
Zvažované farmakoterapeutické přístupy léčby Alzheimerovy choroby
Intended pharmacotherapeutical approaches of Alzheimer’s disease therapy
Alzheimer’s disease is a progressive neurodegenerative disorder mainly manifested by memory loss, personality changes, and cognitive dysfunction. Despite the fact that tireless research is being conducted, up-to-date pharmacotherapy of AD is presented only by two groups diverging in the mechanism of action. The larger one uses acetylcholinesterase inhibitors, and the second group is represented by the N-methyl-D-aspartate antagonist memantine. Even though the etiology of Alzheimer’s disease is unknown, several different therapeutic approaches are being investigated. The aim of this paper is to provide an overview of the present state of intended therapeutics for AD, describing their mechanism of action if known, displaying chemical structures, and the state of clinical trials if any.
Keywords:
Alzheimer’s disease • acetylcholinesterase • therapeutic approaches • beta amyloid • tau protein
Authors:
Jan Korábečný1 ,2; Eva Hrubá 3; Ondřej Soukup 2; Filip Zemek 2; Kamil Musílek 4; Eugenie Nepovímová 1; Katarína Špilovská 2; Veronika Opletalová 1; Kamil Kuča 5
Authors‘ workplace:
Univerzita Karlova v Praze, Farmaceutická fakulta v Hradci Králové, Katedra farmaceutické chemie a kontroly léčiv
1; Univerzita obrany, Fakulta vojenského zdravotnictví, Katedra toxikologie, Hradec Králové
2; Univerzita Karlova v Praze, Farmaceutická fakulta v Hradci Králové, katedra farmakologie a toxikologie
3; Univerzita Hradec Králové, Přírodovědecká fakulta, Katedra chemie
4; Univerzita obrany, Fakulta vojenského zdravotnictví, Centrum pokročilých studií a Fakultní nemocnice Hradec Králové
5
Published in:
Čes. slov. Farm., 2012; 61, 4-10
Category:
Review Articles
Overview
Alzheimerova choroba je progresivní neurologické onemocnění, které se projevuje ztrátou paměti, změnou osobnosti a kognitivní dysfunkcí. Současná farmakoterapie Alzheimerovy choroby je prezentována skupinami dvou typů látek, početnější z nich představují inhibitory acetylcholinesterasy, druhou zastupuje memantin jako antagonista působící na N-methyl-D-aspartátových receptorech. Ačkoliv etiologie Alzheimerovy choroby není dosud zcela plně známá, pro její terapii je zvažováno několik skupin látek. Cílem předkládané práce je poskytnout přehled látek zamýšlených pro léčbu Alzheimerovy choroby, jejich mechanismus účinku, chemickou strukturu a je-li znám, tak i stav klinických studií, ve kterých se právě nachází.
Klíčová slova:
Alzheimerova choroba • acetylcholinesterasa • terapeutické přístupy • beta amyloid • tau protein
Úvod
Alzheimerova choroba (Alzheimer’s disease – AD) je progresivní, neurologické onemocnění, které se manifestuje ztrátou paměti, změnou charakterových vlastností jedince, celkovou kognitivní dysfunkcí a dalšími funkčními změnami. AD je nejběžnější formou stařecké demence. Její podstata, symptomy, rizikové faktory a možnosti terapie však zaznamenaly velký rozvoj až v posledních 30 letech. Ačkoliv od této doby bylo objeveno mnoho nového v souvislosti s AD, příčiny vzniku AD zůstávají stále neobjasněny1–3).
S progresí nemoci následují další příznaky, mezi něž patří ztráta paměti narušující běžný denní režim postiženého, problémy v plánování a organizaci osobního života, obtíže při řešení problémů, běžných úkonů v domácnosti, v práci a volném čase, zmatenost a dezorientace při určování času nebo místa, na kterém se osoba nachází, vizuální a prostorová dezorientace, je narušená mluva i psaný projev, chybí schopnost vlastního úsudku, jsou pozorovány časté změny v náladě i chování a v neposlední řadě se jedná o stažení postiženého ze společenského života4).
Pacienti s AD prochází třemi stadii vývoje onemocnění, přičemž se jednotlivé fáze mohou překrývat. První stadium se projevuje pouze mírnými příznaky (mild stage of AD), kdy dochází ke zhoršení paměti, ztrátě iniciativy, průbojnosti, prostorové a přechodně časové dezorientaci postiženého. Druhé, středně těžké stadium (moderate stage of AD) je prezentováno výraznějšími příznaky a problémy, které již nemocnému znemožňují vykonávat řadu běžných každodenních aktivit. Dochází k výpadkům paměti a halucinacím. Ve třetím stadiu (severe stage of AD) je pacient zcela závislý na svém okolí, má obtíže s chůzí, výrazné poruchy chování a je neschopen poznávat své okolí, přátele i rodinné příslušníky. Neschopnost jakéhokoliv pohybu a upoutání na lůžko zpravidla vyústí v pneumonii, která se ve většině případů AD stává smrtelnou5).
Přibližný počet pacientů s AD je 5,4 milionu obyvatel v USA. V roce 2050 je odhadováno, že incidence AD v USA vzroste na jeden milion obyvatel ročně s celkovým počtem 11–16 milionů zasažených. Odhaduje se, že v současné době trpí tímto onemocněním 36 milionů lidí na celém světě a že počet nemocných v roce 2050 přesáhne 100 milionů. V České republice je postiženo Alzheimerovou chorobou přibližně 50–70 tisíc osob1, 6).
Příčiny AD doposud nejsou zcela známy. Většina vědců se domnívá, že AD stejně jako další chronické nemoci vzniká kombinací několika rizikových faktorů, za hlavní z nich je považován věk. Mezi další rizikové faktory přispívající k rozvoji AD patří dědičnost a genetická podmíněnost (přenos genu ApoE ε4), kardiovaskulární onemocnění související s nedostatečným prokrvením mozku, vysoký cholesterol, diabetes mellitus, hypertenze, fyzická i duševní inaktivita, kouření, obezita a častá traumata v oblasti hlavy (boxeři, hokejisté)7–9).
Současná terapie AD je prezentována dvěma skupinami látek3). První, početnější, jsou inhibitory acetycholinesterasy (AChEIs), které můžeme dále dělit do dvou generací10, 11). První generace je představována takrinem (THA, schválen k užívání v roce 1993 Americkou agenturou Food and Drug Administration, obchodní název Cognex®, USP Sciele Pharm Inc), který byl v roce 1998 stažen z trhu pro své nežádoucí účinky (hepatotoxicita, cholinergní nežádoucí účinky)12). V současné době prožívají takrinové deriváty renesanci, jsou hledány netoxická analoga na bázi 7-methoxytakrinu (7-MEOTA) nebo sloučeniny takrinu s oxidem dusnatým (obr. 1)13–17). Druhá generace AChEIs je zastoupena méně toxickými donepezilem (1997; Aricept®, Eisai Company and Pfizer Inc.), rivastigminem (2000; Exelon®, Novartis Pharmaceuticals) a galantaminem (2001; Hoechst Marion Roussel Inc., Shire Pharmaceutical Group, and Janssen Pharmaceutical, Reminyl® a Nivalin®, pro USA Razadyne®)18, 19).
