
Antagonisty angiotenzínových AT1 receptorov
Antagonists of angiotensin AT1 receptors
The medicinal agents which block AT1 receptors are used for the treatment of hypertension, cardiac failure, and renal diseases. In clinical practice they have been available already for 15 years. All antagonists of AT1 receptors were projected without any knowledge of the space (3D) structure of the AT1 receptor. At present several strategies of projecting antihypertensive drugs are used, which are based on the influence on a number of receptors (e.g. the endothelial ones), not only angiotensin AT1 receptors. With the use of rational methods of drug projecting based on the knowledge of the spatial structure of the active site of the AT1 receptor, a development of new highly effective ligands for the therapy of cardiovascular diseases will be made possible. In the future, a development of drugs can be expected, which will block not only the angiotensin AT1 receptors, but which will be able to influence also other endogenous ligands, e.g. endothelin and nitric oxide.
Key words:
RAAS – AT1 receptors – antagonists of AT1 receptors
Authors:
Pavol Ježko; Milan Remko
Authors‘ workplace:
Univerzita Komenského v Bratislave, Farmaceutická fakulta, Katedra farmaceutickej chémie
Published in:
Čes. slov. Farm., 2011; 60, 171-181
Category:
Review Articles
Overview
Liečivá, ktoré blokujú AT1 receptory sa používajú v liečbe hypertenzie, srdcového zlyhania a ochorení obličiek. V klinickej praxi sú dostupné už 15 rokov. Všetky antagonisty AT1 receptorov boli projektované bez znalosti priestorovej (3D) štruktúry AT1 receptora. V súčasnej dobe sa používajú viaceré stratégie projektovania antihypertenzívnych liečiv, ktoré sú založené na ovplyvnení viacerých receptorov (napr. endotelínových), nielen angiotenzínových AT1 receptorov. S použitím racionálnych metód projektovania liečiv založených na poznaní priestorovej štruktúry aktívneho miesta AT1 receptora bude možný vývoj nových vysoko účinných ligandov pre terapiu kardiovaskulárnych ochorení. V budúcnosti je možné očakávať vývoj liečiv, ktoré budú blokovať nielen angiotenzínové AT1 receptory, ale budú schopné ovplyvniť aj iné endogénne ligandy, napríklad endotelín a oxid dusnatý.
Kľúčové slová:
RAAS – AT1 receptory – antagonisty AT1 receptorov
Úvod
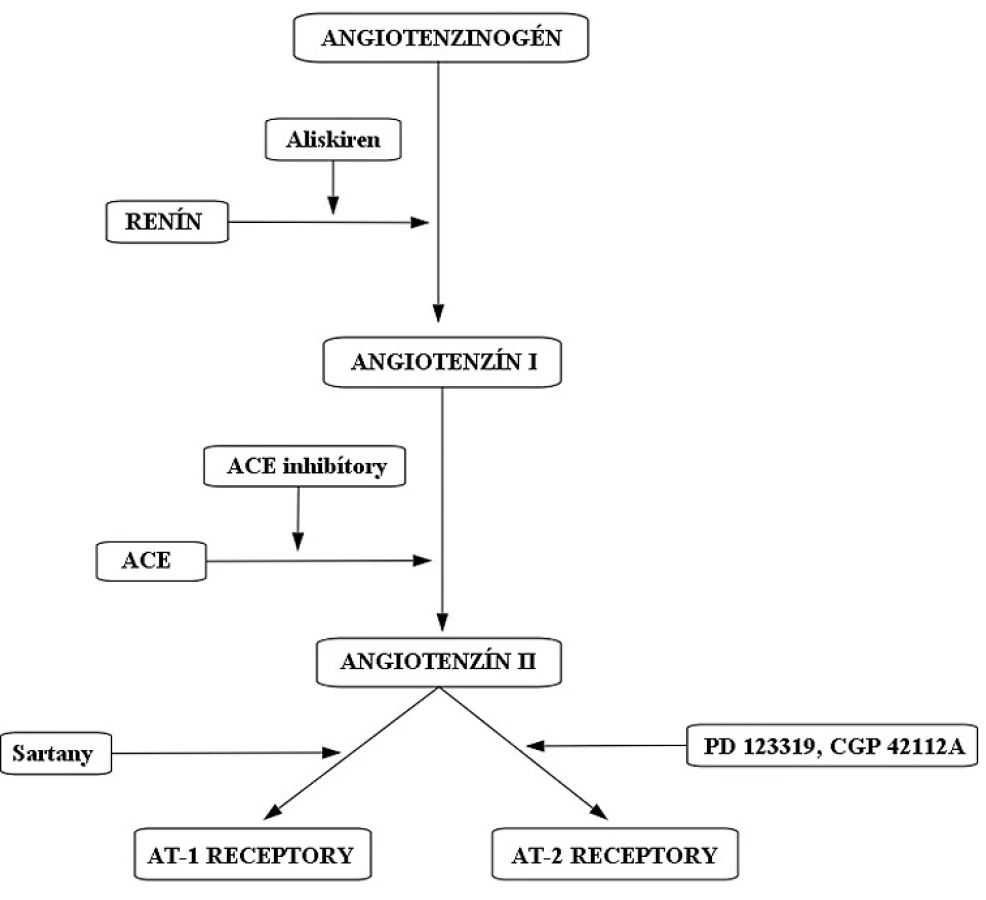
Systém renín-angiotenzín-aldosterón (RAAS) (obr. 1) má kľúčovú úlohu v regulácií hodnôt krvného tlaku (TK). Zasahuje aj do homeostázy tekutín prostredníctvom aldosterónu. Tento systém sa podieľa na patogenéze hypertenzie1), kongestívnom srdcovom zlyhaní2) a chronickom zlyhaní obličiek3). Liečivá, ktoré majú schopnosť ovplyvniť aktivitu RAAS, sú využívané v liečbe kardiovaskulárných ochorení. Na začiatku celej kaskády stojí renín. Ten je do krvného obehu uvoľňovaný na základe viacerých stimulov z juxtaglomerulárnych buniek obličiek. Renín katalyzuje vznik angiotenzínu I z angiotenzinogénu. V klinickej praxi sa na inhibíciu renínu používa liečivo aliskirén4). Angiotenzín konvertujúci enzým (ACE) mení angiotenzín I na angiotenzín II. ACE inhibítory zabraňujú vzniku angiotenzínu II tým, že blokujú enzým ACE (napr. perindopril, ramipril). Angiotenzín II je oktapeptidový primárny mediátor RAAS s krátkym biologickým polčasom (1–2 minúty). Angiotenzín II pôsobí na svoje receptory (AT1 a AT2). Sartany (napr. losartan) špecificky antagonizujú účinok angiotenzínu II na AT1 receptoroch (obr. 1).
Angiotenzínové (AT) receptory
V súčasnosti sú známe dva hlavné typy AT-receptorov: AT1 a AT2 5). Sú to špecifické receptory nachádzajúce sa na povrchu buniek. AT1 receptor má dva podtypy: AT1A a AT1B. AT1A receptory sú postsynaptické a AT1B receptory sú presynaptické6). Stimuláciou AT1 receptorov dochádza k vazokonstrikcii, zvýšeniu aktivity sympatika, hyperplázii a hypertrofii buniek, produkcii aldosterónu. AT2 receptory majú antihypertrofické, vazodilatačné, antifibrotické, neuroprotektívne, renoprotektívne, natriuretické, vazoprotektívne a antiproliferatívne účinky7).
Štruktúra AT1 receptorov
AT1 receptor je integrálny membránový proteín, ktorý pozostáva z 359 aminokyselín8). AT receptory patria do skupiny membránovo viazaných receptorov spriahnutých s G-proteínom (GPCR). GPCR sa nachádzajú na vonkajšej bunkovej membráne všetkých buniek. Pozostávajú zo siedmych transmebránových helixov, s extracelulárnym amino-koncom a intracelulárnym karboxylovým koncom. Zatiaľ je presne známa iba primárna štruktúra AT1 receptora. Priestorové 3D modely AT1 receptora boli zhotovené len na základe kryštálovej štruktúry rodopsínu (napr. PDB: 1F88).
