH 10), přičemž při běžné teplotě 25 °C k degradaci vzorků isoflavonů nedochází.
Stabilita ß-glukosid-malonátů formononetinu a biochaninu A byla také studována 17) v extraktech jetele lučního (T. pratense) technikou RP-HPLC ve spojení s hmotnostním, UV-Vis a fluorescenčním detektorem, přičemž při zvýšené teplotě (83 °C) docházelo k hydrolýze konjugátů až na aglykony. Stabilita cukerných konjugátů v extraktech je primárně ovlivněna teplotou skladování vzorků, jelikož dochází k jejich hydrolýze na aglykony, čemuž lze z velké části zabránit jejich zmrazením.
Teplota je významným faktorem i při zpracování potravin obsahujících isoflavony, kdy například u tepelného zpracování sojového mléka byl pozorován výrazný pokles obsahu genistinu 18). Výše popsané poznatky, týkající se hydrolýzy isoflavonů 19),mají proto značný význam při přípravě vzorků pro analýzu a technologii zpracování potravin a léčiv.
Izolace isoflavonů
Účinná a selektivní isolace cílových analytů ze složitých přírodních matric je jedním z nejdůležitějších kroků na cestě od původního vzorku k analytickému stanovení. Cílem je získat analyty ze vzorku a koncentrovat je v malém množství kapaliny vhodné pro následnou analytickou metodu. Dobrá extrakční metoda by neměla být příliš časově náročná (nejvýše desítky minut). Z hlediska selektivity extrakce je třeba, aby látky extrahované zároveň s analyty s nimi neinterferovaly, a nezkreslovaly tak výsledky. Zcela zásadní je pro dobrou extrakční metodu vysoká výtěžnost.
Dříve běžně používaná kapalinová extrakce je v současné době pro extrakce z rostlinných materiálů nahrazována dvoufázovou kapalinovou extrakcí 20). Pro preparativní účely je využíván také refluxní kapalinový systém 21) methanol/H2O (80 : 20 v/v) a kapalinová extrakce směsným rozpouštědlem (10% n–butanolu s 90% octanu ethylnatého) s výtěžky cca 70 %. U některých typů matrice se dosud využívají i metody s nižšími výtěžky (např. 60 % u extrakcí z krysích tkání), pokud mají dobrou reprodukovatelnost 22).
Kapalinová extrakce v ultrazvukové lázni značně snížila časovou náročnost metod, např. z dvaceti na šest hodin u šalvěje 23). Ultrazvuk o vysoké intenzitě se používá pro extrakci isoflavonů a olejů ze sojových bobů do hexanu, isopropanolu nebo do směsi hexan/isopropanol 3 : 2 24). Nejlepšího výtěžku bylo dosaženo u tříhodinové extrakce (62,3 %). Ze sojových bobů, pomletých v tekutém dusíku, lze v ultrazvuku téměř kvantitativně (výtěžek 80–90 %) vyextrahovat isoflavony: daidzin, glycitin, genistin a malonylgenistin za pouhých 20 min do 50% ethanolu při 60 °C 25). Delší doba extrakce v tomto případě vedla k poklesu výtěžku. Metoda se používá i pro rutinní analýzu 26).
Účinnost přes 90 % vykazuje extrakce daidzeinu, genisteinu, formononetinu a biochaninu A na pevnou fázi (SPE) na patronách Speed ABN 27). Metoda se používá pro rychlé stanovení isoflavonů v rostlinách, potravinách a dalších biologických matricích a byla dále rozpracována na dvoudimenzionální extrakci na pevnou fázi (2D-SPE), která umožňuje současně extrahovat dvacet isoflavonů: čtrnáct glykosidmalonátů a šest acetylglykosidů z jetele 28). Zvláštní pozornost byla věnována specifickým případům koeluce dvou nebo více analytů, což způsobovalo potíže při stanovení sekundárních metabolitů v rostlinné matrici. I pro tento případ bylo nalezeno vhodné řešení 29).
Sedm základních isoflavonů bylo kvantitativně vyextrahováno z různých potravin na bázi sojových bobů 30) extrakcí kapalinou za zvýšené teploty a tlaku (accelerated solvent extraction, ASE). Podle srovnávací studie uvedené v této práci je každá extrakční metoda vhodná pro jinou skupinu látek. ASE v kombinaci s ultrazvukem a SPE předčištěním byla použita 31) pro extrakci a izolaci aglykonů a glykosidů isoflavonů z různých částí sóji s výtěžkem 96–106 %.
Další rychlou a spolehlivou metodou pro extrakci isoflavonů ze sóje je mikrovlnná extrakce (MAE) 32). Výtěžku 97–103 % při extrakci malonyl - a acetylderivátů daidzinu, glycitinu a genistinu a daidzinu, glycitinu, genistinu, daidzeinu, glyciteinu a genisteinu do 50% ethanolu bylo dosaženo při 50 °C již za 20 min. Srovnávací studie extrakčních metod 33) prokázala, že nejvhodnějšími metodami pro stanovení celkového obsahu isoflavonů v burácích jsou MAE a Soxtec, zatímco ultrazvuk a kapalinová extrakce se nejlépe hodí pro stanovení spektra a relativního zastoupení isoflavonů v matrici (aglykonových a glykosidových konjugátů).
Extrakce kapalinou v nadkritickém stavu (supercritical fluid extraction – SFE) se používá pro analytické účely i k přípravě funkčních potravin 34), např. k izolaci sójového proteinu (SPI). Takto připravený sójový extrakt (při 35 MPa a 45 °C, čas 120 min) obsahuje 640 mg isoflavonů na 100 g, přičemž 83,7 % isoflavonů tvoří glycitin, daidzin a genistin. SFE s kontinuálním (on-line) dodáváním modifikátoru do proudu extrakčního média 35) byla použita pro extrakci daidzinu, glycitinu, genisteinu, ononinu, daidzeinu, glyciteinu, sissotrinu, formononetinu a biochaninu A z jetele. Podle srovnávací studie výtěžnosti daidzinu a genisteinu je SFE mírně účinnější než ASE, avšak efektivnější než IKA Soxhlet nebo ultrazvuk.