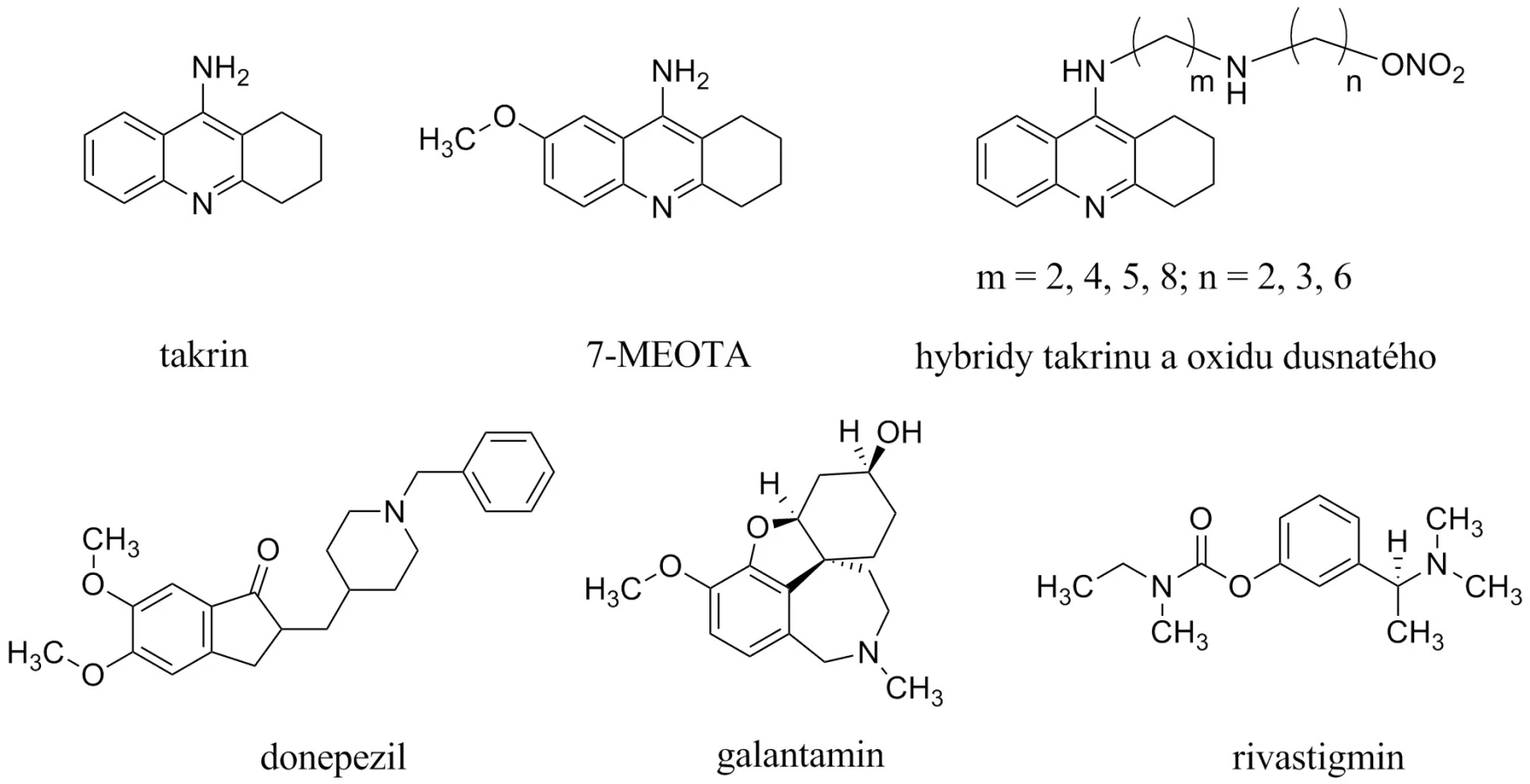
Druhou zavedenou skupinou látek pro terapii AD jsou antagonisté působící na N-methyl-D-aspartátových (NMDA) receptorech, jejichž hlavním představitelem je memantin (Namanda®, Forest). Intenzivně je rovněž studován neramexan patřící do této skupiny (obr. 2)20–26).
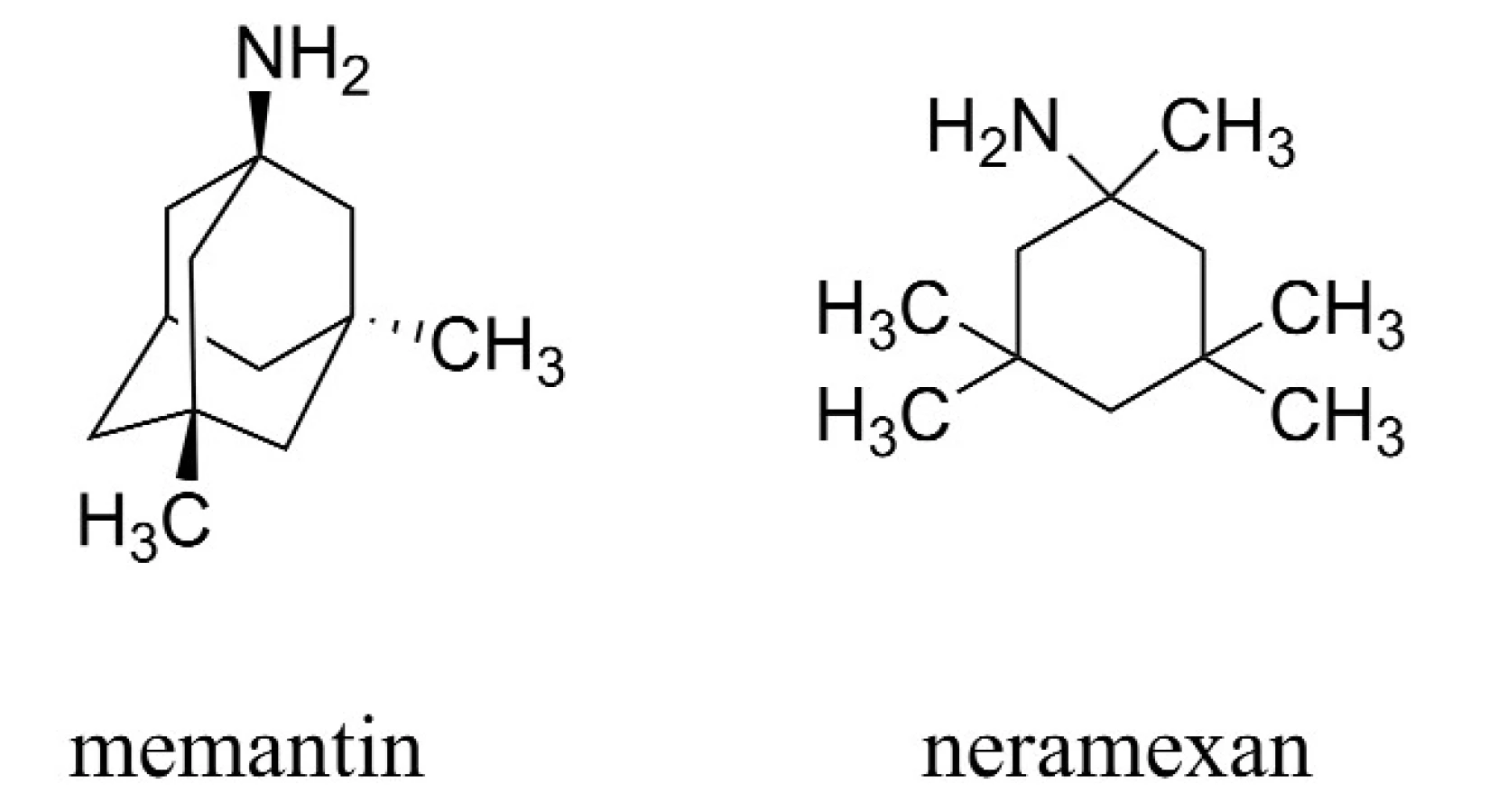
Tato souhrnná práce je zaměřena na terapeutické skupiny léčiv zvažovaných pro terapii AD a popisuje novodobé trendy vývoje skupin látek, u nichž předpokládáme zavedení do praxe. Tato další terapeutika se nachází v různých fázích klinického testování.
Zvažované terapeutické přístupy v oblasti AD
Vakcinace a imunizace
Transgenní myši imunizované beta-amyloidem (Aß) nesoucí prekurzor lidského transmembránového amyloidního proteinu (amyloid precursor protein – APP) vykazují sníženou produkci amyloidních plaků v mozku (jeden z patologických nálezů na mozku u pacientů s AD) a zlepšení behaviorálních funkcí27, 28). Tato zajímavá zjištění vedla k vakcinačním studiím na úrovni klinických testů u pacientů s AD. Po podání syntetického Aß (AN1792) byly záhy tyto studie přerušeny kvůli zvýšenému výskytu meningoencefalitidy u 6 % takto imunizovaných pacientů29). První výsledky analýzy klinického testování podávání AN1792 však ukázaly tvorbu protilátek proti Aß a signifikantní zlepšení v oblasti kognitivních funkcí30). Celkově tato vakcinační studie však neprokázala žádný benefit, a to paradoxně ani u pacientů, kteří vykázali tvorbu protilátek proti Aß. Magnetická rezonance po imunizaci u nich odhalila snížení mozkového objemu31). Z tohoto pohledu se jeví pasivní imunizace lepší volbou v porovnání s aktivní formou. V současné době se nachází v různých fázích klinického testování několik pasivně imunizačních látek využívající selektivní monoklonální Aß protilátky32).
Modulátory sekretas
Aß je tvořen z APP pomocí dvou štěpících enzymů ß sekretasy a γ-sekretasy. Výzkum v této oblasti je zaměřený na hledání inhibitorů těchto sekretas. Bylo prokázáno, že látka s označením KMI-429 inhibuje ß-sekretasu, a tím redukuje tvorbu patologického Aß in vivo u transgenních myší33) (obr. 3). Velikost molekul těchto inhibitorů však nepovoluje prostup přes hematoencefalickou bariéru a navíc po podání myším s deficiencí ß-sekretasy se vyskytly problémy s učením34, 35). V současné době je věnována pozornost vývoji nízkomolekulárních inhibitorů ß sekretasy36).
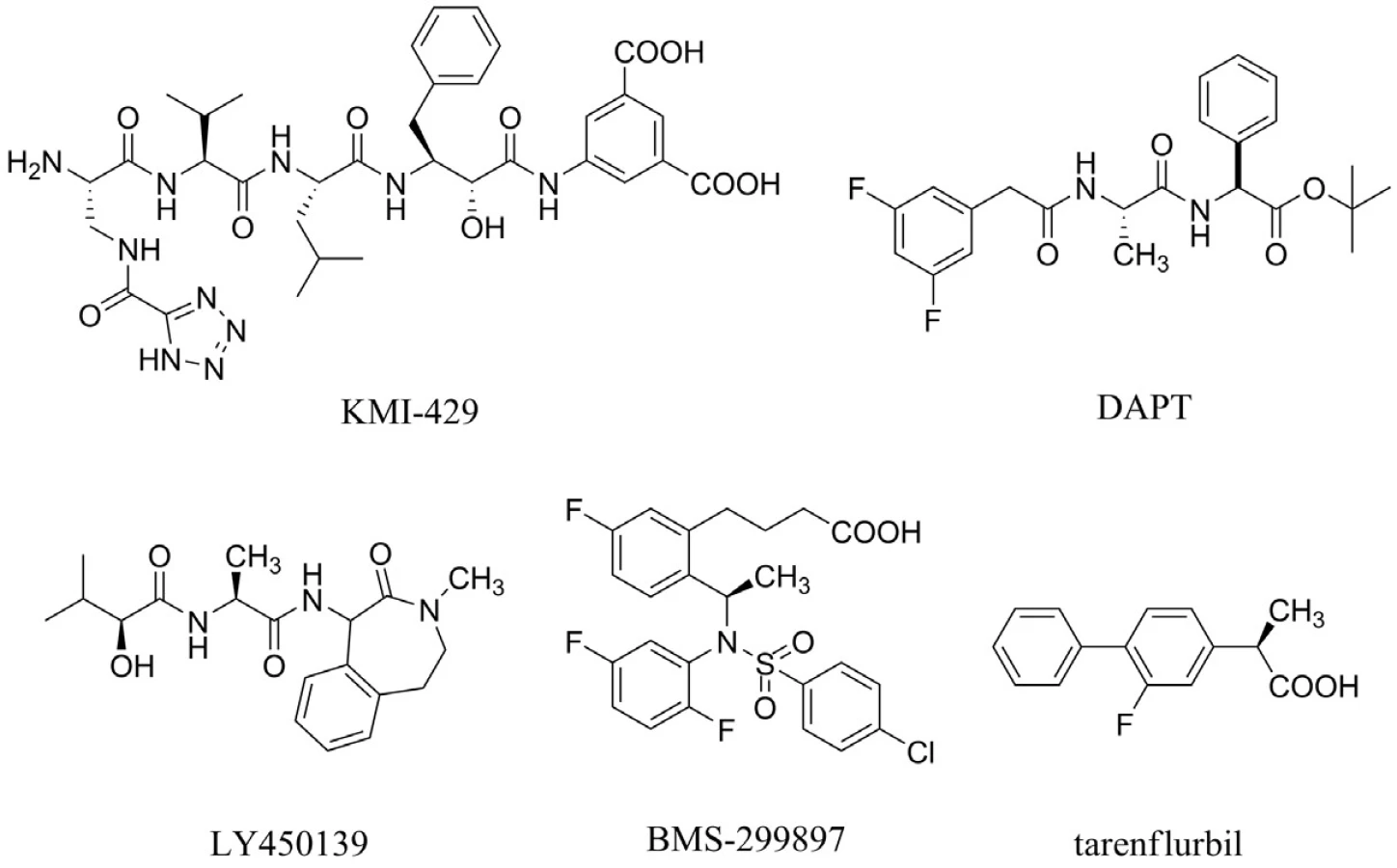
Pokles hladiny Aß v mozku, cerebrospinálním moku (CSF) a plazmě byl pozorován u hlodavců, kterým byly podány inhibitory γ-sekretasy s označením DAPT, LY450139 dihydrát a BMS-29989737–40) (obr. 3). Tyto výsledky vedou k závěru, že Aß může být spojený s kognitivní poruchou u pacientů s AD a že podání inhibitoru γ-sekretasy může vést, zejména v rané fázi AD, k reverzibilním změnám tvorby amyloidních plaků. Povzbudivé studie s těmito inhibitory jsou na druhé straně zpochybňovány jejich nepříliš dobrou selektivitou, která vede například ke škodlivým vlivům v gastrointestinální oblasti, brzlíku či slezině41).
Tarenflurbil (MPC-7869) patří do skupiny modulátorů γ-sekretasy, jehož mechanismem účinku je snížení produkce toxického Aß (Aß42) bez ovlivnění dalších fyziologicky se vyskytujících substrátů42) (obr. 3). Druhá fáze klinického testování tarenflurbilu naznačila jeho možný přínos u pacientů nacházející se v prvním stadiu AD, zejména ve skupině léčené nejvyššími dávkami (800 mg dvakrát denně). V současné době probíhá třetí fáze klinické studie hodnotící potenciální přínos tarenflurbilu43). Dosud zaznamenanými nežádoucími účinky v porovnání s placebem jsou eozinofilie, mírná anémie, hypertenze a vyrážka44).
Anti-agregačně působící skupina látek vůči Aß
Aß tvoří fibrilární shluky, které zapříčiňují smrt neuronů. Tramiprosat, hlavní představitel této skupiny látek, inhibuje tvorbu fibrilárních shluků vazbou na rozpustný Aß, a napodobuje tak účinek glykosaminoglykanů (obr. 4). Celý tento děj má za následek snížení hladiny rozpustného Aß v CSF a redukci tvorby amyloidních plaků. Otevřená studie s tramiprosatem naznačila pozastavení zhoršujících se kognitivních funkcí v raném stadiu vývoje AD45). V současnosti se látka nachází ve třetí fázi klinické studie v Severní Americe a v Evropě, kde je hodnocena z hlediska účinnosti a bezpečnosti.
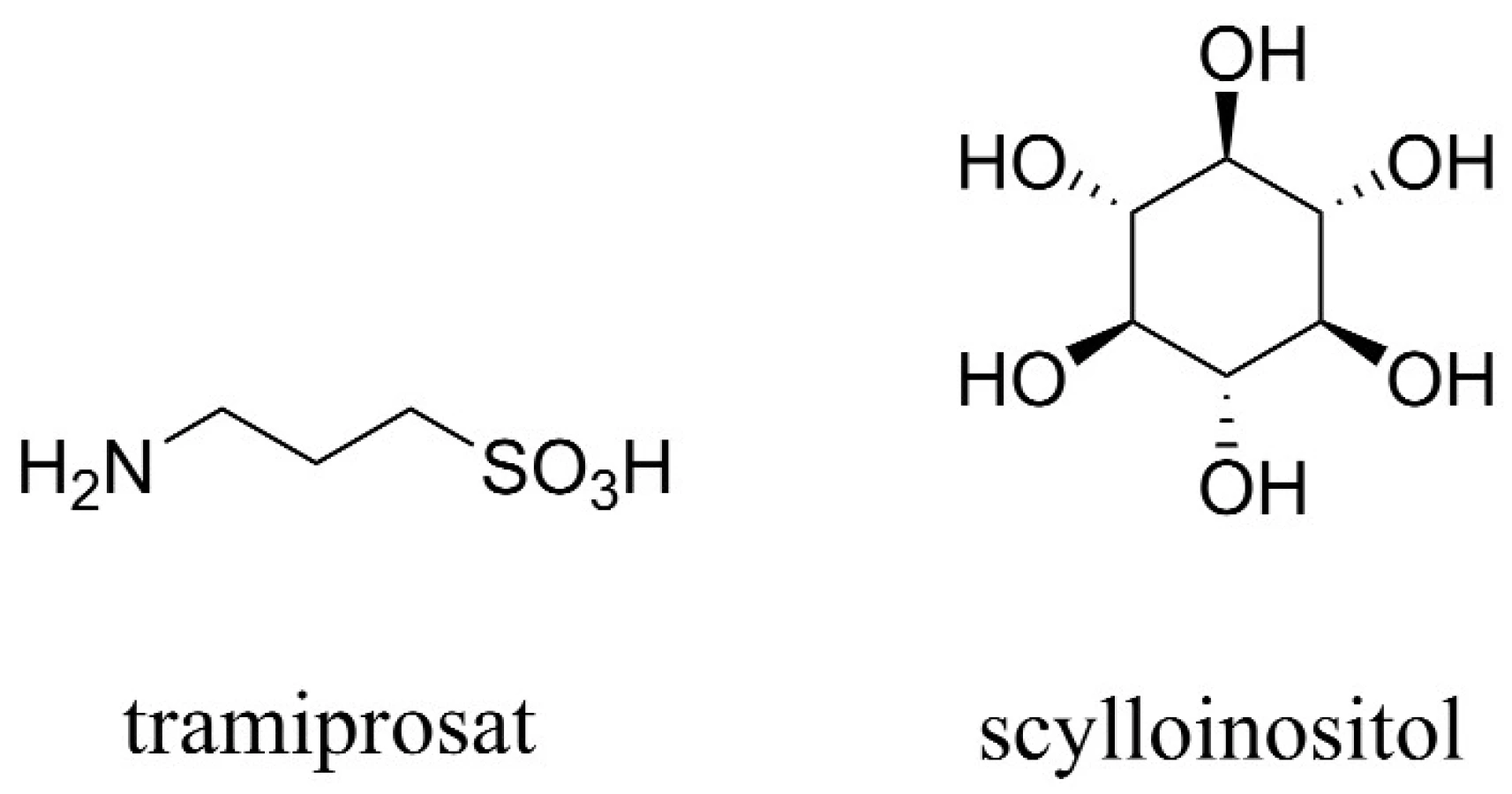
Další látkou patřící mezi anti-agregancia tlumící formování Aß je kolostrinin. Ten vykázal průměrné zlepšení v oblasti kognitivních funkcí u pacientů v raném stadiu AD (pozn.: pro střední stadium byly výsledky testů neprůkazné v porovnání s placebem). Kolostrinin nicméně neprokázal dlouhodobější benefit46, 47).
Zvláštní látkou této skupiny je scylloinositol (AZD-103) (obr. 4). Ten stabilizuje neshlukované, netoxické komplexy Aß, čímž přispívá zejména k redukci škodlivého vlivu shluků Aß, a z hlediska dlouhodobého užívání je dokonce schopen navrátit některé paměťové funkce48).
Statiny v terapii AD
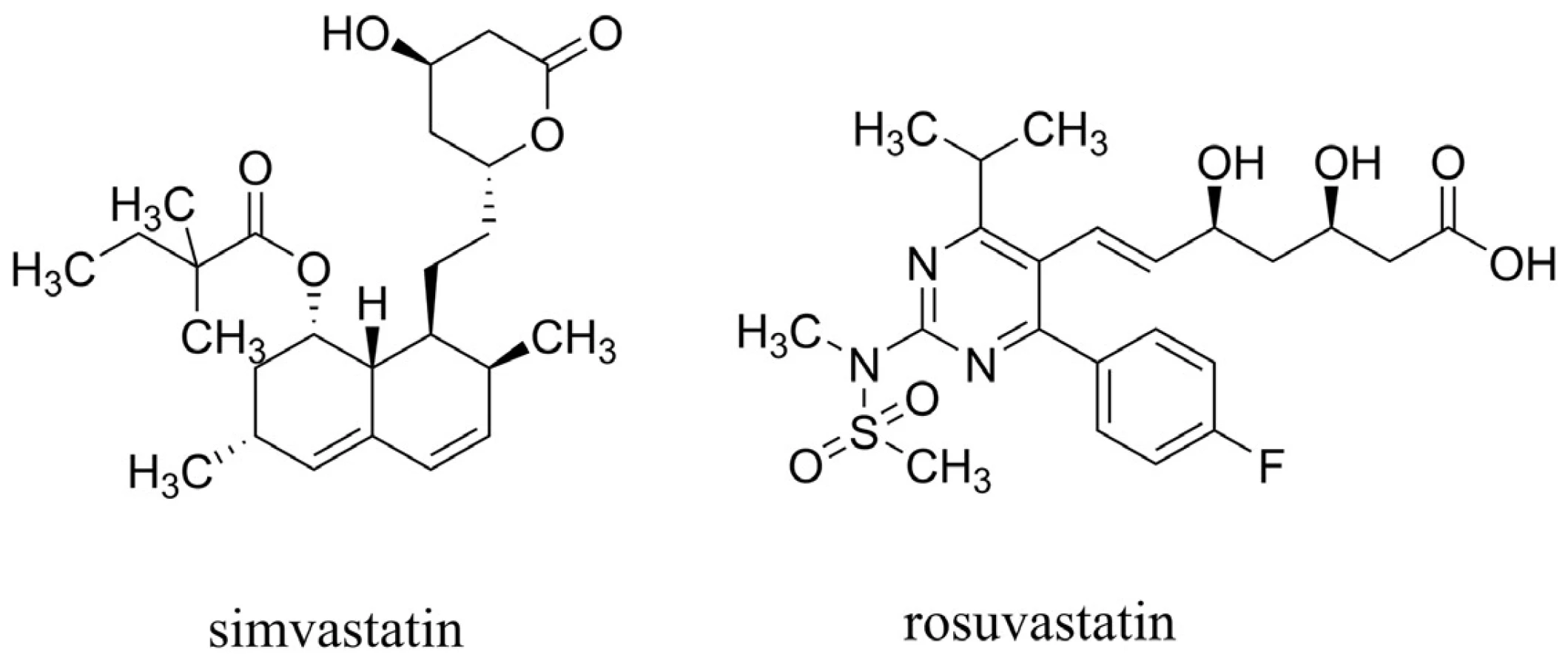
U statinů, látek primárně používaných k léčbě hyperlipidémie působících mechanismem inhibice 3-hydroxy-3-methylglutaryl koenzym A reduktasy (HMG-CoA reduktasa), byl pozorován pokles hladiny Aß in vivo49, 50). Efekt statinů je výrazný zejména při jejich nasazení pacientům před 80. rokem života51). Také několik epidemiologických studií naznačilo jejich přínos vedoucí ke snížení rizika AD52–55). Nicméně velké, kohortové a náhodné, placebem kontrolované studie zaměřené na prevenci koronárních onemocnění nepotvrzují účinek v oblasti kognitivních funkcí u pacientů s AD56–58). Shrnutím těchto poznatků lze říci, že statiny jako léčiva v oblasti AD mohou zpomalit neurodegenerativní procesy, nemohou však pomoci těm, u kterých se již demence AD projevila59). Dva základní strukturní typy statinů jsou vyobrazeny na obrázku 5.
Agonisté na receptorech aktivovaných proliferátory peroxizomů
Abnormality ve struktuře insulinu a insulinová rezistence mohou přispívat k neuropatologii a klinickým symptomům spojeným s AD60). Thiazolidindionový derivát rosiglitazon (pozn.: v roce 2010 stažen z trhu kvůli možným kardiovaskulárním nežádoucím účinkům) periferně zvyšuje insulinovou citlivost agonistickým působením na receptoru aktivovaném proliferátory peroxizomů (PPAR-γ) (obr. 6). Rosiglitazon působí redukci mediátorové RNA, která tvoří insulinově degradabilní enzym (IDE) (pozn.: je tedy zvýšena produkce insulinu). Tento účinek zprostředkovaný PPAR-γ zvyšuje uvolňování IDE, který je primárně využíván k metabolizaci Aß61). Studie prováděná u 30 vybraných jedinců naznačila možný benefit rosiglitazonu in vivo u AD, přičemž byly zlepšeny kognitivní projevy v porovnání s placebo skupinou62). Navíc u jedinců bez genu ApoE ε4 došlo k výraznějšímu zlepšení kognice než u pacientů nesoucích tento gen. Dalším agonistou působícím na PPAR-γ testovaným v klinických studiích v oblasti AD je pioglitazon63) (obr. 6). Rovněž je nutné dodat, že samotné podání insulinu intranazálně zlepšuje paměťové funkce64). Na úzkou souvislost mezi narušenou rovnováhou insulinu a mozkového glukosového metabolismu v mozku u AD poukazují nejnovější studie s metforminem. Ten snižuje insulinovou rezistenci a první testy naznačují jeho možný benefit v terapii AD65).

Chelátory kovů
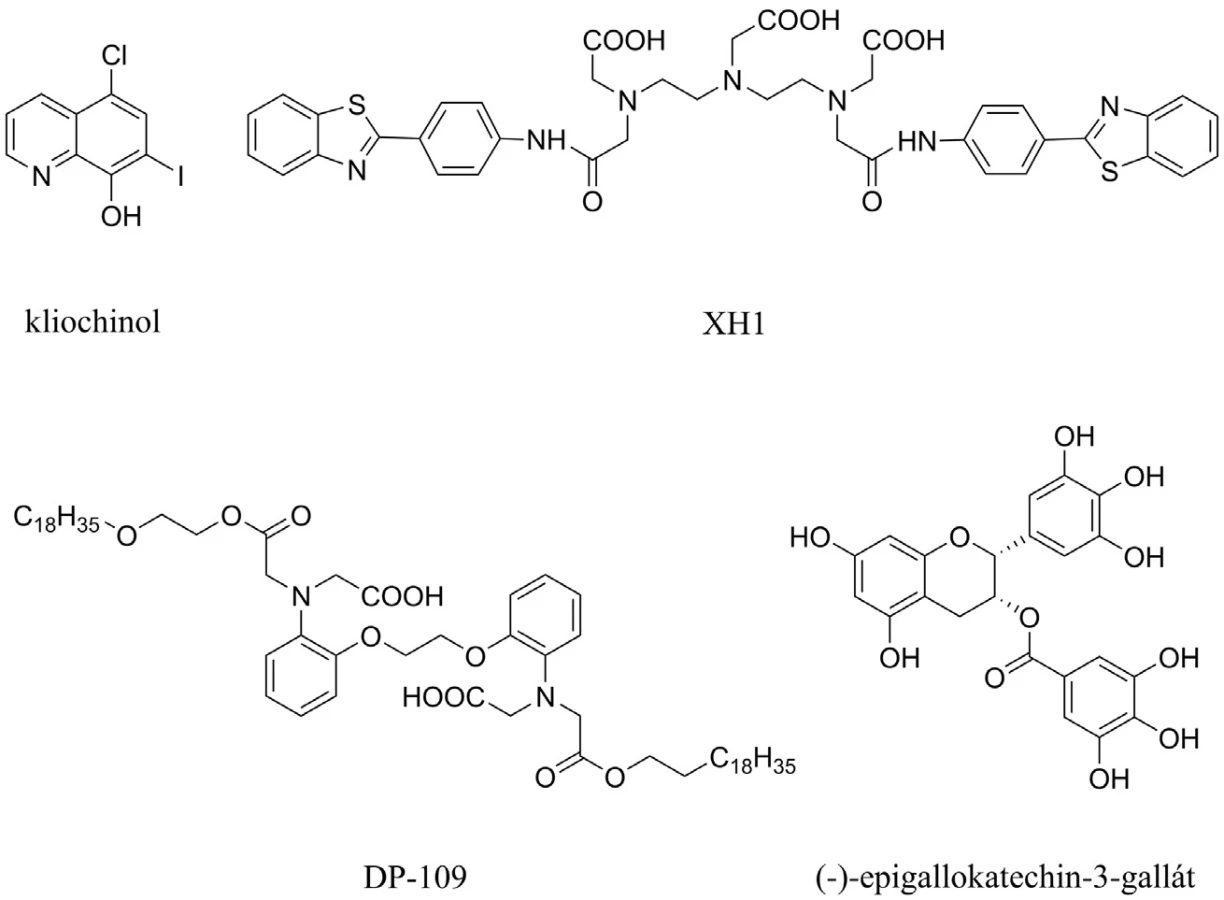
Aß interaguje s biogenními prvky, jako jsou zinek, měď a železo, a tvoří s nimi cytotoxické agregáty66). Kliochinol byl první látkou z této skupiny, který byl testován proti tvorbě Aß (obr. 7). Během 36týdenního užívání prokázal snížení Aß v mozku o 49 %. Dva nežádoucí účinky – synkopální epizody a srdeční arytmie – se objevily během této periody užívání67, 68). Mezi další chelátory zvažované k užití v souvislosti s AD patří látky s označením XH1, DP-109 a ( ) epigallokatechin-3-galát, které všechny shodně redukují poškození mozku způsobené Aß69–71) (obr. 7).
Agonisté M1 muskarinových receptorů
Bylo zjištěno, že muskarinové receptory subtypu 1 (M1) hrají multifaktoriální roli v patogenezi AD. Bylo prokázáno, že M1 muskarinový agonista (AF267B) podpořil v in vivo i in vitro testech neamyloidogenní cestu štěpení APP zprostředkovanou α–sekretasou, která místo toxického Aß dává za vznik rozpustnému APPsα, jež má neuroprotektivní charakter (obr. 8). Dále bylo pozorováno, že tento M1 agonista snižoval hladiny Aß v CSF, snižoval výskyt markerů zánětu a kognitivních poruch72–74). Další studie ukázaly, že AF267B má pozitivní vliv i na tau protein, který není hyperfosforylován, a může tak plnit svou fyziologickou funkci, která spočívá ve stabilizaci mikrotubulů v neuronech CNS a která bývá v případě AD narušena75). Další látkou této skupiny zavedenou do klinického testování je například talsaklidin (obr. 8). Ten, stejně jako AF267B, prokázal redukci hladiny Aß v CSF76). Podobně i xanomelin a jeho deriváty působící stimulaci M1 receptorů snižují hladiny Aß, pozitivně ovlivňují tau protein a navíc potencují účinek inhibitorů AChE, čímž zlepšují kognitivní projevy pacientů s AD77, 78) (obr. 8). Přestože velká komplexnost působení nedává příliš prostoru pro přesné vymezení mechanismu účinku, je zřejmé že aktivace postsynaptických, resp. presynaptických M1 muskarinových receptorů ovlivňuje hned na více úrovních patogenezi této choroby79). A zcela určitě se potom jedná o zajímavý přístup, který se může ukázat jako velký benefit v léčbě AD80).

Ovlivnění RAGE receptorů
RAGE (Receptor for Advanced Glycation Endproducts) je transmembránový receptor patřící do rodiny imunoglobulinů. Poprvé byl popsán roku 1992 Neeperem et al.81). Jeho název je odvozen ze schopnosti vázat koncové produkty glykace odvozené z glykoproteinů. RAGE, v přítomnosti Aß a v kontaktu s endoteliálními buňkami krevních destiček může indukovat migraci monocytů skrze endoteliální buňky mozku. Tato monocytární diapedéza dle všeho hraje důležitou roli v zánětlivém procesu AD82). V současné době jsou vyvíjeny ligandy pro tento receptor, které by mohly potlačovat akumulaci Aß, a tím přispívat ke zlepšení prognózy AD83).
Periferně působící vychytávače Aß
Jak již bylo zmíněno, redukce hladiny Aß je stále považována za jeden z terapeutických cílů AD. Zatímco aktivní imunizací lze docílit přímé snížení hladiny Aß v mozku, periferně působící vychytávače jako gelsolin (protein vázající aktin) mají vysokou afinitu pro periferní Aß, čímž rovněž napomáhají k celkovému zlepšení v prognóze AD a jsou oblastí intenzivního výzkumu84). Zajímavou skupinou látek jsou rovněž dihydropyridinová antihypertenziva. Ve vztahu k AD kromě mírnění produkce Aß snižují rovněž poškození mozku způsobené Aß a současně usnadňují clearance tohoto toxického patologického proteinu85).
Závěr
Současné představení devíti různých terapeutických přístupů v léčbě Alzheimerovy choroby svědčí o významu nalezení efektivnější léčby pro tuto chorobu. Ačkoliv se touto problematikou zabývají přední vědecké týmy z celého světa již několik desetiletí, nepodařilo se do praxe zavést jiné léčivo než symptomaticky působící AChEIs či memantin (NMDA-antagonista). Je to dáno zejména tím, že se dosud nepodařilo přesně určit etiologii této nemoci. I přes toto celosvětové úsilí je AD stále palčivějším problémem lidstva, nejen po stránce zdravotní, ale i po stránce sociální či ekonomické a je možné, že kauzální lék na tuto chorobu nebude nikdy nalezen.
Střet zájmů: žádný.
Došlo 13. října 2011 / Přijato 15. prosince 2011
doc. Ing. Kamil Kuča, Ph.D.
Univerzita obrany, Fakulta vojenského zdravotnictví, Centrum pokročilých studií a Fakultní nemocnice Hradec Králové
Sokolská 581, 500 05 Hradec Králové, Česká republika
e-mail: kucakam@pmfhk.cz
Sources
1. Alzheimer’s Association. 2011 Alzheimer’s disease facts and figures. Alzheimers Dement 2011; 7, 208–244.
2. Herrup K. Re imaginizing Alzheimer’ disease – an age-based hypothesis. J Neurosci 2010; 30, 16755–16762
3. Drtinova L., Pohanka M. Alzheimerova demence: aspekty současné farmakologické léčby. Čes slov Farm 2011; 60, 219–228.
4. Craig D., Mirakhur A., Hart D. J., McIlroy S. P., Passmore A. P. A cross-sectional study of neuropsychiatric symptoms in 435 patients with Alzheimer’s disease. Am J Geriat Psychiat 2005; 13, 460–468.
5. Ballard C., Gauthier S., Corbett A., Brayne C., Aarsland D., Jones E. Alzheimer’s disease. Lancet 2011; 377, 1019–1031.
6. Brodaty H., Breteler M. M., Dekosky S. T., Dorenlot P., Fratiglioni L., Hock C., Kenigsberg P. A., Scheltens P., De Strooper B. The world of dementia beyond 2020. J Am Geriatr Soc 2011; 59, 923–927.
7. Souder E., Beck C. Overview of Alzheimer’s disease. Nurs Clin N Am 2004; 39, 545–559.
8. Kim K. Y., Wood B. E., Wilson M. I. Risk factors for Alzheimer’s disease: An overview for clinical practitioners. Consult Pharm 2005; 20, 224–230.
9. Bateman R. J., Aisen P. S., De Strooper B., Fox N. C., Lemere C. A., Ringman J. M., Salloway S., Sperling R. A., Windisch M., Xiong C. Autosomal-dominant Alzheimer’s disease: a review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res Ther 2011; 3, 1–13.
10. Giacobini E. Do cholinesterase inhibitors have disease-modifying effects in Alzheimer’s disease? CNS Drugs 2001; 15, 85–91.
11. Giacobini E., Mori F., Lai C. C. The effect of cholinesterase inhibitors on the secretion of APPS from rat brain cortex. Ann NY Acad Sci. 1996; 777, 393–398.
12. Knapp M. J., Knopman D. S., Solomon P. R., Pendlebury W. W., Davis C. S., Gracon S. I. A 30-week randomized controlled trial of high-dose tacrine in patients with Alzheimer’s disease. JAMA 1994; 271, 985–991.
13. Dejmek L. 7-Meota. Drug Future 1990; 15, 126–129.
14. Korabecny J., Musilek K., Holas O., Binder J., Zemek F., Marek J., Pohanka M., Opletalova V., Dohnal V., Kuca K. Synthesis and in vitro evaluation of N-alkyl-7-methoxytacrine hydrochlorides as potential cholinestrase inhibitors in Alzhemeir disease. Bioorg Med Chem Lett 2010; 20, 6093–6095.
15. Korabecny J., Holas O., Musilek K., Pohanka M., Opletalova V., Dohnal V., Kuca K. Synthesis and in vitro evaluation of new tacrine derivatives-bis-alkylene linked 7-MEOTA. Lett Org Chem 2010; 7, 327–331.
16. Korabecny J., Musilek K., Holas O., Nepovimova E., Jun D., Zemek F., Opletalova V., Patocka J., Dohnal V., Nachon F., Hroudova J., Fisar Z., Kuca K. Synthesis and in vitro evaluation of N-(bromobut-3-en-2-yl)-7-methoxy-1,2,3,4-tetrahydroacridin-9-amine as a cholinesterase inhibitor with regard to Alzheimer’s disease treatment. Molecules 2010; 15, 8804–8812.
17. Korabecny J., Musilek K., Zemek F., Horova A., Holas O., Nepovimova E., Opletalova V., Hroudova J., Fisar Z., Jung Y.-S., Kuca K. Synthesis and in vitro evaluation of 7-methoxy-N-(pent-4-enyl)-1,2,3,4-tetrahydroacridin-9-amine – new tacrine derivative with cholinergic properties. Bioorg Med Chem Lett 2011; 21, 6563–6556.
18. Fang L, Appenroth D., Decker M., Kiehntopf M., Lupp A., Peng S., Fleck C., Zhang Y., Lehman J. NO-donating tacrine hybrid compounds improve scopolamine-induced cognition impairment and show less hepatotoxicity. J Med Chem 2008; 51, 7666–7669.
19. Trinh, N. H., Hoblyn, J., Mohanty, S., Yaffe, K. Efficacy of cholinesterases inhibitors in the treatment of neuropsychiatric symptoms and functional impairment in Alzheimer disease: a meta-analysis. JAMA 2003; 289, 210–216.
20. Kamal, M. A., Klein, P., Yu, Q. S., Tweedie, D., Li, Y., Holloway, H. W., Tweedie, D., Greig, N. H. Kinetics of human serum butyrylcholinesterase and its inhibition by a novel experimental Alzheimer therapeutic, bisnorcymserine. J Alzheimers Dis 2006; 10, 43–51.
21. Li F., Tsien J. Z. Memory and the NMDA receptors. New Engl J Med 2009; 361, 302–303.
22. Harkany T., Abraham I., Timmerman W., Laskay G., Toth B., Safari M., Konya C., Sebens J. B., Korf J., Nyakas C., Zarandi M., Soos K., Penke B., Luiten P. G. Beta-amyloid neurotoxicity is mediated by a glutamate-triggered excitotoxic cascade in rat nucleus basalis. Eur J Neurosci 2000; 12, 2735–2745.
23. Amadoro G., Ciotti M. T., Costanzi M., Cestari V., Calissano P., Canu N. NMDA receptor mediates tau–induced neurotoxicity by calpain and ERK/MAPK activation. P Natl Acad Sci USA 2006; 103, 2892–2897.
24. Sonkusare S. K., Kaul C. L., Ramarao P. Dementia of Alzheimer’s disease and other neurodegenerative disorders-memantine, a new hope. Pharmacol Res 2005; 51, 1–17.
25. Tariot P. N., Farlow M. R., Grossberg G. T., Graham S. M., McDonald S., Gergel I. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA 2004; 291, 317–324.
26. Wenk G. L., Baker L. M., Stoehr J. D., Hauss-Wegrzyniak B., Danysz W. Neuroprotection by novel antagonists at the NMDA receptor channel and glycineB sites. Eur J Pharmacol. 1998; 347, 183–187.
27. Schenk D., Barbour R., Dunn W., Gordon G., Grajeda H., Guido T., Hu K., Huang J., Johnson-Wood K., Khan K., Kholodenko D., Lee M., Liao Z., Lieberburg I., Motter R., Mutter L., Soriano F., Shopp G., Vasquez N., Vandevert C., Walker S., Wogulis M., Yednock T., Games D., Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999; 400, 173–177.
28. Lavie V., Becker M., Cohen-Kupiec R., Yacoby I., Koppel R., Wedenig M., Hutter-Paier B., Solomon B. EFRH-phage immunization of Alzheimer’s disease animal model improves behavioral performance in Morris water maze trials. J Mol Neurosci 2004; 24, 105–113.
29. Orgogozo J. M., Gilman S., Dartigues J. F., Laurent B., Puel M., Kirby L. C., Jouanny P., Dubois B., Eisner L., Flitman S., Michel B. F., Boada M., Frank A., Hock C. Subacute meningoencephalitis in a subset of patients with AD after A beta 42 immunization. Neurology 2003; 61, 46–54.
30. Hock C., Konietzko U., Streffer J. R., Tracy J., Signorell A., Müller-Tillmanns B., Lemke U., Henke K., Moritz E., Garcia E., Wollmer M. A., Umbricht D., de Quervain D. J., Hofmann M., Maddalena A., Papassotiropoulos A., Nitsch R. M. Antibodies against beta-amyloid slow cognitive decline in Alzheimer’s disease. Neuron 2003; 38, 547–554.
31. Fox N. C., Black R. S., Gilman S., Rossor M. N., Griffith S. G., Jenkins L., Koller M. Effects of A beta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology 2005; 64, 1563–1572.
32. Dodel R. C., Du Y., Depboylu C., Hampel H., Frölich L., Haag A., Hemmeter U., Paulsen S., Teipel S. J., Brettschneider S., Spottke A., Nölker C., Möller H. J., Wei X., Farlow M., Sommer N., Oertel W. H. Intravenous immunoglobulins containing antibodies against beta-amyloid for the treatment of Alzheimer’s disease. J Neurol Neurosur Ps 2004; 75, 1472–1474.
33. Asai M., Hattori C., Iwata N., Saido T. C., Sasagawa N., Szabó B., Hashimoto Y., Maruyama K., Tanuma S., Kiso Y., Ishiura S. The novel beta-secretase inhibitor KMI-429 reduces amyloid beta peptide production in amyloid precursor protein transgenic and wild-type mice. J Neurochem 2006; 96, 533–540.
34. Wong P. BACE. Alzheimers Dement 2005; 1, S3.
35. Citron M. Beta-secretase inhibition for the treatment of Alzheimer’s disease-promise and challenge. Trends Pharmacol Sci 2004; 25, 92–97.
36. Mancini F., de Simone A., Andrisano V. Beta-secretase as a target for Alzheimer’s disease drug discovery: an overview of in vitro methods for characterization of inhibitors. Anal Bioanal Chem 2011; 400, 1979–1996.
37. Lanz T. A., Himes C. S., Pallante G., Adams L., Yamazaki S., Amore B., Merchant K. M. The gamma-secretase inhibitor N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester reduces A beta levels in vivo in plazma and cerebrospinal fluid in young (plaque-free) and aged (plaque-bearing) Tg2576 mice. J Pharmacol Exp Ther 2003; 305, 864–871.
38. El Mouedden M., Vandermeeren M., Meert T., Mercken M. Reduction of A beta levels in the Sprague Dawley rat after oral administrativ of the functional gamma-secretase inhibitor, DAPT: a novel non-transgenic model for A beta production inhibitors. Curr Pharm Design 2006; 12, 671–676.
39. May P. C., Yang Z., Li W., Hyslop P. A., Siemers E., Boggs L. N. Multi-compartmental pharmacodynamic assessment of the functional gamma-secretase inhibitor LY450139 dihydrate in PDAPP transgenic mice and non-transgenic mice. Neurobiol Aging 2004; 25, S65.
40. Barten D. M., Guss V. L., Cosa J. A., Loo A., Hansel S. B., Zheng M., Munoz B., Srinivasan K., Wang B., Robertson B. J., Polson C. T., Wang J., Roberts S. B., Hendrick J. P., Anderson J. J., Loy J. K., Denton R., Verdoorn T. A., Smith D. W., Felsenstein K. M. Dynamics of {beta}-amyloid reductions in brain, cerebrospinal fluid, and plasma of {beta}-amyloid precursor protein transgenic mice treated with a {gamma}-secretase inhibitor. J Pharmacol Exp Ther 2005; 312, 635–643.
41. Imbimbo B. P. