
Využitie rádionuklidovej röntgenofluorescenčnej analýzy pri stanovení obsahu ťažkých kovov v dextránoch v tuhom a kvapalnom skupenstve
Use radionuclide x-ray fluorescence analysis for determination of heavy metals content in dextrans in solid and liquid state
The aim of this study was to determine heavy metals in dextrans samples and to observe the influence of sample preparation before the analysis on the detection limit by radionuclide x-ray fluorescence analysis (XRF). The content of elements Mn, Fe, Cu, Co, Zn, Pb, As, Hg and Cd was determined in dextran tablets, their ashes after the mineralization and in solutions after the preconcetration by precipitation with thioacetamide and after following filtration of created sulphides through the membrane filter. Calculated detection limits proved the advantage of preconcentration of elements from the liquid state. The results suggest the possible use of combination of XRF with this preconcentration method in quality control of drugs.
Key words:
heavy metals – dextran – preconcentration – thioacetamide – X-ray fluorescence
Authors:
V. Jánošová; O. Štroffeková; M. Sýkorová
Authors‘ workplace:
Univerzita Komenského Bratislava, Farmaceutická fakulta, Katedra farmaceutickej analýzy a nukleárnej farmácie
Published in:
Čes. slov. Farm., 2009; 58, 83-87
Category:
Review Articles
Overview
Práca je zameraná na stanovenie ťažkých kovov vo vzorkách dextránov a na sledovanie vplyvu úpravy vzoriek pred analýzou na detekčný limit pri ich rádionuklidovej röntgenofluorescenčnej analýze (RRFA). Obsah vybraných prvkov Mn, Fe, Cu, Co, Zn, Pb, As, Hg a Cd bol stanovený v tabletách dextránov, v ich popoloch po mineralizácii a v roztokoch po zakoncentrovaní pomocou zrážania s tioacetamidom a po následnej filtrácii vzniknutých sulfidov cez membránový filter. Vypočítané detekčné limity preukázali výhodnosť zakoncentrovania prvkov z kvapalneho skupenstva. Výsledky poukázali na možné využitie spojenia RRFA s touto metódou zakoncentrovania pri kontrole kvality liečiv.
Kľúčové slová:
ťažké kovy – dextrán – zakoncentrovanie – tioacetamid – röntgenofluorescenčná analýza
Úvod
Ťažké kovy patria medzi významnú skupinu kontaminatov, ktoré pri dlhodobej expozícii môžu negatívne ovplyvniť zdravotný stav obyvateľstva a negatívne ovplyvniť stabilitu liečiva (jeho účinnej zložky), alebo spôsobiť jej celkové znehodnotenie. Preto všetky látky používané vo farmaceutickom priemysle, liečivá a lieky musia spĺňať kritéria kvality a nezávadnosti, môžu obsahovať len povolený limit obsahu ťažkých kovov, ktorý je presne definovaný v SL1 1, 2).
Farmaceutický priemysel v snahe zvyšovať kvalitu a bezpečnosť liekov, využíva moderné analytické metódy a kladie vysoké požiadavky na ich analytické parametre. Vývoj metód je zameraný na ich zefektívnenie a zjednodušenie (AAS, AES ...) 3–7).
Vzorky, ktoré sú predložené k analýze sú často vo forme zmesí, jednotlivé časti je nutné vhodne upraviť, oddeliť pomocou vhodne zvolenej separačnej metódy (filtrácia, odparenie, mineralizácia suchou a mokrou cestou 8,9). Medzi efektívne metódy úpravy vzoriek patria komplexotvorné a zrážacie reakcie, ktoré vyžadujú vhodnú voľbu skúmadla (NaDDTC, APDC, EDTA ...) 10–13) SL1 odporúča a využíva na stanovenie obsahu anorganických nečistôt tioacetamid. Výsledky zrážania pomocou tohto skúmadla sú hodnotené vizuálne, nie je zrejmé zastúpenie jednotlivých prvkov v analyzovanej vzorke. Kvantitatívne vyhodnotenie je možné realizovať v spojení s inštrumentálnymi metódami, ktorými sa zakoncentrované sulfidy prvkov na filtroch s výhodou stanovia v stopových množstvách.