V této oblasti v zásadě převládají dva základní přístupy: 1. kombinace několika metod přinášející vysoký výtěžek a selektivitu při malém objemu extraktu a 2.výběr jedné ne zcela kvantitativní, ale reprodukovatelné a nepříliš pracné metody, kterou je možné z větší části automatizovat. V prvním případě se jako optimální jeví spojení ultrazvuku nebo některé z metod schopných uvolnit analyty z matrice a následné přečištění SPE. Předzpracování vzorku v ultrazvukové lázni příznivě ovlivňuje matrici a urychluje následný přenos hmoty z matrice do extrakčního média. Po aplikaci ultrazvuku je třeba použít rozpouštědlo s dobrými solvatačními schopnostmi a dostatečnou difuzivitou (schopností difundovat dovnitř matrice), které uvolní analyt z aktivních center matrice – zejména metody, jako jsou SFE, MAE, ASE, nebo různě modifikované Soxhletovy extrakce. Surový extrakt pak zpracujeme SPE, která v koloně zadrží případné nežádoucí ko-extraktanty a zároveň sníží objem extraktu na potřebnou úroveň (cca 3 ml). Tento způsob je poměrně pracný a nákladný, ale poskytuje potřebný výtěžek, selektivitu a přesnost.
Během posledního desetiletí byla v oblasti extrakce isoflavonů vyvinuta řada nových metod. Současnou úroveň vědeckých poznatků (do jara 2006) přehledně shrnuje souhrnný referát od de Rijke 5). Koncentrovanější a čistší extrakty následně umožnily stanovení těchto analytů i v rostlinném materiálu s velmi nízkým obsahem isoflavonů, jakož i prozkoumání širší skupiny těchto látek.
Kapalinová chromatografie
Pro studium isoflavonů se původně využívaly planární techniky, jako jsou separace na vrstvě polyamidu a dalších sorbentech. Nyní je k separaci isoflavonů používána moderní kolonová instrumentace HPLC a celá řada jejích modifikací. Separace isoflavonů pomocí chromatografie byla nedávno shrnuta polskými autory 36) a řadu dalších informací může čtenář nalézt v souhrnech 5–8).
Mezi nejrozšířenější patří separace na nepolárních reverzních fázích (C18). Separace isoflavonů je založena na hydrofobní interakci jednotlivých isoflavonů se stacionární fází kolony. Retenční čas separovaných látek je primárně závislý na jejich rozpustnosti ve vodě. Bylo prokázáno, že se zvyšující se hydrofobicitou látek roste na RP kolonách jejich retenční čas. Ten lze obecně ovlivňovat obsahem organického podílu v mobilní fázi. V případě jednotlivých isoflavonů jsou nejméně hydrofobní ß D-glykosidy, následují malonyl a acetyl deriváty a nejhydrofobnější jsou samotné aglykony 37, 38).
Retenční časy separovaných isoflavonů jsou řízeny celou řadou dalších faktorů, jako je jejich samotná afinita k stacionární fázi, která může být modifikována různými dalšími funkčními skupinami, volbou složení mobilní fáze a volbou profilu gradientové eluce, teplotou kolony atd. HPLC separace flavonoidů na reverzních fázích v potravinářských vzorcích byla shrnuta v přehledném článku 39).
Ve většině případů jsou v jednom chromatografickém záznamu na RP separovány aglykony i glykosidy společně a jejich deriváty zároveň. K tomuto účelu ve většině případů nepostačuje izokratická eluce, ale je nutné vhodně zvolit profil pro eluci gradientovou. Separace je obvykle zahájena při nízkém obsahu organického modifikátoru (nejčastěji acetonitril nebo methanol) v mobilní fázi 39), jehož zastoupení se postupně zvyšuje. Se zvyšujícím se podílem organického modifikátoru dochází k rozdělení glykosidů. Aglykony jsou uvolněny ze stacionární fáze až při vyšším zastoupení organického modifikátoru. Pomocí optimalizované lineární gradientové eluce acetonitrilu a 0,2% kyseliny mravenčí bylo možné rozdělit daidzin, glycitin, genistin, ononin, daidzein, glycitein, sissotrin, genistein, formononetin a biochanin A do 6 40) a později s mobilní fází složené z 0,3% kyseliny octové a acetonitrilu do 4 minut 35).
Chromatografická separace isoflavonů dnes směřuje k aplikaci kolon s menším vnitřním průměrem a ke zmenšování velikosti částic sorbentu. Kolony plněné sorbentem s RP o zrnitosti menší jak 2 μm se výrazně osvědčily při separaci isoflavonů. Zároveň umožnily redukci retenčních časů současně separovaných aglykonů a ß-glykosidů pod 60 s a analýzu reálných vzorků sóji a rostlin Trifolium pratense, Iresine herbstii a Ononis spinosa do 2 minut 41). Podobná moderní instrumentace (tzv. U-HPLC) ve spojení s MS detektorem (QTOF) byla použita pro analýzu hormonů a isoflavonů: genistein, daidzein a biochanin A (s retenčními časy 2,39; 2,05 a 3,25 min 42). Vývoj nových U-HPLC technik a jejich kombinace s MS detekcí je uveden v práci 43).
Své uplatnění v separaci isoflavonů našly také kolony s monolitickou stacionární fází. Monolity jsou separační média, která nemají interpartikulární prostory. Stacionární fázi monolitické kolony si tedy lze představit jako jednu separační částici tvořenou polymerním materiálem44,45). Pomocí monolitické kolony byly separovány isoflavony v extraktech ze sóji 24) a jako mobilní fáze byl použit acetonitril s kyselinou octovou. Mobilní fáze složená z kyseliny octové a methanolu byla použita při separaci daidzinu, genistinu, glyciteinu a jejich glykosidů a acetyl - a malonyl - derivátů na C18 monolitické stacionární fázi v potravinářských vzorcích sóji po předchozí extrakci. Separace 12 isoflavonů travala cca 10 min a bylo dosaženo velmi dobrého rozlišení chromatografických píků 26). Mezi nejnovější výsledky také patří aplikace monolitické reverzní fáze pro separaci 11 flavonoidů s celkovou dobou separace kratší než 15 minut 46).
Komplikace při separaci fenolických látek může být řešena také dvojrozměrnou (2-D) kapalinovou chromatografií, která je založena na využití dvou separačních systémů. Tato strategie nachází uplatnění pro separaci fenolických antioxidantů 47) a v analýzách složitých matric. HPLC s neporézní stacionární fází byla taktéž aplikována pro separaci flavonoidů ve vzorcích z Filipendula ulmaria 48). V posledních letech nachází uplatnění v separaci fenolických látek protiproudá chromatografie (resp. urychlená protiproudá chromatografie, HSCCC) v kombinaci s klasickou HPLC. Ta byla použita pro analýzy fenolů v rostlinách, které jsou využívány v tradiční čínské medicíně 49).