Konformačné stavy AT1 receptora
Predpokladá sa, že aj AT1 receptor existuje v rôznych konformačných stavoch. Na aktívnu konformáciu sa viažu agonisty s vysokou afinitou. Tým dochádza k aktivácii signálnych ciest a ďalších procesov. Inverzné agonisty (sartany) sa viažu na neaktívnu konformáciu s vysokou affinitou9).
Vývoj antagonistov AT1 receptorov
Ako prvý antagonista AT receptorov bol testovaný saralazín. Saralazín (Sar1-ALA8-angiotenzín II) bol objavený v sedemdesiatych rokoch dvadsiateho storočia. Je to prvý oktapeptidový analóg angiotenzínu II, u ktorého boli aminokyseliny ASP-1 a PHE-8 zmenené za sarkozín-1 (N-metylglicín) a ALA-8. Tento peptid mal limitovanú terapeutickú hodnotu, kvôli nízkej biologickej dostupnosti, krátkej dobe účinku a významným agonistickým vlastnostiam10, 11).
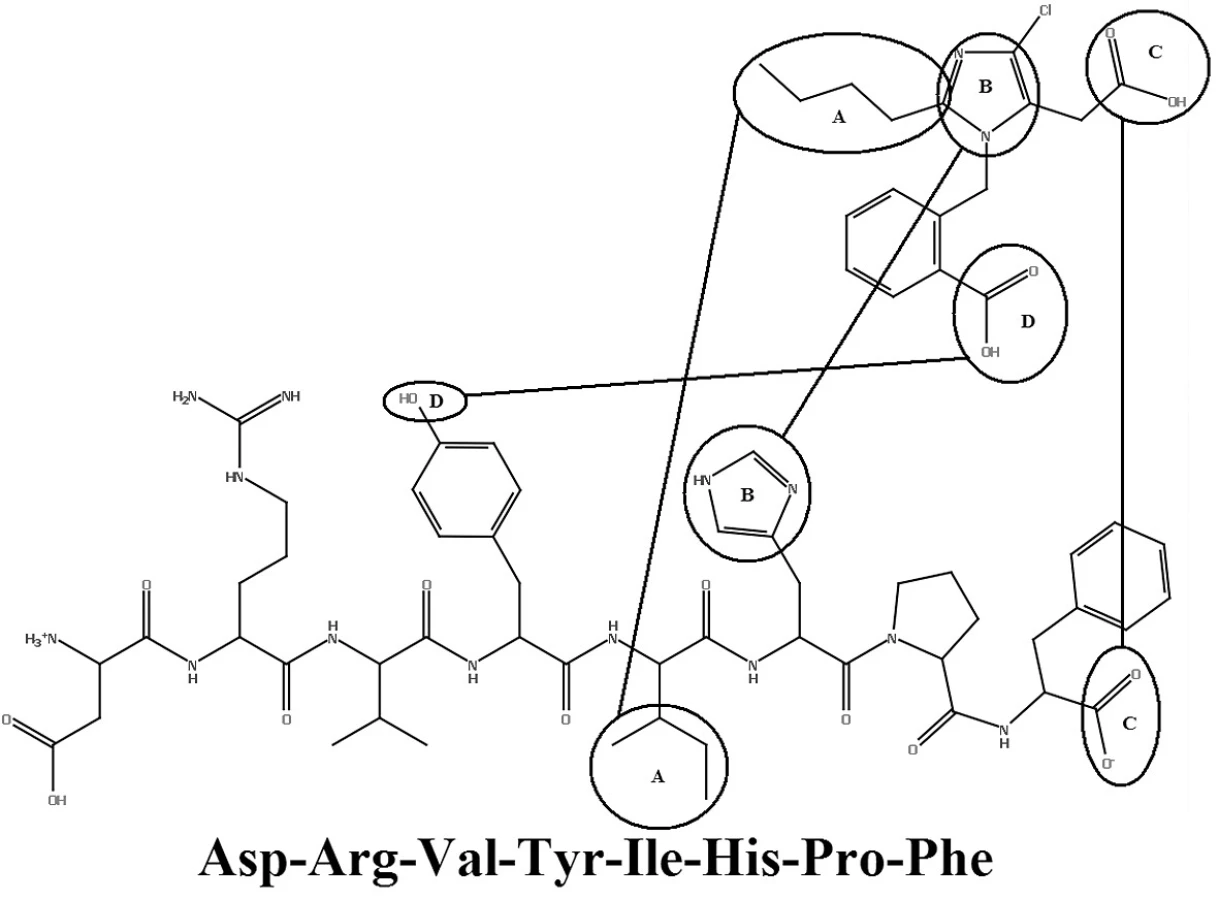
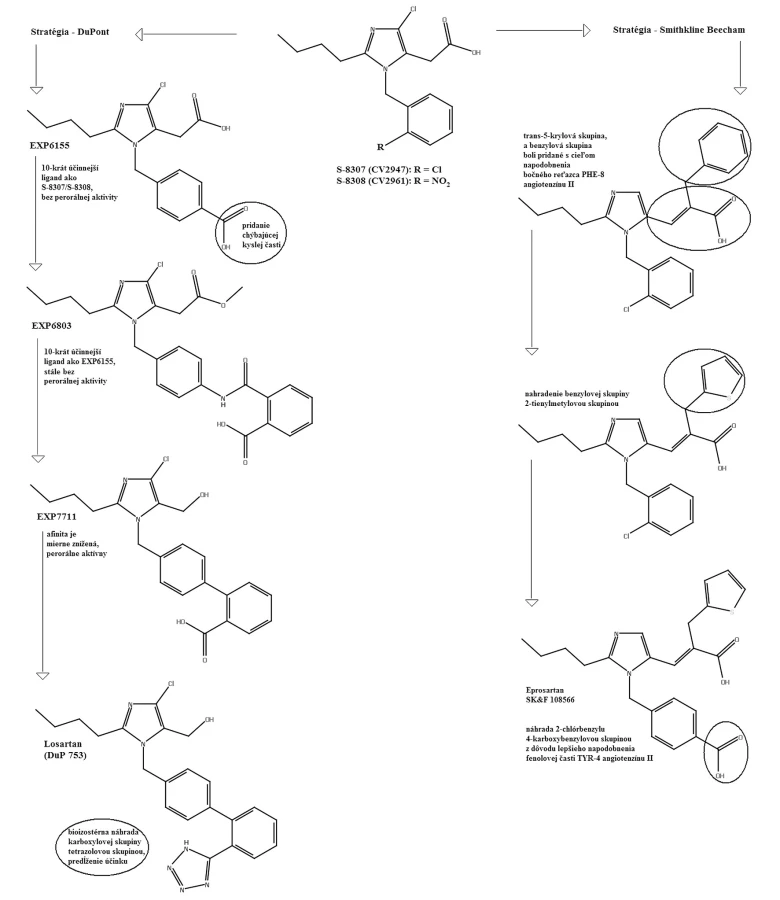
V roku 1982 Furukawa a jeho spolupracovníci z Takeda Chemical Industries objavili sériu derivátov 1-benzylimidazol-5-octovej kyseliny12, 13). V týchto patentových publikáciách bolo niekoľko zlúčenín, ktoré však neobsahovali informácie o selektivite týchto látok. Dve látky z Takedovej série, S-8307 (CV2947) a S-8308 (CV2961), boli pripravené a dôkladne študované skupinou DuPont. Potvrdilo sa, že sú to slabé ale selektívne kompetitívne antagonisty AT1 receptorov bez agonistického účinku14, 15). Zlepšenie účinnosti Takedových 1-benzylimidazol-5-octových kyselín, derivátov S-830716) bolo možné s použitím Fermandjianovho modelu konformácie angiotenzínu II. Zo štruktúrnych vlastností angiotenzínu II bolo známe, že C-koncový segment tohto peptidu je dôležitým elementom pri väzbe na receptor, čo dalo základ hypotéze, že malá molekula môže napodobniť interakciu tejto časti peptidu prostredníctvom karboxylovej skupiny kyseliny octovej. Jadrom superpozície bolo prekrytie medzi imidazolovou skupinou S-8307 s HIS-6 angiotenzínu II. Lipofilná n-butylová skupina v pozícii 2 imidazolu sa zhodovala s bočným reťazcom ILE-5 angiotenzínu II. Benzylová skupina molekuly S‑8307 bola obmenená v para polohe, čo mohlo zvýšiť účinnosť tým, že sa simuloval bočný reťazec TYR-4 angiotenzínu II. Tento proces viedol k príprave ligandu EXP 6155, ktorý bol 10 krát účinnejší ako pôvodná zlúčenina S-830717). Tento počiatočný úspech viedol k vývoju derivátov ligandu EXP 6155. U zlúčeniny EXP 771118) bola amidová spojovacia skupina nahradená jednoduchou väzbou. Zvýšila sa tak jej perorálna aktivita. Na ďalšie zvýšenie perorálnej aktivity bifenylových derivátov boli kyslé skupiny, ako karboxylová skupina, systematicky bioizostérne nahradené tetrazolovou skupinou. Táto skupina značne zvýšila účinnosť a dobrú perorálnu antihypertenzívnu aktivitu.