Požiadavka identifikovať a stanoviť vo vzorkách dextránov dodaných výrobcom Biotika a. s. Slovenská Ľupča vybrané prvky Mn, Fe, Cu, Zn, Pb, As, Hg a Cd vyplynula z požiadaviek praxe. Keďže roztoky dextránov z galenického hľadiska zaraďujeme medzi parenterálne prípravky, musia spĺňať zvýšené požiadavky na akosť, ktoré uvádzajú liekopisné články 1, 2). Kontaminácia ťažkými kovmi v hotových infundibíliách je neprípustná, a to nielen z hľadiska možných inkompatibilít ale aj z možného toxického pôsobenia na organizmus.
Nakoľko sa dextrány v praxi nachádzajú vo forme tekutých infundibílií alebo v tuhej forme určenej na prípravu roztokov, je optimálne nájsť metódu, ktorá umožní rýchle, selektívne a polykomponentné stanovenie obsahu prvkov v oboch skupenstvách. V práci bola na tento účel vybraná nukleárna analytická metóda rádionuklidová röntgenofluorescenčná analýza (RRFA) 14). Táto metóda je založená na interakcii nízkoenergetického žiarenia gama a X (s energiou menšou ako 150 keV) s látkou. Celá energia gama-fotónu je odovzdaná jednému orbitálovému elektrónu, ktorý pre nadbytok energie opustí atóm. Atóm sa zo vzbudeného stavu vracia do základného stavu vyžiarením kvanta fluorescenčného žiarenia. Energia vzniknutého fluorescenčného žiarenia je charakteristická pre emitujúci (stanovovaný) prvok. Intenzita je úmerná množstvu stanovovanej zložky v analyzovanej vzorke 15).
POKUSNÁ ČASŤ
Materiál a metódy
Na analýzu boli použité vzorky dextránov určených na prípravu parenterálnych prípravkov (Biotika a. s. Slovenská Ľupča, SR). Vzorka dextránu A bol dextrán 40 č. šarže 041103 a vzorka dextránu B bol dextrán 40 č. šarže 050701. Vo vzorkách boli metódou RRFA stanovné prvky Mn, Fe, Cu, Zn, Pb, As, Hg a Cd, pričom boli k excitácii uvedených prvkov použité primárne zdroje žiarenia 241Am (s energiou 27–33 keV, aktivitou 740 MBq a dobou premeny 470 rokov) a 238Pu (s energiou 12 až 22 keV, aktivitou 1110 MBq a dobou premeny 86,4 roka). Meracie zariadenie tvoril polovodičový Si/Li detektor, mnohokanálový analyzátor EG & G ORTEC s vyhodnocovacím programom MEASTRO-32®. Bola použitá bočná reflexná geometria vzorka – zdroj – detektor. Čas meria bol 4000 s 14).
Analýza dextránov v tuhom skupenstve
Pre prípravu štandardov bola použitá matrica kyselina benzoová p. a. (Lachema a. s., Brno, Česká republika) 1,0000 g, na ktorú sa nanášali štandardné roztoky Fe, Cu, Zn, Pb, As, Hg a Cd (v 2 % HNO3, c = 1,000 ± 0,002 g/l, certifikovaný referenčný materiál, Slovenský metrologický ústav Bratislava, SR). Pre zostrojenie kalibračných kriviek bolo na matricu – kys. benzoovú nanesené prvky Fe, Cu, Zn a Pb v rozsahu obsahov 0–50 μg. Pre stanovenie obsahu prvkov Pb, As, Hg a Cd vo vzorkách dextránov boli pripravené štyri štandardy. Štandard č. 1 obsahoval 50 μg Pb, štandard č. 2 50 μg As, štandard č. 3 50 μg Pb a 50 μg As a štandard č. 4 50 μg Pb, As, Hg a Cd. Po vysušení pri laboratórnej teplote a zhomogenizovaní sa z každého štandardu lisovali tri tablety pod tlakom 9 MPa, v hmotnosti cca 0,3000 g a priemere 20 mm. Tablety boli podrobené RRFA s použitím rádionuklidového zdroja 238Pu a 241Am.