Další separační techniky
V posledních letech byl pozorován výrazný nárůst počtu prací zaměřených na elektromigrační metody separace přírodních látek. Tyto metody jsou ve velké míře využívány pro studium flavonoidů či isoflavonů 49). Výhodou elektromigračních technik je možnost jejich spojení se senzitivními elektrochemickými detektory (ED). To je možné především díky tomu, že se jako základní elektrolyty (BGE) používají pufrované roztoky (často borátový pufr v mM koncentraci). Aplikace elektrochemických detektorů je u chromatografických technik limitována díky obsahu organických modifikátorů v jejich mobilní fázi. Kromě konvenční kapilární zónové elektroforézy (CZE) jsou využívány i další techniky, jako je kapilární elektrochromatografie (CEC) a micelární elektrokinetická kapilární chromatografie (MEKC) popřípadě kapilární izotachoforéza (ITP).
Z elektromigračních metod se nejvíce využívá CZE a již v 90. letech minulého století byly navrženy postupy pro efektivní separaci isoflavonů 50). Výsledky dosažené CZE byly srovnávány s výsledky HPLC separace v práci 51). Pomocí obou separačních technik a UV-VIS DAD detektoru byly analyzovány isoflavony v sóji, vlčím bobu a hrachu. CZE separace byla výrazně rychlejší než HPLC. Oproti tomu však HPLC separace byla více selektivní k analyzovaným isoflavonům, a to především v jednotlivých biologických matricích. CZE ve spojení s ED byla také aplikována pro analýzu puerarinu, daidzeinu a rutinu v rostlinách Pueraria lobata, což je rostlina využívaná v tradiční medicíně k léčbě nachlazení a chřipky 52). Jako pracovní elektroda byl použit uhlíkový disk o průměru 300 μm, který byl umístěn proti ústí kapiláry dlouhé 40 cm, na kterou bylo vkládáno stejnosměrné napětí 9 kV. Na základě hydrodynamických voltamogramů byl zvolen jako vhodný pracovní potenciál elektrody 0,9 V. Uvedenou technikou bylo možné kvantifikovat nanomolární koncentrace isoflavonů. Stejný elektrochemický detektor byl použit 53) pro MEKC separaci puerarinu a daidzeinu v rostlinách a farmako-chemických preparátech z Puerariae radix (směs rozdrcených kořenů P. lobata a P. thomsonii). Jako surfaktant pro pseudostacionární fázi byl použit dodecylsulfát sodný (SDS) a separace probíhala při napětí 18 kV. MEKC v poslední době našla celou řadu aplikací pro separaci ve vodě málo rozpustných polyfenolů nebo jim podobných látek. Ve spojení s UV-detektorem lze pomocí MEKC analyzovat biochanin A, formononetin, genistein a daidzein v jeteli 54). V přítomnosti SDS lze efektivnějšího rozdělení látek dosáhnout přidáním organického modifikátoru (ethanol 5 % v/v) do mobilní fáze (30 mM borátový pufr). Separace výše uvedených isoflavonů technikou MEKC ve většině prací nepřesáhla 20 min. Na rozdíl od CZE je možné pomocí MEKC efektivně separovat i hydrofobní aglykony, které jsou v pufrech používaných pro CZE poměrně málo rozpustné. Tento problém lze částečně odstranit přídavkem dimethylsulfoxidu (DMSO) anebo použitím alkalických pufrů 55).
CEC je další metodou, kterou je možné využít pro separaci isoflavonů. Tato technika je založena na separaci analytu v kapiláře obsahující sorbent; na rozdíl od HPLC se však separuje v elektricky nabitém poli. Velmi dobrých výsledků bylo také dosaženo pomocí CEC s monolitickou kolonou se stacionární fáze na bázi lauryl akrylátu. Touto technikou byl separován daidzein, genistein a glycitein a jejich konjugáty v produktech ze sóji 55).
Kromě UV-VIS a ED detektorů je možné spojit elektromigrační kolonové separace s hmotnostními 56) nebo fluorescenčními detektory. Obecně lze říci, že elektromigrační techniky nacházejí řadu aplikací nejen při identifikaci isoflavonů v různých rostlinných materiálech, ale jsou využívány například i pro stanovení disociačních konstant 57) a studium mechanismu vlivu UV-B záření na jejich stabilitu 58). Disociační konstanty pKa genisteinu (9,5), daidzeinu (9,55), glyciteinu (9,73) a dalších isoflavonů byly určeny ze závislostí jejich elektroforetické pohyblivosti na pH základního elektrolytu. Další detaily týkající se elektromigračních technik a jejich aplikace pro analýzu isoflavonů, ale i dalších flavonoidních látek lze nalézt v přehledných prácích 59, 60).
Spektrometrické metody
Mezi nejčastěji používané detektory napojené na chromatografickou nebo elektromigrační separaci jsou detektory UV-Vis, především detektory diodového pole (DAD), které mohou pracovat v širokém spektru vlnových délek 29). UV-Vis spektra řady isoflavonů jsou však velmi podobná. Vzhledem k tomu, že je v současnosti známo asi 700 isoflavonů, které se vyskytují v různých rostlinných materiálech, je využití UV-Vis detekce pro identifikaci isoflavonů z velké části omezené. K účelům detailnějšího studia separovaných látek a k jejich identifikaci je vhodné používat MS detektory, které mohou postihnout jednotlivé aglykony a jejich konjugáty či deriváty. Kromě selektivní analýzy je možné využít hmotnostní spektrometrii i pro strukturní analýzu 61). V případě určení struktury isoflavonů může být použita kombinace kolonových separací s MS a nukleární magnetickou rezonancí 62) (NMR) nebo tandemové (MS/MS) uspořádání hmotnostních spektrometrů 63).