Pomocou jednoduchých štruktúrnych modelov v molekulovom projektovaní (obr. 2) sa demonštrovala možnosť použitia nepeptidových derivátov antagonistov AT1 receptorov. Celý proces vývoja vyvrcholil objavením losartanu (COZAAR®, tiež označovaný DuP 753 a MK-954) (obr. 3), ktorý je prototypom skupiny účinných, perorálne aktívnych, nepeptidových antagonistov AT1 receptorov20).
Skupina pracujúca vo farmaceutickej spoločnosti Smith-Kline Beecham tiež založila svoju projektovaciu stratégiu na kombinácii molekulového modelovania a štruktúrnych vlastností peptidov20). Päťnásťnásobné zvýšenie väzbovej aktivity bolo realizované rozšírením reťazca v polohe 5-imidazolu prostredníctvom trans-5-akrylovej kyslej skupiny. Následne bola pridaná benzylová skupina, ktorá mala lepšiu podobnosť s bočným reťazcom PHE-8. Nahradenie benzylovej skupiny 2-tienylmetylovou skupinou a následná náhrada –2-chlórbenzylu 4-karboxybenzylovou skupinou lepšie napodobnila fenolovú časť TYR-4 angiotenzínu II, čo viedlo k vývoju eprosartanu (SK&F 108566) (obr. 3).
Molekulový mechanizmus účinku antagonistov AT1 receptorov
Mechanizmus väzby antagonistov na ľudský AT1 receptor ešte nie je úplne objasnený. Literatúra uvádza dôležité údaje o častiach aktívneho miesta, kde prebieha interakcia s rôznymi antagonistami AT1 receptorov21–24). Viaceré štúdie vychádzajú z homológnych bielkovín.
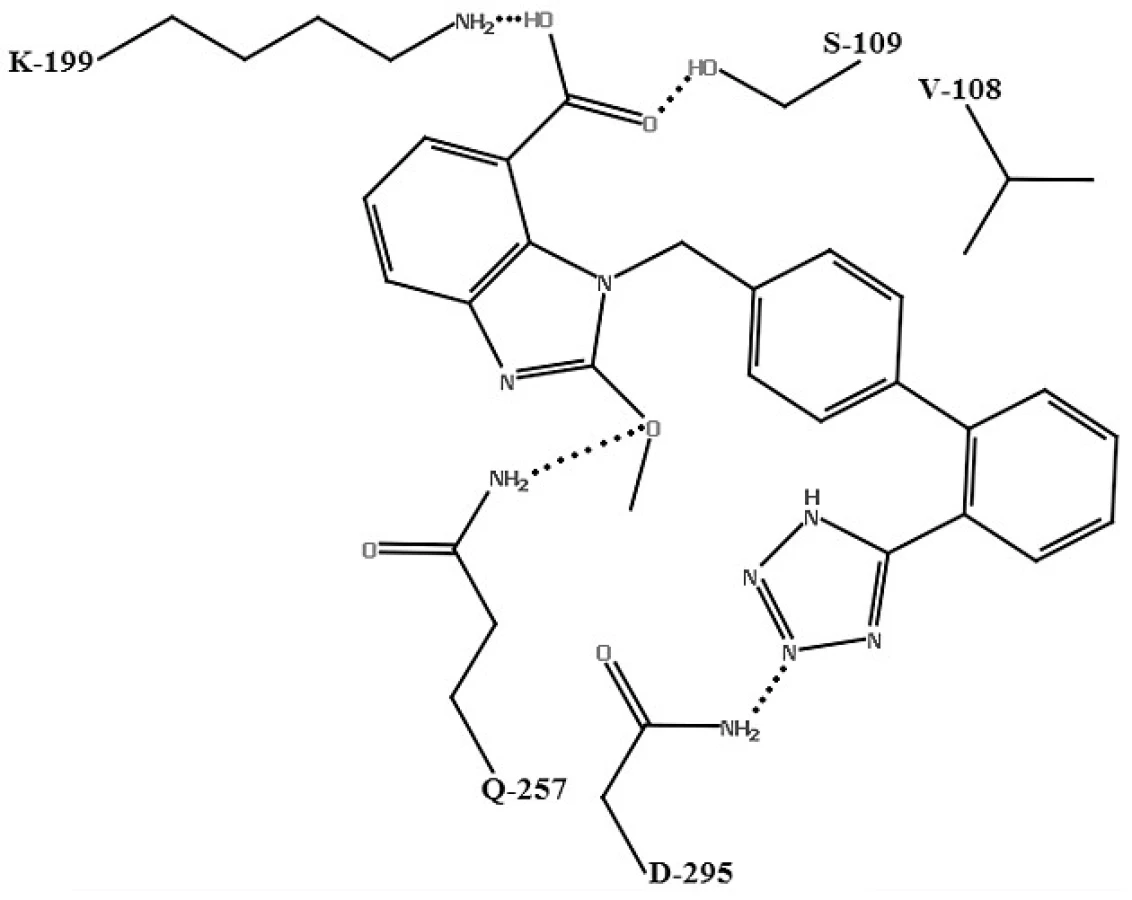
Pre kandesartan, valsartan a losartan25) bol navrhnutý nasledovný molekulový mechanizmus účinku: dva atómy dusíka na tetrazolovom kruhu sartanov tvoria vodíkové väzby s ASN-295 AT1 receptora. Karboxylová skupina interaguje so SER-109 a LYS-199. LYS-199 je dôležitý pre interakciu s karboxylovou skupinou antagonistov AT1 receptora. Atóm kyslíka v etoxy-skupine na benzimidazolovej časti candesartanu môže naviac tvoriť ďalšiu vodíkovú väzbu s GLN-257. Počet interakcií medzi aktívnym miestom a kandesartanom je v zhode s jeho vyššou väzbovou affinitou oproti valsartanu. Keďže losartan nemá vo svojej štruktúre karboxylovú skupinu, môže tvoriť vodíkovú väzbu iba so SER-109. Hydroxylová skupina v hydroxymetylovej časti losartanu má slabšiu interakciu so SER-109 ako karboxylová skupina kandesartanu a valsartanu. Losartan vykazuje najmenší počet vodíkových interakcií s aktívnym miestom, čo je v zhode s jeho najnižšou väzbovou affinitou k receptoru (obr. 4).