Zo vzoriek sa lisovalo päť tabliet za rovnakých podmienok ako tomu bolo u štandardov. Pripravené vzorky tabliet dextránov boli podrobené RRFA s použitím rádionuklidového zdroja 238Pu a 241Am pri dobe merania 4000 s.
Analýza popolov
Do vyžíhaných porcelánových téglikov bolo navážené po 1,00 g skúšanej látky – dextránu. Sušili sa 1 hod. pri teplote 100 °C a vyžíhali do konštantnej hmotnosti v muflovej peci pri teplote 600 °C ± 25 °C. Zo spojených popolov po homogenizácii boli pripravené tablety pod tlakom 9 MPa, v hmotnosti cca 0,3000 g a priemere 20 mm, ktoré boli podrobené RRFA s použitím rádionuklidového zdroja 238Pu a 241Am.
Pre kvantitatívne vyhodnotenie bola použitá metóda štandardného prídavku. Na tabletu popola bolo pridané po 50 μg Fe, Cu a Zn a tableta bola podrobená RRFA.
Analýza dextránov v kvapalnom skupenstve
Pre stanovenie obsahu Mn, Fe, Co, Cu, Zn, Hg, Pb a Cd v kvapalných vzorkách, bol zvolený modifikovaný postup liekopisnej skúšky na ťažké kovy – metódy A so zachytením vzniknutých sulfidov na membránovom filtri. Namiesto predpísaného roztoku olova, ktorý uvádza SL 1, boli pre stanovenie jednotlivých prvkov použité štandardné roztoky Mn, Fe, Co, Cu a Zn. K 12,0 ml demineralizovanej vody sa pridalo 2,0 ml roztoku octanu amónneho (upraveného na pH 3,5; 4,5; 5,5; 6,5; 7,5; 8,5; 9,5; 10,5 a 11,5 prídavkom NaOH a HNO3) a 1,20 ml tioacetamidového skúmadla R. K uvedenej zmesi sa pridal definovaný objem štandardného roztoku prvku. Zmes sa po 4 minútach filtrovala cez membránový filter Pragopor 4 (priemer 35 mm, veľkosť pórov 3 μm, aktívna plocha 4,91 cm2, Pragochema spol. s r.o.) na filtračnom zariadení. Filter so zachytenými sulfidmi bol po vysušení podrobený RRFA. Rovnakým spôsobom za rovnakých podmienok boli pripravované slepé pokusy bez prídavku štandardných roztokov prvkov. Pre stanovenia jednotlivých prvkov boli vybrané optimálne pH zrážania (najvyšší počet impulzov).
Kalibračné krivky boli zostrojené na základe meraní štandardov obsahujúcich 0–50 μg prvkov pri optimálnom pH (Mn, Co, Cu, Pb pri pH 11,5 a Fe, Zn, Hg a Cd pri pH 8,5) zrážania sulfidov. Vzorky boli pripravené podľa SL1: skúšaný roztok S obsahoval 5,0 g dextránu doplnených do 50 ml demineralizovanou vodou. K 12,0 ml skúšaného roztoku bolo potom pridaných 2,0 ml octanového roztoku o pH 8,5 resp. 11,5 a 1,20 ml tioacetamidového skúmadla R. Po dvoch minútach bola zmes prefiltrovaná cez Pragopor 4 a filter bol po vysušení analyzovaný RRFA.