K ionizaci vzorku se nejčastěji používá elektrosprej, který je možné napojit na LC nebo CE. Mezi další ionizační techniky patří laserová desorpce/ionizace za účasti matrice (MALDI), chemická ionizace za atmosférického tlaku (APCI), termosprej 64) a další 65). Velmi dobrých výsledků bylo dosaženo pomocí ESI-MS v kombinaci s HPLC po předchozí SFE 35) izolaci s detekčními limity (LOD) pro aglykony 0,2–1,0 fmolů a pro glykosidy 1,3–3,6 fmolů na nástřik. Stejný MS detektor se už dříve osvědčil pro určení profilu isoflavonů u T. pratense a při identifikaci jejich glykosidů 28) nebo při detekci isoflavonů ve vzorcích potravin ze sóji. LOD byl 1,2 a 1,6 fmolů pro daidzin, genistin a 1–3 fmoly pro daidzein, genistein, formononetin, biochanin A a ononin 30). MS je velmi účinný nástroj pro identifikaci isoflavonů v rostlinných materiálech, přičemž každý isoflavon je charakteristický nejen svým molekulárním ionem, ale také specifickými produkty jeho fragmentace (retro-Diels-Alderova fragmantace). Studie pojednávající o fragmentaci isoflavonů byla uveřejněna Kangem et al. 66) a údaje o fragmentaci cukerných konjugátů byly publikovány v práci 9). Mezi další moderní techniky patří nedávno uveřejněná práce týkající se stanovení acetylglukosidů a jejich metabolitů v lidské moči spojením LC s APCI/MS 67). Velmi selektivní jsou také nově vyvinuté techniky MS/MS, které je možné použít pro určení profilu isoflavonů 68) a v celé řadě dalších aplikací 61, 65, 69). Kombinace U-HPLC/QTOF-MS byla použita pro analýzu fytoestrogenů ve vzorcích vody. Detekční limity stanovovaných isoflavonů se pohybovaly v rozmezí 5 až 30 ng v litru vody 42).
Pořizovací i provozní náklady pro MS jsou ve srovnání s ostatními detekčními technikami poměrně vysoké. Selektivitu klasických spektrofotometrických technik je možné zvýšit použitím derivatizačního činidla, které se cíleně naváže na analyzované isoflavony. Kromě klasické UV-Vis detekce je používána fluorescenční detekce derivatizačního činidla nebo i nativní fluorescence samotných isoflavonů, kterou lze např. u puerarinu a daidzeinu pozorovat při pH 8–9 70). Pokud mobilní fáze obsahuje kyselinu, je vhodné použít postkolonové přidání alkalického pufru, které výrazně zvyšuje citlivost analýzy. Limit kvantifikace (LOQ) byl pro puerarin 3,48 a pro daidzein 1,16 ng . ml-1. Vzhledem k tomu, že isoflavony vykazují nativní fluorescenci, může být jejich derivatizace fluorescenčním činidlem v řadě případů značně limitována.
Pro senzitivní analýzu isoflavonů mohou být také využity chemiluminiscenční techniky. Kombinace chemiluminescence a průtokové injekční analýzy (FIA) byla aplikována pro analýzu puerarinu v různých vzorcích s limitem detekce 0,1 ng . ml-1. Pomocí peristaltických pump byl smísen vzorek obsahující puerarin s luminolem, KIO4 a roztokem NaOH a směs byla po proběhnutí reakce dávkována do cely s luminometrem. Uvedenou technikou bylo možné analyzovat při průtoku 2 ml . min-1 180 vzorků za hodinu 71). Chemiluminiscenční reakce představují účinnou alternativu pro optickou detekci isoflavonů, která může být využita v řadě klinických nebo potravinářských či farmaceutických provozech.
Imunochemické metody
Mezi metody vhodné pro rychlou identifikaci isoflavonů v rostlinných, ale i potravinářských vzorcích patří imunochemická analýza. Původně vyvinuté techniky založené na radioaktivním značení (RIA: radioimunoanalýza) 72), jsou dnes nahrazovány technikou heterogenní enzymové imunoanalýzy (ELISA) 73–76) nebo časově modulované detekce fluorescence (TR-FIA, viz níže).
V případě techniky ELISA jsou nejprve vytvořeny konjugáty mezi isoflavony a telecím sérovým albuminem (BSA) nebo jiným vysokomolekulárním nosičem a proti vzniklým konjugátům (antigenům) jsou získány (monoklonální nebo polyklonální) protilátky 74). Vzorky jsou nanášeny do jamek mikrotitrační destičky a na antigeny, které jsou ve vzorku obsaženy se vážou získané protilátky. Po promývacích procedurách je potom množství navázaných protilátek kvantifikováno pomocí další protilátky značené peroxidázou. Po přidání substrátu a vytvoření vhodných podmínek pro průběh enzymové reakce je kvantifikován její produkt, jehož koncentrace odpovídá množství hledaného isoflavonu ve vzorku. ELISA byla použita pro identifikaci biochaninu A, daidzeinu a genisteinu s limity detekce (LOD), které se pohybovaly v rozmezí 1,1–5,3 pg isoflavonů na jamku. Pomocí imunochemických technik je možné zjišťovat isoflavony v homogenátech rostlinných vzorků, nebo jejich extraktech 73).
Dále byla ELISA použita pro analýzu genisteinu a daidzeinu v potravinách a lidských tělních tekutinách (plazma a moč), přičemž byla pomocí ELISA studována distribuce daidzeinu a genisteinu v čase, a to u pacientů, kterým bylo podáno 100 mg isoflavonů 75, 76). Maximum daidzeinu i genisteinu bylo v plazmě nalezeno po 6 až 8 hod. a v moči po 10 hod. od jejich perorální aplikace. Další možností stanovení isoflavonů je použití metody časově modulované detekce fluorescence (TR-FIA), kde je protilátka konjugovaná s Eu, Tb, Sm chelátem přechodného kovu, který se v průběhu imunochemické reakce přemění ve fluoreskující značku. Pomocí imunochemických technik je možné kvantifikovat isoflavony v homogenátu rostlinných vzorků, nebo jejich extraktech, v potravinách i v klinických vzorcích. Využití imunoanalytických metod v klinické chemii má své opodstatnění vzhledem k jednoduchosti a rychlosti těchto analýz 77).
Elektrochemické metody
Vzhledem k tomu, že isoflavony jsou elektroaktivní látky, byla vyvinuta řada technik zaměřených na jejich elektrochemickou detekci nebo spojení elektrochemických detektorů s kolonovými separacemi. Jako pracovní elektrody se osvědčily elektrody vyrobené z uhlíku a jeho různých modifikací, na kterých je možné pozorovat oxidační signály isoflavonů. Jako elektrodový materiál je možné použít skelný a pyrolytický uhlík nebo tzv. uhlíkovou pastu.