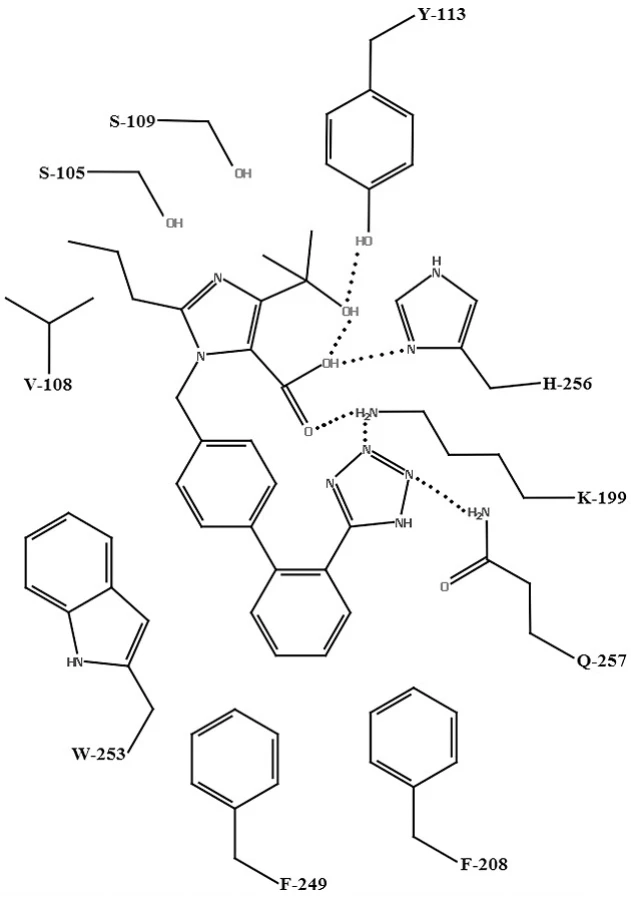
Na vytvorenie homológneho modelu AT1 receptora sa použila RTG štruktúra hovädzieho rodopsínu. Tento model bol použitý na vysvetlenie molekulového mechanizmu účinku olmesartanu (obr. 5), kryštálová štruktúra PDB: 1F88, a telmisartanu (obr. 6), kryštálová štruktúra PDB: 1U19. Hydroxylová skupina olmesartanu21) interaguje s TYR-113 AT1 receptora. Tetrazolová skupina olmesartanu interaguje s karbamoylovou skupinou GLN‑257. Medzi karboxylovou skupinou na imidazole olmesartanu je elektrostatická interakcia s LYS-199 AT1 receptora. Interakcia medzi karboxylovou skupinou a HIS-256 nie je nevyhnutná, pretože strata tejto väzby spôsobená mutáciou HIS-256 môže byť vykompezovaná interakciou medzi karboxylovou skupinou a LYS-199. LYS-199 je tiež v blízkosti tetrazolovej skupiny (vzdialenosť 2,8 Ā), rovnakej vzdialenosti ako medzi LYS-199 a karboxylovou skupinou. Táto vzdialenosť naznačuje prítomnosť elektrostatickej interakcie medzi LYS-199 a tetrazolovou skupinou. Bifenylová časť olmesartanu sa nachádza v lipofilnej oblasti pozostávajúcej z PHE-208, PHE-249, a TRP-253 tohto modelu (3,7–4,5 Å), ktorá sa nachádza v triede A rodine GPCR26).
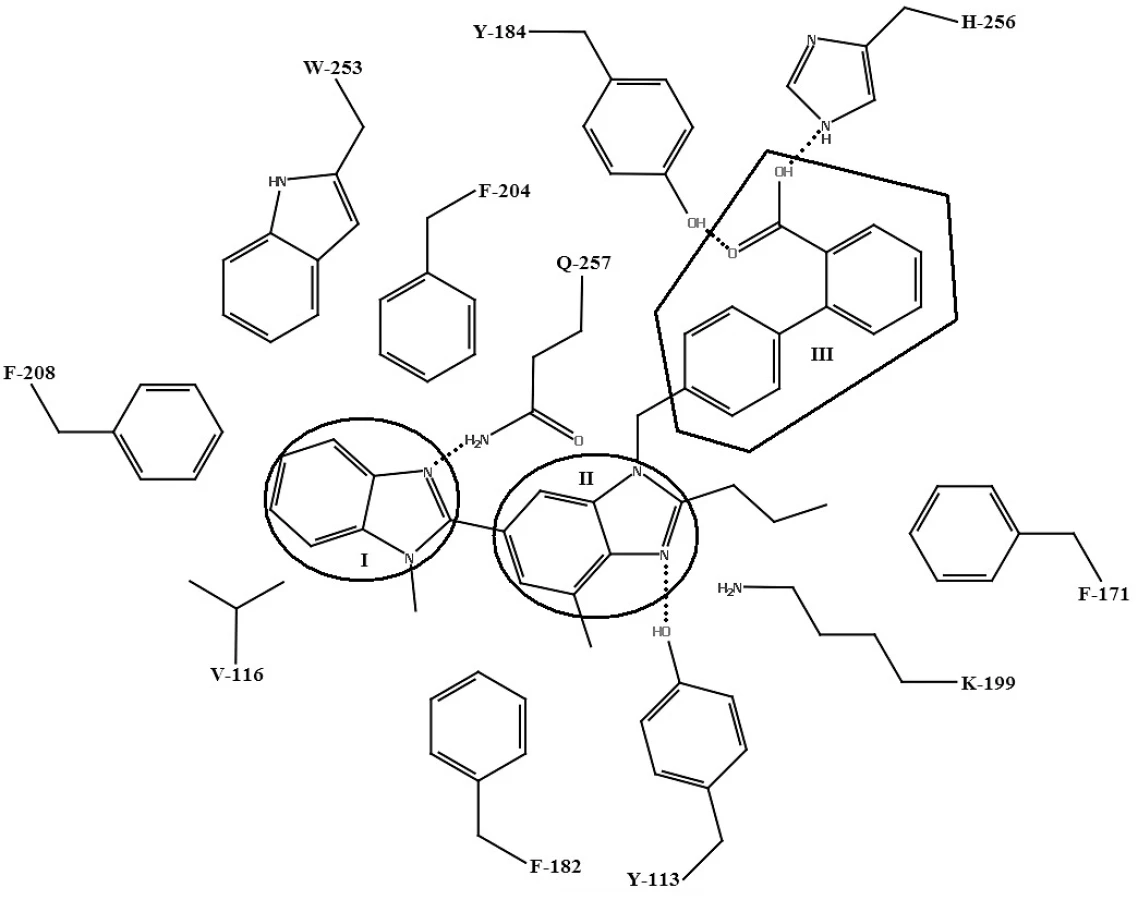
Telmisartan27) podlieha viacerým interakciám s aktívnym miestom, ktoré nie sú prítomné pri väzbe s inými sartanmi. Ide o interakcie s hydrofóbnymi bočnými aminokyselinovými reťazcami AT1 receptora: VAL-116, PHE-204, PHE-208 a TRP-253. Tieto aminokyselinové zvyšky v lipofilnej časti aktívneho miesta interagujú s benzimidazolovou časťou telmisartanu (I). Centrálna benzimidazolová časť telmisartanu (II) interaguje s TYR-113 (vodíková väzba), PHE-171, PHE-182 (hydrofóbne interakcie) a LYS-199 (katión-π interakcia). Bifenylová časť s karboxylovou kyselinou (III) interaguje s TYR-184 (vodíková väzba) a HIS-256 (iónová interakcia) (obr. 7)27).
Klinicky používané liečivá
Typický antagonista AT1 receptorov obsahuje vo svojej štruktúre tieto funkčné skupiny:
- centrálnu imidazolovú časť, na ktorú môže byť naviazaný benzénový kruh,
- bifenylovú časť viazanú na imidazolový kruh,
- kyslú skupinu, a to tetrazol alebo karboxyl, ktorý interaguje so zvyškom aminokyseliny GLN. Orto-pozícia tejto kyslej skupiny je dôležitá pre vytočenie bifenylovej časti z planárneho usporiadania (nekoplanárna konformácia)28).
- 3 - až 4-uhlíkový alkylový reťazec alebo alkénový reťazec20),
- karboxylovú skupinu, alebo skupinu, ktorá je akceptorom vodíkovej väzby. Kyslý karboxyl spôsobuje dlhodobý antagonismus, zvyšuje väzbu na plazmatické bielkoviny20) a spôsobuje rozdiely medzi in vitro a in vivo pokusmi.
Losartan (I) (COZAAR®, MERCK) bol prvým liečivom zavedeným do klinickej praxe. Hlavný účinok losartanu je pripisovaný jeho metabolitu EXP 3174 (II)29). Je to imidazol-5-karboxylová kyselina, výsledný produkt oxidácie imidazol 5-hydroxymetylovej skupiny losartanu. Bol identifikovaný ako hlavný metabolit losartanu v plazme potkanov30).


Valsartan (III) (DIOVAN®, NOVARTIS) nie je heterocylický antagonista AT1 receptorov. Imidazolový kruh, prítomný v losartane, je nahradený u valsartanu acylovanou aminokyselinou.

Základný skelet irbesartanu (IV) (AVAPRO®, SANOFI AVENTIS US) tvorí 4-spirocyklopentánimidazolín-5-ón. Karbonylová skupina irbesartanu je akceptorom vodíkovej väzby.

Eprosartan (V) (TEVETEN®, ABBOTT) je odlišný od ostatných antagonistov, pretože vo svojej štruktúre neobsahuje bifenyl-tetrazolový kruh. Namiesto tejto časti má karboxybenzyl. Karboxylová skupina, prítomná v losartane, je nahradená 2-(tiofén-2-ylmetyl)prop-2-énovou kyselinou.

Viaceré antagonisty AT1 receptorov obsahujú vo svojej štruktúre dve kyslé skupiny. Kandesartan (VI), olmesartan (VIII) a azilsartan (X) sa v tejto forme zle vstrebávajú, preto boli pripravené ich prodrug formy: kandesartan cilexetil (VII), olmesartan medoxomil (IX), azilsartan medoxomil (XI) so zlepšenou perorálnou dostupnosťou.






Kandesartan (ATACAND®, ASTRAZENECA) je benzimidazolový analóg losartanu. V porovnaní s losartanom má namiesto butylovej skupiny etoxy-skupinu.
Telmisartan (XII) (MICARDIS®, BOEHRINGER INGELHEIM) má v porovnaní s ostatnými antagonistami vo svojej štruktúre karboxylovú kyselinu naviazanú na bifenylovú časť. Táto karboxylová skupina je účinnejšia ako jej tetrazolový analóg31). Imidazolová časť losartanu je zmenená za benzimidazol.

Olmesartan (olmesartan medoxomil, BENICAR®, DAIICHI SANKYO) obsahuje vo svojej štruktúre hydroxyizopropyl.
Azilsartan (azilsartan medoxomil, EDARBI®, TAKEDA PHARMACEUTICALS) je analóg kandesartanu. V porovnaní s kandesartanom má namiesto tetrazolu 4,5-dihydro-1,2,4-oxadiazol-5-ón.
Antagonisty AT1 receptorov v pokročilom štádiu vývoja
Embusartan (XIII) (BAY 10-6734), s dihydropyridínónovým kruhom, je novo vyvinutý, perorálne účinný AT1 antagonista32). BAY 10-6735 (XIV) je terapeuticky účinná látka, vytvorená hydrolýzou embusartanu. Je účinnejší ako losartan33). Embusartan vykazuje kompetetívny, ale BAY 10-6735 nekompetitívny antagonizmus34). Doba pôsobenia embusartanu je 24 hodín33). V porovnaní s losartanom, ľahko prekračuje hematoencefalickú bariéru vzhľadom k vysokému lipofilnému charakteru (log P = 5,49)35). KRH-594 (XV), acyliminotiadiazolín, je selektívny ARB a rovnako účinný ako EXP 317436). Nemá žiadny aktívny metabolit a viaže sa silne na AT1 receptor dlhodobým spôsobom. Zlepšuje komplikácie spôsobené hypertenziou, ako je zlyhanie obličiek, srdcová hypertrofia a zhrubnutie steny tepny. KRH-594 má pozitívne výsledky u diabetických komplikácií, ako je nefropatia a hyperlipidémie u spontánne hypertenzných potkanov (DM-1K-SHR)37). KT3-671 (XVI) (KD3-671) má sedem-členný kruh pripojený na imidazolový kruh. Je to silný, kompetetívny, selektívny antagonista AT1 receptora38). Má asi 7× vyššiu afinita k AT1 receptoru v membráne pečeni potkana ako losartan. U potkanov39) a psov40) KT3-671 vytvára trvalý pokles krvného tlaku po perorálnom podaní, nemá vplyv na normálnu funkciu obličiek a znižuje hypertenziou-indukované patologické zmeny v obličkách41). Nedávno bola zistená jeho cievna sympatoinhibičná aktivita42).




Duálne antagonisty angiotenzínu II a endotelínu
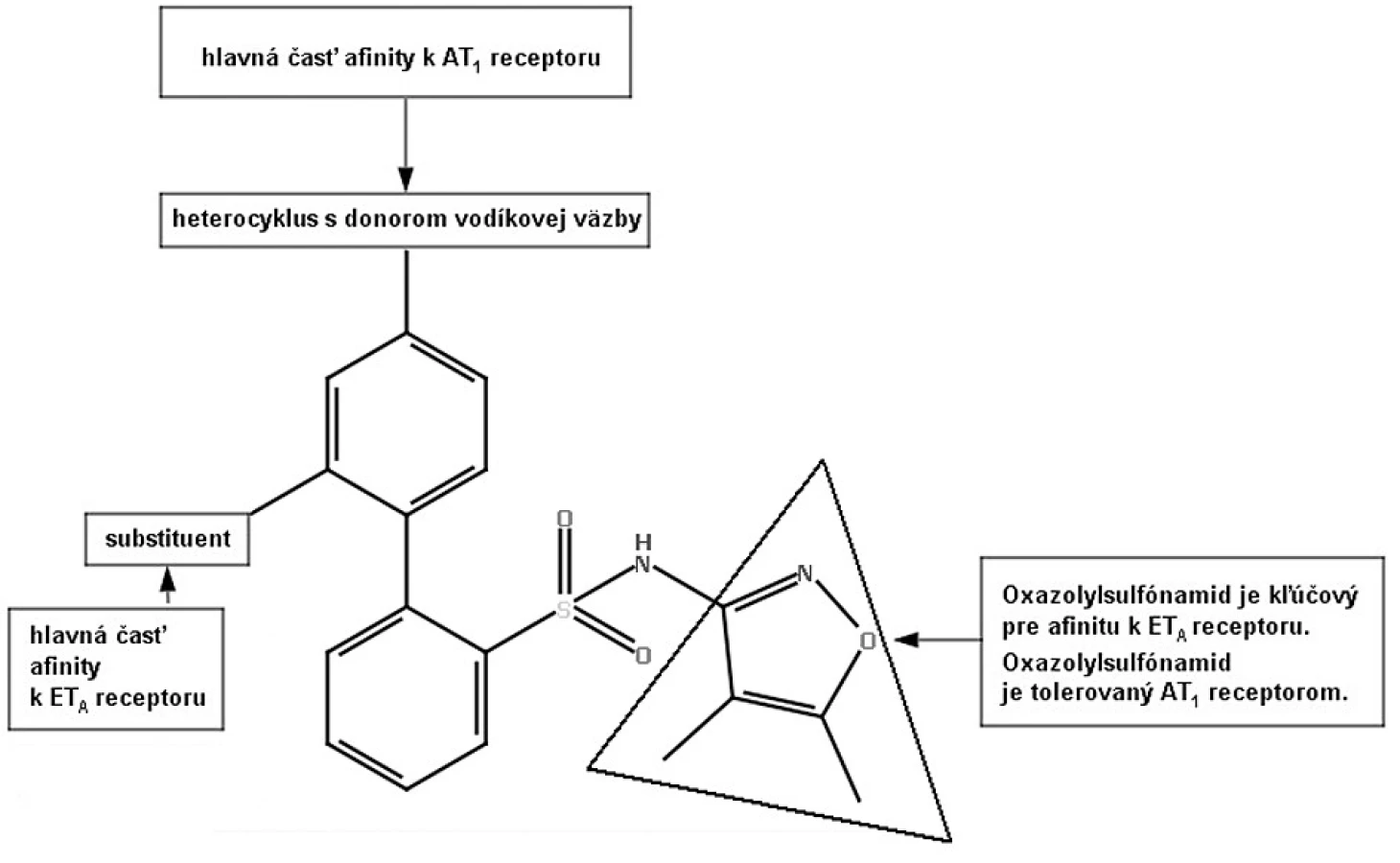
Na liečbu hypertenzie sa používa viacero liečiv, ale približne jedna tretina pacientov stále nie je adekvátne liečená43, 44). Zvýšené hladiny angiotenzínu II podporujú syntézu a vazokonstrikčný účinok endotelínu I (ET-1), a naopak, zvýšená hladina ET-1 zvyšuje syntézu a vazokonstrikčný účinok angiotenzínu II. Tým sa vytvára pozitívny dvojitý mechanizmus spätnej väzby. Zároveň je to aj veľmi dobrý cieľ pre terapiu hypertenzie45, 46). Štúdie na zvieratách ukázali, že súčasná blokáda AT1 a ET-A receptorov produkuje vyšší terapeutický benefit ako antagonizovanie samotného AT1 alebo ET-A receptora47). Na modeli renovaskulárnej hypertenzie u psov sa ukázalo, že kombinácia losartanu a bosentanu (neselektívny antagonista ET-A, ET-B receptora) znížila krvný tlak o 40 mm Hg v porovnaní so samotnou liečbou losartanu, ktorý znížil TK o 20 mm Hg48). Racionálnym projektovaním liečiv bol bifenylový blokátor ET-A receptora modifikovaný tak, že sa stal aj antagonistom AT1 receptora (viz obr. 7).
Duálna inhibícia AT1 a ET-1 receptorov je efektívnym postupom kontroly hypertenzie v predklinických modeloch. Zlúčenina PS-433540 (XVII) zo série 2’-substituovaných N-3-isoxazolyl bifenylsulfónamidov mala lepší účinok ako irbesartan na normálnom modeli hypertenzie u spontánne hypertenzných potkanov. Tým sa ukázala synergická blokáda AT1 a ET-A receptorov v jednej zlúčenine49, 50). Zlúčenina PS-433540 kombinuje silný antagonizmus na obidvoch AT1 aj ET-A receptoroch. Môže byť veľmi užitočná pri liečbe hypertenzie a iných kardiovaskulárnych ochorení, ktoré vyplývajú z vysokého krvného tlaku (napr. srdcové zlyhanie).

Antagonisty AT1 receptorov s NO donornou skupinou
Oxid nusnatý (NO) je signálna molekula, ktorá má komplexné chemické a biologické vlastnosti. U ľudí hrá NO hlavnú rolu v modulácii tonusu ciev. NO má výraznú antitrombotickú aktivitu. Má tiež protektívne účinky na srdce a obličky51). Porucha regulácie systému NO je spájaná s kardiovaskulárnymi ochoreniami. Znížená aktivita NO sa považuje za dôležitý determinant dysfunkcie endotelu a hypertenzie52).
Objav a vývoj hybridných liečiv, donorov NO, bol založený na pripojení NO-donornej skupiny na už existujúcu molekulu liečiva, ktorej farmakologické vlastnosti sú dobre známe53).
Jedným zo zástupcov tejto skupiny ligandov je analóg losartanu54, 55). Zlúčenina 3-[(nitrooxy)metyl]benzoát losartanu (XVIII), výrazné znížila systolický krvný tlaku u spontánne hypertenzných potkanov po 4-týždňom dávkovaní (orálnom alebo subkutánnom). Tento ligand mal ekvivalentný efekt ako losartan. Na modeloch myokardiálnej ischémie reperfúzie má táto látka kardioprotektívne účinky. Losartan však takúto aktivitu nevykazuje. Ligand inhiboval agregáciu krvných doštičiek vo väčšej miere než losartan. Spomínané vlastnosti môžu byť spájané s prídavnou NO-uvoľňujúcou skupinou bifunkčnej zlúčeniny, na rozdiel od monofunkčného ARB.
![(XVIII) 3-[(nitrooxy)metyl]benzoát losartan](https://pl-master.mdcdn.cz/media/image/0fc580cbf4dcdeea30564976d195b150.jpeg?version=1537791899)
Ďalším ligandom, ktorý dokáže uvoľniť NO-skupinu je derivát telmisartanu. WB1106 (XIX)56) znižuje krvný tlak u spontánne hypertenzných potkanov v rovnakej miere ako telmisartan. Na rozdiel od telmisartanu, WB1106 zvyšuje hladinu cGMP v aorte, v podobnom rozsahu ako ACE inhibítor fosinopril. U vysoko obéznych potkanov, ktorý mali stravu bohatú na cukor, WB1106 účinne redukoval získanú telesnú hmotnosť. Ďalej redukoval hladiny glukózy v sére nalačno a výrazne zlepšoval orálnu glukózovú toleranciu. Látka WB1106 bola v ekvivaletných dávkach účinnejšia ako telmisartan. To pravdepodobne odráža synergické účinky NO a telmisartanu na metabolizmus glukózy.

ZÁVER
Vývoj nových antagonistov AT1 receptorov sa bude pravdepodobne zameriavať na ovplyvnenie viacerých farmakologických cieľov. Okrem blokády AT1 receptorov je výhodné ovplyvniť aj ďalšie receptory, napríklad endotelínové. Tým sa dosiahne u pacientov dostatočná kontrola krvného tlaku, zníži sa riziko ochorení kardiovaskulárneho systému a cukrovky. Dnes je možné projektovať ligandy, ktoré majú multifunkčnú farmakologickú aktivitu. Vývoj a vývin takýchto multifunkčných ligandov je komplexnou problematikou. Pri návrhu multifunkčných ligandov je potrebné nájsť optimálny podiel účinnosti pre rozdielne farmakologické ciele. Zjednotenie farmakokinetiky môže dovoliť lepšie prispôsobiť farmakodynamickú aktivitu pre viaceré ciele, v porovnaní s jednoduchou terapeutickou kombináciou. Okrem toho, multifunkčný ligand je možné použiť vo fixnej dávke v kombinácii s inými liečivami. To umožňuje pomocou jednej liekovej formy ovplyvniť tri alebo viac terapeutických cieľov. V súčastnosti je stále veľký problém s kryštalizáciou membránovo viazaných receptorov spriahnutých s G-proteínom. Po vyriešení kryštálovej štruktúry AT1 receptora bude možné použiť štruktúrne dáta na projektovanie nových ligandov, antagonistov AT1 receptorov, pomocou metód molekulového modelovania. Ďalšia generácia antagonistov AT1 receptorov tak môže mať významnú klinickú hodnotu a zároveň môže poskytnúť širší terapeutický profil ako liečivo, ktoré dokáže blokovať iba AT1 receptor.
Došlo 10. května 2011
Přijato 24. června 2011
Adresa pro korespondenci:
PharmDr. Pavol Ježko
Katedra farmaceutickej chémie FaF UK
Odbojárov 10, 832 32 Bratislava, Slovenská republika
e-mail: jezko@fpharm.uniba.sk
Sources
1. Frohlich, E. D.: Renin-angiotensin system inhibition improves coronary flow reserve in hypertension. J. Cardiovasc. Pharmacol. 2001; 37, S35–S39.
2. Opie, L. H., Sack, M. N.: Enhanced angiotensin II activity in heart failure: reevaluation of the counterregulatory hypothesis of receptor subtypes. Circ. Res. 2001; 88, 654–658.
3. Ruster, C., Wolf, G.: Renin-angiotensin-aldosterone system and progression of renal disease. J. Am. Soc. Nephrol. 2006; 17, 2985–2991.
4. Triller, D. M., Evang, S. D., Tadrous, M., Yoo, B. K.: First renin inhibitor, aliskiren, for the treatment of hypertension. Pharm. World. Sci. 2008; 30, 741–749.
5. De Gasparo, M., Alexander, W., Bernstein, K. E., Catt, K. J., Goodfriend, T. L., Horiuchi, M., Husain, A., Inagami, T., Timmermans, P. B., Unger, T.: Angiotensin receptors. Last modified on 2010-12-22. Accessed on 2011-05-02. IUPHAR database (IUPHAR-DB). http://www.iuphar-db.org/DATABASE/FamilyMenu Forward?familyId=6..
6. Guimaraes, S., Carneiro, C., Brandao, F., Pinheiro, H., Albino-Teixeira, A., Moura, D.: A pharmacological differentiation between postjunctional (AT1A) and prejunctional (AT1B) angiotensin II receptors in the rabbit aorta. Naunyn Schmiedebergs Arch. Pharmacol. 2004; 370, 262–269.
7. Jones, S. E, Vinh, A., McCarthy, C. A., Gaspari, T. A., Widdop R.E.: AT2 receptors: Functional relevance in cardiovascular disease. Pharmacol. Ther. 2008; 120, 292–316.
8. Oro, C., Qian, H., Thomas, W. G.: Type 1 angiotensin receptor pharmacology: Signaling beyond G proteins. Pharmacol. Ther. 2007; 113, 210–226.
9. Gether, U.: Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr. Rev. 2000; 21, 90–113.
10. Streeten, D. H. P., Anderson, G. H. Jr., Freiberg, J. M., Freiberg, J. M., Dalakos, T. G. N.: Use of an angiotensin II antagonist (Saralasin) In: The recognition of „angiotensin-ogenic” hypertension. Engl. J. Med. 1975; 292, 657-662.
11. Moore, A. F., Fulton, R. W.: Angiotensin II antagonists-saralasin. Drug Dev. Res. 1984; 4, 331-349.
12. Furukawa, Y., Kishimoto, S., Nishikawa, K.: Hypotensive imidazole derivatives. United States Patent 4340598, 1982.
13. Furukawa, Y., Kishimoto, S., Nishikawa, K.: Hypotensive imidazole-5-acetic acid derivatives. United States Patent 4355040, 1982.
14. Wong, P. C., Chiu, A. T., Price, W. A., Thoolen, M. J., Carini, D. J., Johnson, A. L., Taber, R. I., Timmermans, P. B.: Nonpeptide angiotensin II receptor antagonists. I. Pharmacological characterization of 2-n-butyl-4-chloro-1-(2-chlorobenzyl) imidazole-5-acetic acid, sodium salt (S-8307). J. Pharmacol. Exp. Ther. 1988; 247, 1–7.
15. Chiu, A. T., Carini, D. J., Johnson, A. L., McCall, D. E., Price, W. A., Thoolen, M. J. M. C., Wong, P. C., Taber, R. I., Timmermans, P. B.: Nonpeptide angiotensin II (AII) receptor antagonists II. Pharmacology of S-8308. Eur. J. Pharmacol. 1988; 157, 13–21.
16. Duncia, J. V., Chiu, A. T., Carini, D. J., Gregory, G. B., Johnson, A. L., Price, W. A., Wells, G. J., Wong, P. C., Calabrese, J. C., Timmermans, P. B.: The discovery of potent nonpeptide angiotensin II receptor antagonists: A new class of potent antihypertensives. J. Med. Chem. 1990; 33, 1312–1329.
17. Chiu, A. T., Duncia, J. V., McCall, D. E., Wong, P. C., Price, W.A. Jr., Thoolen, M. J., Carini, D. J., Johnson, A. L., Timmermans, P. B.: Nonpeptide angiotensin II receptor antagonists. III. Structure-function studies. J. Pharmacol. Exp. Ther. 1989; 250, 867–874.
18. Carini, D. J., Duncia, J. V., Aldrich, P. E., Chiu, A. T., Johnson, A. L., Pierce, M. E., Price, W. A., Santella, J. B., Wells, G. J.: Nonpeptide angiotensin II receptor antagonists: the discovery of a series of N-(biphenylylmethyl)imidazoles as potent, orally active antihypertensives. J. Med. Chem. 1991; 34, 2525–2547.
19. Aulakh, G. K., Sodhi, R. K., Singh, M.: An update on non-peptide angiotensin receptor antagonists and related RAAS modulators. Life Sci. 2007; 81, 615–639.
20. Wexler, R. R., Greenlee, W. J., Irvin, J. D., Goldberg, M. R., Prendergast, K., Smith, R. D., Timmermans, P. B.: Nonpeptide Angiotensin II Receptor Antagonists: The Next Generation in Antihypertensive Therapy. J. Med. Chem. 1996; 39, 625–656.
21. Miura, S., Fujino, M., Hanzawa, H., Kiya, Y., Imaizumi, S., Matsuo, Y., Tomita, S., Uehara, Y., Karnik S. S., Yanagisawa, H., Koike, H.,Komuro, I., Saku, K.: Molecular mechanism underlying inverse agonist of angiotensin II type I receptor. J. Biol. Chem. 2006; 281, 19288–19295.
22. Underwood, D. J., Strader, C. D., Rivero, R., Patchett, A. A., Greenlee, W., Prendergast, K.: Structural model of antagonist and agonist binding to the angiotensin II, AT1 subtype, G protein coupled receptor. Chem. Biol. 1994; 1, 211–221.
23. Verheijen, I., Fierens, F. L. P., De Backer, J. P., Vauquelin, G., Vanderheyden, P.: Interaction between the partially insurmountable antagonist valsartan and human recombinant angiotensin II type 1 receptors. Fundam. Clinic. Pharmacol. 2000; 14, 577–585.
24. Verheijen, I., De Backer, J. P., Vanderheyden, P., Vauquelin, G.: A two-state model of antagonist–AT1 receptor interaction: further support by binding studies at low temperature. Biochem. Pharmacol. 2003; 65, 1339–1341.
25. Bhuiyan, M. A., Ishiguro, M., Hossain, M., Nakamura, T., Ozaki, M., Miura, S., Nagatomo, T.: Binding sites of valsartan, candesartan and losartan with angiotensin II receptor 1 subtype by molecular modeling. Life Sci. 2009; 85, 136–140.
26. Bondensgaad, K., Ankersen, M., Thogersen, H., Hansen, B. S., Wulff, B. S., Bywater, R. P.: Recognition of Privileged Structures by G-Protein Coupled Receptors. J. Med. Chem. 2004; 47, 888–899.
27. Ohno, K., Amano,Y., Kakuta, H., Niimi, T., Takakura, S., Orita, M., Miyata, K., Sakashita, H., Takeuchi, M., Komuro, I., Higaki, J.,Horiuchi, M., Kim-Mitsuyama, S., Mori, Y., Morishita, R., Yamagishi, S.: Unique “delta lock’’ structure of telmisartan is involved in its strongest binding affinity to angiotensin II type 1 receptor. Biochem. Biophys. Res. Commun. 2010; 404, 434–737.
28. Krovat, E. M., Langer, T.: Non-Peptide Angiotensin II Receptor Antagonists: Chemical Feature Based Pharmacophore Identification. J. Med. Chem. 2003; 46, 716–726.
29. Wong, P. C., Price, W. A. Jr., Chiu, A.T., Duncia, J. V., Carini, D. J., Wexler, R. R., Johnson, A. L., Timmermans, P. B.: An active metabolite of DuP 753, an orally active antihypertensive agent. J. Pharmacol. Exp. Ther. 1990; 255, 211–217.
30. Christ, D. D., Kilkson, T., Wong, N., Lam, G. N.: Formation and disposition of EXP3174, a pharmacologically active metabolite of the novel angiotensin II receptor antagonist DuP 753. In.: International Society for the study of Xenobiotica. San Diego 1990; 137.
31. Ries, U. J., Mihm, G., Narr, B., Hasselbach, K. M., Wittneben, H., Entzeroth, M., Van Meel, J. C. A., Wienen, W., Hauel, N. H.: 6-Substituted benzimidazoles as new nonpeptide angiotensin II receptor antagonists: synthesis, biological activity, and structure-activity relationships. J. Med. Chem. 1993; 36, 4040–4051.
32. Stasch, J.-P., Knorr, A., Hirth-Dietrich, C., Kramer, T., Hubsch, W., Dressel, J., Fey, P., Beuck, M., Sander, E., Frobel, K., Kazda, S.: Long-term blockade of the angiotensin II receptor in renin transgenic rats, salt-loaded Dahl rats, and stroke-prone spontaneously hypertensive rats. Arzneimittelforschung/Drug Research. 1997; 47, 1016–1023.
33. Breithaupt-Grogler, K., Malerczyk, C., Belz, G. G., Butzer, R., Herrmann, V., Stass, H., Wensing, G.: Pharmacodynamic and pharmacokinetic properties of an angiotensin II receptor antagonist characterization by use of Schild regression technique in man. Int. J. Clin. Pharmacol. Ther. 1997; 35, 434–441.
34. Knorr, A., Stasch, J. P., Beuck, M., Bisschoff, F., Fröbel, F., Hubch, C., Hirth-Dietrich, S., Kramer, T.: Pharmacology of BAY 10-6734, an AT1-selective angiotensin II receptor antagonist. Naunyn Schmeidebergs Arch. Pharmacol. 1996; 353, R69.
35. Wang, J. M., Tan, J., Leenen, F. H.: Central nervous system blockade by peripheral administration of AT1 receptor blockers. J. Cardiovasc. Pharmacol. 2003; 41, 593–599.
36. Tamura, K., Okuhira, M., Amano, H., Inokuma, K.-I., Hirata, T., Mikoshiba, I., Hashimoto, K.: Pharmacological profiles of KRH-594, a novel nonpeptide angiotensin II-receptor antagonist. J. Cardiovasc. Pharmacol. 1997; 30, 607–615.
37. Inada, Y., Murakami, M., Tazawa, S., Akahane, M.: KRH-594, a new angiotensin AT1 receptor antagonist, ameliorates nephropathy and hyperlipidaemia in diabetic spontaneously hypertensive rats. Clin. Exp. Pharmacol. Physiol. 2000; 27, 270–276.
38. Mochizuki, S., Sato, T., Furata, K., Hase, K., Ohkura, Y., Fukai, C., Kosakai, K., Wakabayashi, A., Tomiyama, A.: Pharmacological properties of KT3-671, a novel nonpeptide angiotensin II receptor antagonist. J. Cardiovasc. Pharmacol. 1995; 25, 22–29.
39. Yanagisawa, T., Ueyama, N., Kawai, T., Sonegawa, M., Baba, H., Mochizuki, S., Kozakai, K., Tomiyama, T.: 4,5,6,7-Tetrahydro-8-oxo-cycloheptimidazoles: a new class of potent nonpeptide angiotensin II receptor antagonist. Bioorg. Med. Chem. Lett. 1993; 3, 1559–1564.
40. Takata, Y., Tajima, S., Mochizuki, S., Suzaka, H., Tomiyama, A., Kato, H.: Antihypertensive activity and pharmacokinetics of KD3-671, a nonpeptide AT1-receptor antagonist, in renal hypertensive dogs. J. Cardiovasc. Pharmacol. 1998; 32, 834–844.
41. Amano, H., Fujimoto, K., Suzuki, T., Fujii, T., Mochizuki, S., Tomiyama, A., Kawashima, K.: Antihypertensive effect of chronic KT3-671, a structurally new nonpeptide angiotensin AT1-receptor antagonist in strokeprone spontaneously hypertensive rats. Jpn. J. Pharmacol. 1995; 69, 215–222.
42. Takata, Y., Kurihara, J., Yoda, T., Suzuki, S., Matsuoka, Y., Okubo, Y., Koto, H.: KT3-671, an angiotensin AT1 receptor antagonist, attenuates vascular but not cardiac responses to sympathetic nerve stimulation in pithed rats. J. Cardiovasc. Pharmacol. 2001; 37, 427–436.
43. Waeber, B.: Achieving blood pressure targets in the management of hypertension. Blood Press. Suppl. 2001; 2, 6–12.
44. Gavras, H. P.: Issues in hypertension: drug tolerability and special populations. Am. J. Hypertens. 2001; 14, 231S–236S.
45. Imai, T., Hirata, Y., Emori, T., Yanagisawa, M., Masaki, T., Marumo, F.: Induction of endothelin-1 gene by angiotensin and vasopressin in endothelial cells. Hypertension. 1992; 19, 753–757.
46. Chua, B. H., Chua, C. C., Diglio, C. A., Siu, B. B.: Regulation of endothelin-1 mRNA by angiotensin II in rat heart endothelial cells. Biochim. Biophys. Acta. 1993; 1178, 201–206.
47. Ikeda, T., Ohta, H., Okada, M., Kawai, N., Nakao, R., Siegl, P. K. S., Kobayashi, T., Miyauchi, T., Nishikibe, M.: Antihypertensive effects of a mixed endothelin-A - and -B-receptor antagonist, J-104132, were augmented in the presence of an AT1-receptor antagonist, MK-954. J. Cardiovasc. Pharmacol. 2000; 36, S337–S341.
48. Massart, P. E., Hodeige, D. G., Van Mechelen, H., Charlier, A. A., Ketelslegers, J. M., Heyndrickx G. R., Donckier, J.E.: Angiotensin II and endothelin-1 receptor antagonists have cumulative hypotensive effects in canine Page hypertension. J. Hypertens. 1998; 16, 835–841.
49. Murugesan, N., Tellew, J, E,, Gu, Z,, Kunst, B. L., Fadnis, L., Cornelius, L.A., Baska, R. A., Yang, Y., Beyer, S. M., Monshizadegan, H., Dickinson, K. E., Panchal, B., Valentine, M.T., Chong, S., Morrison, R. A., Carlson, K. E., Powell, J.R., Moreland, S., Barrish, J. C., Kowala, M. C., Macor, J. E.: Discovery of N-Isoxazolyl Biphenylsulfonamides as Potent Dual Angiotensin II and Endothelin A Receptor Antagonists. J. Med. Chem. 2002; 45, 3829–3835.
50. Murugesan, N., Gu, Z., Fadnis, L., Tellew, J. E., Baska, R. A., Yang, Y., Beyer, S. M., Monshizadegan, H., Dickinson, K.E., Valentine, M.T., Humphreys, W.G., Lan, S. J., Ewing, W. R., Carlson, K. E., Kowala, M. C., Zahler, R., Macor, J. E.: Dual Angiotensin II and Endothelin A Receptor Antagonists: Synthesis of 2’-Substituted N-3-Isoxazolyl Biphenylsulfonamides with Improved Potency and Pharmacokinetics. J. Med. Chem. 2005; 48, 171–179.
51. Loscalzo, J., Welch G.: Nitric oxide and its role in the cardiovascular system. Prog. Cardiovasc. Dis. 1995; 38, 87–104.
52. Giles, T. D.: Aspects of nitric oxide in health and disease: a focus on hypertension and cardiovascular disease. J. Clin. Hypertens. (Greenwich). 2006; 8, 2–16.
53. Martelli, A., Breschi, M. C., Calderone, V.: Pharmacodynamic hybrids coupling established cardiovascular mechanisms of action with additional nitric oxide releasing properties. Curr. Pharm. Des. 2009; 15, 614–636.
54. Breschi, M. C., Calderone, V., Digiacomo, M., Macchia, M., Martelli, A., Martinotti, E., Minutolo, F., Rapposelli, S., Rossello, A., Testai, L., Balsamo, A.: New NO-Releasing Pharmacodynamic Hybrids of Losartan and Its Active Metabolite: –Design, Synthesis, and Biopharmacological Properties. J. Med. Chem. 2006; 49, 2628–2639.
55. Breschi, M. C., Calderone, V., Digiacomo, M., Martelli, A., Martinotti, E., Minutolo, F., Rapposelli, S., Balsamo, A.: NO-Sartans: A New Class of Pharmacodynamic Hybrids as Cardiovascular Drugs. J. Med. Chem. 2004; 47, 5597–5600.
56. Li, Y. Q., Ji, H., Zhang, Y. H., Shi, W. B., Meng, Z. K., Chen, X. Y., Du, G. T., Tian, J.: WB1106, a novel nitric oxide-releasing derivative of telmisartan, inhibits hypertension and improves glucose metabolism in rats. Eur. J. Pharmacol. 2007; 577, 100–108.
57. Kurtz, T. W., Klein, U.: Next generation multifunctional angiotensin receptor blockers. Hypertension Res. 2009; 32, 826–834.
58. Baleanu-Gogonea, C., Karnik, S.: Model of the whole rat AT1 receptor and the ligand-binding site. J. Mol. Model. 2006; 12, 325–337.
59. Liefde, I. V., Vauquelin, G.: Sartan-AT1 receptor interactions: in vitro evidence for insurmountable antagonism and inverse agonism. Mol. Cell Endocrinol. 2009; 302, 237–243.
Labels
Pharmacy Clinical pharmacologyArticle was published in
Czech and Slovak Pharmacy

2011 Issue 4
Most read in this issue
- K dějinám farmaceutického průmyslu v Českých zemích. Interpharma
- Antagonisty angiotenzínových AT1 receptorov
- Význam genetického polymorfizmu enzýmov cytochrómu P450 – časť II. Cytochróm P450 2C9
- Antiflogistické hydrogély na báze ľalie bielej