VÝSLEDKY A DISKUSIA
Vlastnej príprave štandardov predchádzalo overenie charakteru vzorky, najmä jej absorpčných vlastností. Pri RRFA nestanovované časti vzorky môžu ovplyvniť výsledok stanovenia. Lisovaním vzoriek do tabliet definovaného tvaru a hmotnosti bola zabezpečená konštantná hrúbka vzorky. Pretože dextrány sú hygroskopické látky, pred analýzou boli tablety sušené v exsikátore. Pri overovaní charakteru vzoriek nebola zistená prítomnosť prvkov, ktoré by v značnej miere ovplyvnili absorpčné vlastnosti vzorky. Z toho sa vychádzalo i pri príprave štandardov.
Pre overenie lineárnej závislosti nameranej početnosti od obsahu prvku vo vzorke boli pripravené štyri štandardy s obsahom Fe, Cu, Zn a Pb v rozsahu obsahov 0–50 μg prvkov na 1,0000 g kyseliny benzoovej. Nalisované tablety dextránov boli podrobené RRFA za použitia rádionuklidového zdroja 238Pu.
Korelačné koeficienty zostrojených kalibračných priamok potvrdili lineárnu závislosť početnosti od obsahu Fe, Cu, Zn, Pb. Parametre kalibračnej priamky pre jednotlivé prvky sa nachádzajú v tabuľke 1.
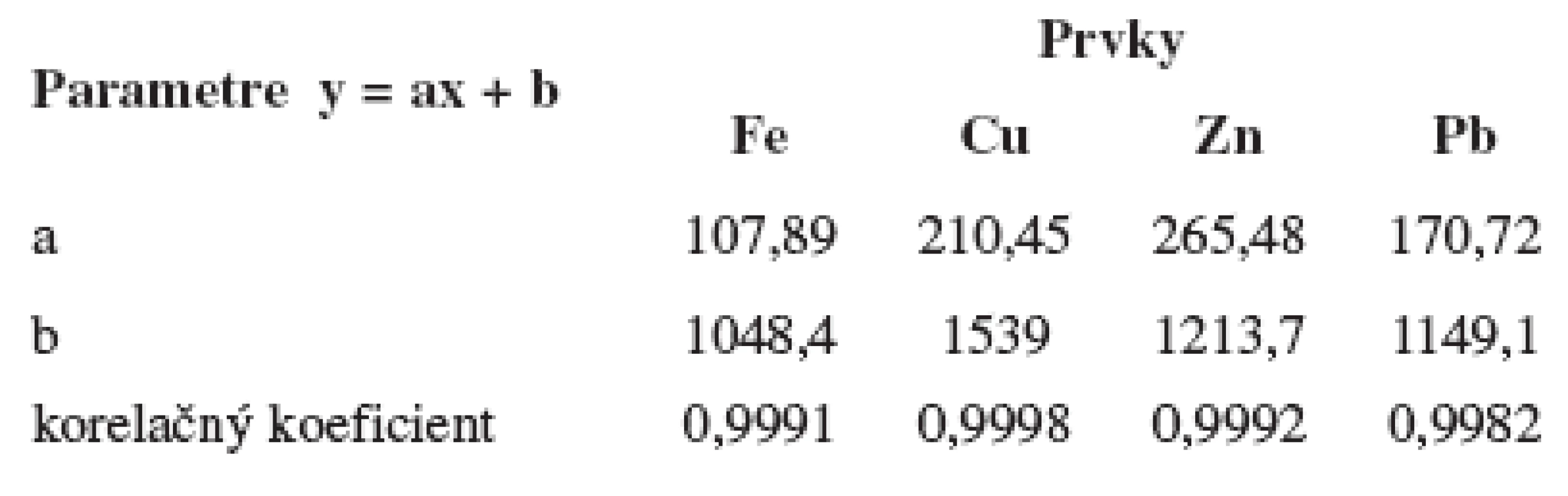
Pre zistenie prítomnosti Pb, As, Hg a Cd vo vzorkách bol použité rádionuklidové zdroje 238Pu a 241Am. Pretože energia Lα čiary Pb (10,549 keV) a energia Kα čiary As (10,543) ležia v tej istej energetickej línii, bolo nevyhnutné pripraviť štandardy obsahujúce len Pb, len As a štandard obsahujúci oba prvky a použitie oboch rádionuklidových zdrojov 238Pu a 241Am. Z nameraných výsledkov sa zistilo, že za použitia zdroja žiarenia 238Pu nebolo možné od seba odlíšiť píky As a Pb pretože energia Lα čiary Pb je 10,549 keV a energia Kα čiary As je 10,543 keV. Po zmeraní spektier zdrojom 241Am by bolo možné tieto prvky odlíšiť na základe Lß čiary Pb, ako je vidno na obrázku 1. Literatúra uvádza pomer Lα a Lß čiary 3 : 4, na základe čoho by sa dal určiť príspevok Pb v spoločnom píku Lα Pb a Kα As.

Na základe nameraných početností (počet impulzov/4000 s) neanalytického signálu (kyseliny benzoovej) meraného pri energiách zodpovedajúcich stanovovaným prvkov za použitia rádionuklidových zdrojov 238Pu a 241Am boli vypočítané detekčné limity pre Fe (1,01 μg/g), Cu (1,26 μg/g), Zn (0,72 μg/g), Pb (2,35 μg/g), As (0,97 μg/g), Hg (2,18 μg/g) a Cd (1,83 μg/g). Keďže Cd má absorpčný prah vyšší než je energia zdroja 238Pu, pre identifikáciu a stanovenie tohto prvku je možné použiť len 241Am. Pre Pb As a Hg je vhodnejší zdroj 238Pu, ktorý umožňuje dosiahnutie nižších detekčných limitov.
Po zmeraní spektier žiarenia oboch rádionuklidových zdrojov po interakcii s tabletami štandardov a vzoriek a po odpočte neanalytického signálu (kyselina benzoová) bol vypočítaný obsah prvkov v tabletách vzoriek dextránov, ktorý bol následne prepočítaný na 1,0000 g vysušenej vzorky. Výsledný obsah jednotlivých prvkov uvádzá tabuľka 2. Obsah olova, arzénu, ortuti a kadmia bol u obidvoch vzoriek pod detekčným limitom.

Medzi používané spôsoby zakoncentrovania tuhých biologických látok patrí aj mineralizácia suchou cestou. Popol získaný mineralizáciou vzoriek dextránov bol podrobený RRFA za obvyklých podmienok. Z výsledku analýzy vyplynulo, že signál v energetických oblastiach prvkov Pb, As, Hg a Cd vykazoval početnosti zhodné s neanalytickým signálom, čo potvrdzuje predpoklad prchania týchto prvkov pri vysokých teplotách. Významné početnosti sa vyskytovali v energetických oblastiach prvkov Fe, Cu a Zn. Kvantitatívne hodnotenie bolo realizované metódou štandardného prídavku stanovovaných prvkov priamo do popola. Boli vypočítané detekčné limity pre jednotlivé prvky ako početnosti zodpovedajúce obsahu 1 μg prvku po korekcii o neanalytický signal. Výsledky sú uvedené v tabuľke 3.

V ďalšej časti experimentu bol stanovený obsah ťažkých kovov v dextránoch vo vodných roztokoch. Bolo potrebné prvky z roztoku zakoncentrovať, za tým účelom bola použitá modifikovaná Limitná skúška na ťažké kovy (reakcia s tioacetamidovým skúmadlom). Vyhodnotenie pomocou RRFA bolo realizované po filtrácii vzniknutých sulfidov cez nitrocelulózový filter Pragopor 4.