Elektroda ze skelného uhlíku (GCE) byla použita pro analýzu flavonoidů a fenolických kyselin diferenční pulzní voltametrií s limity detekce v mikromolárních jednotkách 78), a pro stanovení celkového obsahu isoflavonů ve vzorcích sóji 79). Uhlíková pastová elektroda (CPE) byla použita pro stanovení daidzeinu a genisteinu 80), přičemž byly pozorovány dva oxidační píky (pík I kolem 0,4 V a pík II kolem 0,7 V). Pro analýzu vybraných flavonoidů byla použita adsorptivní rozpouštěcí voltametrie s CPE 81). Elektrodový materiál byl připraven smícháním nujolu nebo difenyletheru s uhlíkovým práškem a elektrochemická detekce pobíhala v průtokovém injekčním analyzátoru (FIA). Flavonoidy byly akumulovány na povrchu uhlíkové elektrody při potenciálu 0,2 V a následně provedeno elektrochemické měření v rozsahu potenciálu od 0 do 1 V. Oxidační signály flavonoidů bylo možné pozorovat kolem 0,4 V. V případě rutinu byly pozorovány dobře vyvinuté píky v koncentračním rozsahu od 10 do 100 nM 81). Uvedená technika byla použita pro analýzy celkového obsahu flavonoidů v nápojích 82). Elektrochemické detektory mají významné uplatnění především v separacích CE (viz kapitola Ostatní separační techniky) a mohou být využity i v chromatografické separaci. Detekčního limitu 51 fmolů (21 pg) na nástřik bylo dosaženo s coulometrickým detektorem 80).
Interakce isoflavonů
Molekulární podstata fyziologického účinku isoflavonů nespočívá pouze v jejich estrogenní aktivitě, ale také v jejich interakci s celou řadou buněčných substrátů. Především je zájem vědců soustředěn na studium interakcí isoflavonů s nukleovými kyselinami 83, 84), proteiny 85, 86) a receptory v cytoplazmatické membráně buněk 87). Předpokládáme, že právě získáváním poznatků o interakci isoflavonů s biomakromolekulami bude možné objasnit jejich fyziologické účinky, a vnést tak více světla do kontroverzní otázky jejich pozitivního vs. negativního vlivu na zdraví člověka.
Bylo prokázáno, že isoflavony mohou interagovat s DNA a vytvářet komplexy s bázemi, které se v DNA vyskytují. Studium interakcí 84) mezi telomerickou DNA (která je bohatá na guaninové zbytky) a daidzinem metodou ESI-MS, PAGE, metodou cirkulárního dichroismu a molekulárního modelování napovídá, že isoflavony mohou nejenom interagovat s DNA, ale jsou také schopny stabilizovat jejich struktury – jako je např. G-kvadruplex. Předpokládá se, že daidzin by tak mohl být využit jako protinádorové léčivo inhibující telomerázovou aktivitu 84). Interakce guaninů a adeninů v jednořetezcové chromozomální DNA s kvercetinem a rutinem byly pozorovány pomocí voltametrie s vnuceným pravoúhlým napětím a uhlíkovou elektrodou. Dá se předpokládat, že tato technika bude použitelná pro rychlé a senzitivní sledování interakce flavonoidních látek s DNA 83). Kromě DNA interagují isoflavony i s bílkovinami. Byla například studována interakce lidského sérového albuminu s genisteinem a dalšími isoflavony metodou spektrofluorimetrie, kde byla prokázána interakce genisteinu s tryptofanovými zbytky v molekule bílkoviny 86). Pomocí „kompetičních experimentů” (competitition binding assay) byla studována vazba genisteinu, biochaninu A, formononetinu, kumestrolu a dalších na estrogenní receptor α a ß 87). Velmi důležité jsou také studie zaměřené na interakci isoflavonů s cytoplazmatickou membránou buňky, která je tvořena dvouvrstvou lipidů. Studována je především adsorpce isoflavonů na lipidy membrány a jejich vliv na peroxidaci lipidů a na integritu a propustnost buněčných membrán 88). Získané výsledky bude možné využít pro studium farmakokinetických vlastností isoflavonů obsažených v léčivech.
Příjem isoflavonů
Jedny z prvních fyziologických účinků isoflavonů (u živočichů) byly zaznamenány u hospodářských zvířat, která konzumovala plodiny s vysokou koncentrací isoflavonů (nebo nadměrné množství plodin obsahujících isoflavony), což vedlo k poruchám reprodukce (viz estrogenní aktivita isoflavonů). V rostlinné produkci je proto žádoucí eliminovat nadměrný příjem isoflavonů u hospodářských zvířat a monitorovat výskyt sekundárních metabolitů v rostlinných materiálech. Pomocí RP HPLC/UV-Vis DAD byl sledován výskyt isoflavonů a fenolických kyselin v semenech hrachu (Pisum sativum). Obsah isoflavonů v semenech byl ovlivňován speciálními technologickými procesy 89). Ukazuje se, že podmínky skladování a vhodné fyzikální nebo chemické ošetření může cíleně měnit obsah isoflavonů v rostlinných produktech. Naproti tomu byly vyvinuty techniky genového inženýrství umožňující ovlivňovat obsah isoflavonů (daidzein, glycitein, genistein) v semenech sóji na základě exprese C1 a R transkripčních faktorů z kukuřice, které aktivují fenypropanoidovou metabolickou dráhu. K identifikaci isoflavonů byla použita HPLC-DAD, které předcházela extrakce isoflavonů do methanolu 90).
Lidský organismus přijímá isoflavony nejčastěji potravou (tzv. funkční potraviny), která byla vyrobena z luštěnin nebo sójových bobů. Pojem funkční potravina byl poprvé použit již v 80. letech minulého století v Japonsku 59). V současnosti lze funkční potraviny definovat jako potraviny, které mají pozitivní efekt na jednu nebo více fyziologických funkcí lidského organismu nebo snižují, popřípadě odstraňují, některé poruchy organismu*. Kromě sóji a jejich produktů (např. tofu – sójový sýr, miso – sójová rýže) byly fytoestrogeny a isoflavony analyzovány v celé řadě potravinářských doplňků, jako je např. kudzu (bílý škrobový prášek z kořene keře Pueraria lobata) nebo i ve chmelu (Humulus lupulus) a v alkoholických nápojích (pivo, bourbon) a mnoha dalších zdrojích (ovoce atd.) 91). Analýzy sekundárních metabolitů (a to nejenom isoflavonů) v různých rostlinách potvrdily, že distribuce těchto látek se v pletivech rostliny výrazně mění v závislosti na době sklizně a podmínkách pěstování. Možnosti, které v současnosti nabízejí techniky genového inženýrství, by mohly být v budoucnosti využity k pěstování plodin, které by měly definovaný obsah isoflavonů. V této souvislosti je nutné zohlednit nejenom pozitivní vliv isoflavonů na zdraví člověka, ale také skutečnost, že konzumace zvýšeného množství isoflavonů může vyvolat řadu zdravotních problémů 3).
Závěr
Vzhledem k tomu, že isoflavony, ale i další flavonoidy, mají často podobnou chemickou strukturu a mnohdy i fyzikálně-chemické vlastnosti, je jejich vzájemné rozlišení v rostlinných matricích značně ztíženo. Objev celé řady fyziologických účinků isoflavonů na zdraví člověka podmiňuje vývoj v oblasti moderních instrumentálních metod analýzy těchto látek. Dá se předpokládat, že budou zdokonalovány především techniky přípravy vzorku a techniky isolace, jako je ASE a SPE a mikroextrakční techniky 92). V oblasti kapalinové chromatografie najdou své uplatnění monolitické kolony, kolony plněné sorbenty s velikostí částic pod 2 μm (díky kterým je možné výrazně snížit dobu separace) a také kapilární kolony. Z hlediska detekce isoflavonů a jejich identifikace ve složitých rostlinných matricích jsou s úspěchem aplikovány různé MS detektory, přičemž mezi nejpoužívanější zdroje ionizace patří elektrosprej. Jako velmi perspektivní se také jeví nové nanoESI zdroje 93) a taktéž nedávno vyvinutá ionizace desorpcí elektrosprejem (DESI). Ta představuje přechod mezi ESI a MALDI a umožňuje poměrně senzitivní analýzu biologicky významných látek v podmínkách in situ, které vyžadují minimální čas pro přípravu vzorků 94). V práci diskutované imunoanalytické techniky (např. ELISA a TR-FIA) naleznou uplatnění v klinické praxi při vyšetření tělních tekutin a velkého množství vzorků 95). Naproti tomu byly uveřejněny práce, kde se imunochemické techniky osvědčily při identifikaci isoflavonů v různých rostlinných pletivech a potravinářských produktech. Elektrochemická detekce isoflavonů je velmi často používána ve spojení s elektromigračními technikami 96, 97). Pro tyto účely byly navrženy různé konstrukce elektrochemických detektorů. Jako elektrodový materiál se nejvíce osvědčil skelný uhlík a v klasickém tříelektrodovém voltametrickém zapojení také uhlíková pasta. V budoucnosti lze předpokládat, že budou používány i další materiály a že bude docházet k miniaturizaci detekčních cel a pracovních elektrod a využití nano-materiálů 98) pro jejich výrobu. Pomocí moderních analytických metod lze v současnosti provádět separace celého spektra isoflavonů do několika minut a kvantifikovat jejich ng množství v různých rostlinných materiálech, klinických vzorcích a v potravinách, což je klíčové z hlediska kontroly kvality zemědělských produktů a léčiv.
Použité zkratky
APCI – atmospheric pressure chemical ionization (chemická ionizace za atmosférického tlaku)
ASE – accelerated–solvent extraction (extrakce v urychleném toku rozpouštědla)
BSA – bovine serum albumin (telecí sérový albumin)
CE – capillary electrophoresis (kapilární elektroforéza)
CEC – electrochromatography (elektrochromatografie)
CPE – carbon paste electrode (uhlíková pastová elektroda)
CZE – capillary zone electrophoresis (kapilární zónová elektroforéza)
DAD – diode-array detection (detekce diodovým polem)
ED – electrochemical detector (elektrochemický detektor)
ELISA – enzyme-linked immunosorbent assay (heterogenní enzymová imunoanalýza)
ESI – electrospray ionisation (ionizace elektrosprejem)
FIS – flow injection system (průtokový injekční systém)
GCE – glyssy carbon electrode (elektroda ze skelného uhlíku)
HPLC – heigh performance liquid chromatography (vysokoúčinná kapalinová chromatografie)
HSCCC – high-speed counter-current chromatography (urychlená protiproudá chromatografie)
LC – liquid chromatography (kapalinová chromatografie)
LOD – limit of detection (limit detekce)
LOQ – limit of quantification (limit kvantifikace)
MALDI – matrix-assisted laser desorption (desorpce/ionizace laserem za účasti matrice)
MEKC – micellar electrokinetic capillary chromatography (micelární elektrokinetická kapilární chromatografie)
MS – mass spectrometry (hmotnostní spektrometrie)
MS/MS – tandem MS (tandemové uspořádání MS)
NMR – nuclear magnetic resonance (nukleární magnetická rezonance)
RIA – radioimunoassay (radioimunoanalýza)
RP – reversed phase (reverzní fáze)
SFE – supercritical-fluid extraction (extrakce kapalinou v nadkritickém stavu)
SPE – solid-phase extraction (extrakce pevnou fází)
TR–FIA – time-resolved fluorescence immunoanalysis (časově modulovaná detekce fluorescence)
Došlo: 22. ledna 2008 / Přijato: 12. února 2008
Adresa pro korespondenci:
prof. RNDr. Vlastimil Kubáň, DrSc.
Ústav chemie a biochemie, Mendelova zemědělská a lesnická univerzita
Zemědělská 1, 613 00 Brno
e-mail: kuban@mendelu.cz
Sources
1. Setchell, K. D. R.: Am. J. Clin. Nutr., 1998; 68, 1333S-1346S.
2. Ososki, A. L., Kennelly, E. J.: Phytother. Res., 2003; 17, 845–869.
3. Wanibuchi, H., Kang, J. S., Salim, E. I. et al.: Pure Appl. Chem., 2003; 75, 2047–2053.
4. Fritsche, S., Steinhart, H.: Eur. Food Res. Technol., 1999; 209, 153–179.
5. de Rijke, E., Out, P., Niessen, W. M. A. et al.: J. Chromatogr. A, 2006; 1112, 31–63.