Vlastnú analýzu vzoriek predchádzalo sledovanie podmienok tvorby sulfidov (pH) zo štandardných roztokov prvkov v rozpätí pH 3,5–11,5. Z výsledkov vyplynulo, že kvôli žiaducej polykomponentnej analýze, vybrané prvky musia byť rozdelené na dve skupiny: prvky, ktoré sa zrážajú s tioacetamidovým skúmadlom pri optimálnom pH 8,5 ako Fe, Zn, Hg a Cd a prvky, ktoré sa optimálne zrážajú pri pH 11,5 ako Mn, Co, Cu a Pb.
Po optimalizácii podmienok analýzy boli zhotovené kalibračné priamky pre jednotlivé prvky. Lineárna závislosť nameraných početností od obsahu prvku bola potvrdená vypočítanými korelačnými koeficientami, ktoré sú uvedené v tabuľke 4.
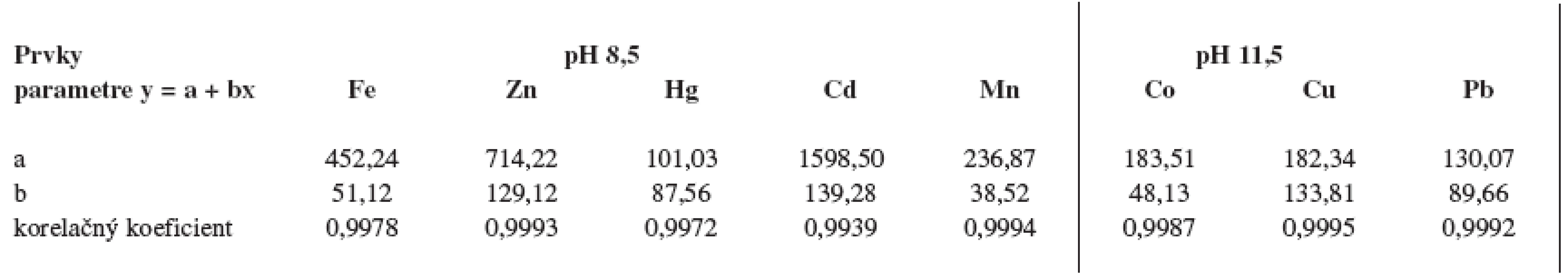
Po analýze vzoriek boli vypočítané detekčné limity ako početnosti zodpovedajúce obsahu 1 μg prvku po korekcii o neanalytický signál. Detekčné limity a obsah prvkov boli vypočítané v 1 g vzorky. Výsledky sú uvedené v tabuľke 5.

Z uvedených vysledkov vyplýva, že analýza dextránov v tuhom a kvapalnom skupenstve RRFA metódou je v uvedených prípadoch rýchla, selektívna a citlivá. Vyžaduje si jednako prípravu vhodných štandardov pre tuhú vzorku a vhodné skúmadla na zakoncentrovanie z kvapalnej vzorky ako aj vhodnú voľbu rádionuklidového zdroja žiarenia. Z výsledkov analýz tuhých vzoriek vyplynulo, že je možné s výhodou využiť mineralizáciu suchou cestou, pri ktorej dochádza k zakoncentrovaniu niektorých prvkov (Fe, Cu a Zn). Nevýhodou je prchanie niektorých prvkov počas mineralizácie, ktoré znemožňuje ich stanovenie v popole. Porovnaním výsledkov obsahov stanovovaných prvkov tuhej a kvapalnej vzorky vyplynulo, že zakoncentrovaním kvapalnej vzorky sa dosiahne vyššia citlivosť (zníženie detekčných limitov). Pri predpokladaných veľmi nízkych obsahov prvkov vo vzorkách je teda výhodné zakoncentrovanie z kvapalného média, pri vyšších obsahoch sú použiteľné oba spôsoby, ako vidno z výsledkov stanovania obsahu zinku.

Bola vypracovaná dostatočne selektívna a citlivá metóda, ktorou je možné doplniť skúšku na ťažké kovy. Uvedené výsledky potvrdili opodstatnenosť zaradenia RRFA medzi liekopisné metódy nakoľko umožňuje rýchlu, nedeštruktívnu a polykomponentnú analýzu vzoriek bez nutnosti zložitej predúpravy vzorky pred samotnou analýzou. Vyššiu citlivosť a selektívnosť danej metódy je možné dosiahnuť zmenou skúmadla (zrážacie, komplexotvorné ...), podmienok analýzy (typ filtrov ...) a ich optimalizáciou (pH, objem pridávaného skúmadla...), ako i zmenou technických parametrov inštrumentálnerj metódy (variabilita zdrojov, geometria ...). Z porovnania vypočítaných detekčných limitov pri jednotlivých prípravách vzoriek na analýzu je zrejmé, že využitie zrážacích reakcií (tioacetamidové skúmadlo) na zakoncentrovanie vybraných prvkov je výhodné, čo vyplýva z dosiahnutých nízkych detekčných limitov.
Táto práca je zahrnutá do výskumných projektov grantovej agentúry MŠ SR VEGA č. 1/4299/07, 1/0003/08 a grantu UK/97/2008.
Došlo 23. března 2009 / Přijato 20. dubna 2009
Adresa pre korešpondenciu:
PharmDr. Veronika Jánošová
Katedra farmaceutickej analýzy a nukleárnej farmácie FaF UK
Odbojárov 10, 832 32 Bratislava, SR
e-mail: janosova@fpharm.uniba.sk
Sources
1. Slovenský liekopis. 1. vyd., zv. I., Bratislava: Herba, 1997; s. 657.
2. Slovenský liekopis. 1. vyd., zv. II., Bratislava: Herba, 1999; s. 1400.
3. Sofikitis, A.M. et al.: Anal. Bioanal. Chem., 2004; 378, 460–464.
4. Budič, B.: Anal. Bioanal.Chem., 2000; 368, 371–377.
5. Yang, G. et al.: J. Anal. Chem., 2005; 60, 480–485.
6. Zaichick, V., Dyatlov, A., Zaichick, S.: J. Radioanal. Nucl. Chem., 2000; 244, 189–193.
7. Lartigue, J. et al.: J. Radianal. Nucl. Chem, 2007; 273, 759–762.
8. Lalor, G. C., Onianwa, P. C., Vutchkov, M. K.: Int. J. Environ. Anal. Chem, 2003; 83, 367–374.
9. Baranowska, I. et al.: Pol. J. Environ. Stud., 2002; 11, 467–471.
10. Jiang, Y.: Microchim. Acta, 2008; 161, 137–142.
11. Soylak, M. et al.: Environ. Monit. Assess., 2007; 127, 169–176.
12. Gordeeva,V.P. et al.: J. Anal. Chem., 2002; 57, 701–707.
13. Giokas, D. L., Eksperiandova, L. P., Blank, A. B.: Anal. Chim. Acta, 2004; 505, 51–58.
14. Štroffeková O. et al.: J. Radioanal. Nucl. Chem., 2008; 275, 659–664.
15. Tölgyessy, J., Havránek, E., Dejmková, E.: Rádionuklidová röntgenofluorescenčná analýza zložiek životného prostredia. Alfa, Bratislava, 1983, s. 203
Labels
Pharmacy Clinical pharmacologyArticle was published in
Czech and Slovak Pharmacy

2009 Issue 2
Most read in this issue
- Beta-blokátory
- K pojmům farmaceutická péče, lékárenská péče a správná lékárenská praxe
- Příznivé účinky rutinu, kvercitrinu a kvercetinu na nespecifické střevní záněty
- Využitie rádionuklidovej röntgenofluorescenčnej analýzy pri stanovení obsahu ťažkých kovov v dextránoch v tuhom a kvapalnom skupenstve