6. Wu, Q. L., Wang, M. F., Simon, J. E.: J. Chromatogr. B, 2004; 812, 325–355.
7. Akhtar, M. H., Abdel-Aal, E. S. M.: Curr. Pharm. Anal., 2006; 2, 183–193.
8. Delmonte, P., Rader, J. I.: J. AOAC Int., 2006; 89, 1138–1146.
9. D’Alessandro, T. L., Boersma-Maland, B. J., Peterson, T. G. et al.: Methods Enzymol., 2005; 400, 316–342.
10. Macková, Z., Koblovská, R., Lapčík, O.: Phytochem., 2006; 67, 849–855.
11. Yokosuka, A., Haraguchi, M., Usui, T. et al.: Bioorg. Med. Chem. Lett., 2007; 17, 3091–3094.
12. Moriyasu, M., Igi, Y., Ichimaru, M. et al.: J. Nat. Med., 2007; 61, 329–333.
13. Chen, S. G., Chen, J. J., Gao, K.: Chem. Pharm. Bull., 2007; 55, 1181-1184.
14. Hegnauer, R., Grayerbarkmeijer, R. J.: Phytochem., 1993; 34, 3–16.
15. Dunford, C. L., Smith, G. J., Swinny, E. E., Markham, K. R.: Photochem. Photobiol. Sci., 2003; 2, 611–615.
16. Mathias, K., Ismail, B., Corvalan, C. M., Hayes, K. D.: J. Agric. Food Chem., 2006; 54, 7495–7502.
17. de Rijke, E., Zafra-Gomez, A., Ariese, F. et al.: J. Chromatogr. A, 2001; 932, 55–64.
18. Shimoni, E.: J. Food Sci., 2004; 69, R160–R166.
19. Delmonte, P., Perry, J., Rader, J. I.: J. Chromatogr. A, 2006; 1107, 59–69.
20. Xu, H. N., He, C. H.: Sep. Purif. Technol., 2007; 56, 85–89.
21. Wu, C. Y., Lai, S. M.: J. Liq. Chromatogr. Relat. Technol., 2007; 30, 1617–1640.
22. Yu, Z. G., Gao, X. X., Zhao, Y. L., Bi, K. S.: Biomed. Chromatogr., 2007; 21, 577–584.
23. Velickovic, D. T., Nikolova, M. T., Ivancheva, S. V. et al.: J. Serb. Chem. Soc., 2007; 72, 73–80.
24. Apers, S., Naessens, T., Van Den Steen, K. et al.: J. Chromatogr. A, 2004; 1038, 107–112.
25. Rostagno, M. A., Palma, M., Barroso, C. G.: J. Chromatogr. A, 2003; 1012, 119–128.
26. Rostagno, M. A., Palma, M., Barroso, C. G.: Anal. Chim. Acta, 2007; 582, 243–249.
27. Klejdus, B., Vitamvásová, D., Kubáň, V.: J. Chromatogr. A, 1999; 839, 261–263.
28. Klejdus, B., Vitamvásová-Štěrbová, D., Kubáň, V.: Anal. Chim. Acta, 2001; 450, 81–97.
29. Klejdus, B., Štěrbová, D., Stratil, P., Kubáň, V.: Chem. Listy, 2003; 97, 530–539.
30. Klejdus, B., Mikelová, R., Adam, V. et al.: Anal. Chim. Acta, 2004; 517, 1–11.
31. Klejdus, B., Mikelová, R., Petrlová, J. et al.: J. Agric. Food Chem., 2005; 53, 5848–5852.
32. Rostagno, M. A., Palma, M., Barroso, C. G.: Anal. Chim. Acta, 2007; 588, 274–282.
33. Chukwumah, Y. C., Walker, L. T., Verghese, M. et al.: J. Agric. Food Chem., 2007; 55, 285–290.
34. Yu, J., Liu, Y. F., Qiu, A. Y., Wang, X. G.: LWT-Food Sci. Technol., 2007; 40, 800–806.
35. Klejdus, B., Lojková, L., Lapčík, O. et al.: J. Sep. Sci., 2005; 28, 1334–1346.
36. Grynkiewicz, G., Ksycinska, H., Ramza, J., Zagrodzka, J.: Acta Chromatogr., 2005; 15, 31–65.
37. Lee, S. K., Row, K. H.: Bull. Korean Chem. Soc., 2003; 24, 1265–1268.
38. Zheng, J. Z., Row, K.: Chin. J. Chem. Eng., 2007; 15, 291–295.
39. Merken, H. M., Beecher, G. R.: J. Agric. Food Chem., 2000; 48, 577–599.
40. Klejdus, B., Mikelová, R., Petrlová, J. et al.: J. Chromatogr. A, 2005; 1084, 71–79.
41. Klejdus, B., Vacek, J., Benešová, L. et al.: Anal. Bioanal. Chem., 2007; 389, 2277–2285.
42. Farre, M., Kuster, M., Brix, R. et al.: J. Chromatogr. A, 2007; 1160, 166–175.
43. Churchwell, M. I., Twaddle, N. C., Meeker, L. R., Doerge, D. R.: J. Chromatogr. B, 2005; 825, 134-143.
44. Sýkora, D., Tesařová, E., Vosmanská, M., Zvolánková, M.: Chem. Listy, 2007; 101, 190–199.
45. Švec, F.: Chem. Listy, 2004; 98, 232–238.
46. Repolles, C., Herrero-Martinez, J. M., Rafols, C.: J. Chromatogr. A, 2006; 1131, 51–57.
47. Cacciola, F., Jandera, P., Bláhová, E., Mondello, L.: J. Sep. Sci., 2006; 29, 2500–2513.
48. Pemp, E., Reznicek, G., Krenn, L.: J. Anal. Chem., 2007; 62, 669–673.
49. Ma, X. F., Tu, P. F., Chen, Y. J. et al.: J. Chromatogr. A, 2003; 992, 193–197.
50. Shihabi, Z. K., Kute, T., Garcia, L. L., Hinsdale, M.: J. Chromatogr. A, 1994; 680, 181–185.
51. Mellenthin, O., Galensa, R.: J. Agric. Food Chem., 1999; 47, 594–602.
52. Chen, G., Zhang, J. X., Ye, J. N.: J. Chromatogr. A, 2001; 923, 255–262.
53. Cao, Y. H., Lou, C. G., Zhang, X. et al.: Anal. Chim. Acta, 2002; 452, 123–128.
54. Zhang, Y., Chen, J., Zhao, L., Shi, Y-P.: Biomed. Chromatogr., 2007; 21, 987–992.
55. Starkey, J. A., Mechref, Y., Byun, C. K. et al.: Anal. Chem., 2002; 74, 5998–6005.
56. Aramendia, M. A., Garcia, I., Lafont, F., Marinas, J. M.: J. Chromatogr. A, 1995; 707, 327–333.
57. McLeod, G. S., Shepherd, M. J.: Phytochem. Anal., 2000; 11, 322–326.
58. Dinelli, G., Aloisio, I., Bonetti, A. et al.: J. Sep. Sci., 2007; 30, 604–611.
59. Herrero, M., Ibanez, E., Cifuentes, A.: J. Sep. Sci., 2005; 28, 883–897.
60. Li, P., Li, S. P., Wang, Y. T.: Electrophoresis, 2006; 27, 4808–4819.
61. Cuyckens, F., Claeys, M.: J. Mass. Spectrom., 2004; 39, 1–15.
62. Elipe, M. V. S.: Anal. Chim. Acta, 2003; 497, 1–25.
63. Cuyckens, F., Ma, Y. L., Pocsfalvi, G., Claeys, M.: Analusis, 2000; 28, 888–895.
64. Dewald, H. D., Worst, S. A., Butcher, J. A., Saulinskas, E. F.: Electroanal., 1991; 3, 777–782.
65. Prasain, J. K., Wang, C. C., Barnes, S.: Free Rad. Biol. Med., 2004; 37, 1324–1350.
66. Kang, J. G., Hick, L. A., Price, W. E.: Rapid Commun. Mass Spectrom., 2007; 21, 857–868.
67. Chen, L. J., Zhao, X., Fang, L. Y., Games, D. E.: J. Chromatogr. A, 2007; 1154, 103-110.
68. Cavaliere, C., Cucci, F., Foglia, P. et al.: Rapid Commun. Mass Spectrom., 2007; 21, 2177–2187.
69. Muller, A., Steinhart, H.: Food Chem., 2007; 102, 436–444.
70. Zhai, X-J., Qu, H-B., Shao, Q., Cheng, Y-Y.: Chromatographia, 2007; 66, 43–47.
71. Wang, C., Song, Z.: Bioorg. Med. Chem. Lett., 2004; 14, 4127–4130.
72. Lapčík, O., Hampl, R., Hill, M. et al.: J. Steroid Biochem. Mol. Biol., 1998; 64, 261–268.
73. Vítková, M., Macková, Z., Fukal, L., Lapčík, O.: Chem. Listy, 2004; 98, 1135-1139.
74. He, J. T., Shi, Z. H., Yan, J. et al.: Talanta, 2005; 65, 621–626.
75. Bennetau-Pelissero, C., Arnal-Schnebelen, B., Lamothe, V. et al.: Food Chem., 2003; 82, 645–658.
76. Bennetau-Pelissero, C., Le Houerou, C., Lamothe, V. et al.: J. Agric. Food Chem., 2000; 48, 305–311.
77. Talbot, D. C. S., Ogborne, R. M., Dadd, T. et al.: Clin. Chem., 2007; 53, 748–756.
78. Blasco, A. J., González, M. C., Escarpa, A.: Anal. Chim. Acta, 2004; 511, 71–81.
79. Escarpa, A., González, M. C., Blasco, A. J. et al.: Electroanal., 2007; 19, 952–957.
80. Klejdus, B., Vacek, J., Adam, V. et al.: J. Chromatogr. B, 2004; 806, 101–111.
81. Volikakis, G. J., Efstathiou, C. E.: Talanta, 2000; 51, 775–785.
82. Volikakis, G. J., Efstathiou, C. E.: Anal. Chim. Acta, 2005; 551, 124–131.
83. Hodek, P., Hanuštiak, P., Křížková, J. et al.: Neuroendocrinol. Lett., 2006; 27, 14–17.
84. Li, W., Zhang, M., Zhang, J. L. et al.: Febs Lett., 2006; 580, 4905–4910.
85. Maliar, T., Jedinak, A., Kadrabová, J., Šturdik, E.: Eur. J. Med. Chem., 2004; 39, 241–248.
86. Mahesha, H. G., Singh, S. A., Srinivasan, N., Rao, A. G. A.: FEBS J., 2006; 273, 451–467.
87. Morito, K., Aomori, T., Hirose, T. et al.: Biol. Pharm. Bull., 2002; 25, 48–52.
88. Erlejman, A. G., Verstraeten, S. V., Fraga, C. G., Oteiza, P. I.: Free Rad. Res., 2004; 38, 1311–1320.
89. Dvořák, R., Pechová, A., Pavlata, L. et al.: Czech J. Anim. Sci., 2005; 50, 519–527.
90. Yu, O., Shi, J., Hession, A. O. et al.: Phytochem., 2003; 63, 753–763.
91. Rader, J. I., Delmonte, P., Trucksess, M. W.: Anal. Bioanal. Chem., 2007; 389, 27–35.
92. Lord, H. L.: J. Chromatogr. A, 2007; 1152, 2–13.
93. Wickremsinhe, E. R., Singh, G., Ackermann, B. L. et al.: Curr. Drug Metab., 2006; 7, 913–928.
94. Takats, Z., Wiseman, J. M., Gologan, B., Cooks, R. G.: Science, 2004; 306, 471–473.
95. Zhao, M. P., Zhou, S., Yan, J., Li, L.: Curr. Pharm. Anal., 2007; 3, 25–38.
96. Escarpa, A., Gonzalez, M. C., Crevillen, A. G., Blasco, A. J.: Electrophoresis, 2007; 28, 1002-1011.
97. Bachmann, S., Huck, C. W., Bakry, R., Bonn, G. K.: Electrophoresis, 2007; 28, 799–805.
98. Pumera, M., Sanchez, S., Ichinose, I., Tang, J.: Sens. Actuator B Chem., 2007; 123, 1195–1205.
Labels
Pharmacy Clinical pharmacologyArticle was published in
Czech and Slovak Pharmacy

2008 Issue 2
Most read in this issue
- Polosyntetické deriváty celulosy jako základ hydrofilních gelových systémů
- Laktobacily a ich probiotické vlastnosti
- Standardní receptura pro přípravu léčivých přípravků v lékárnách I. Suspenze k aplikaci na kůži
- Léčivé rostliny a diabetes mellitus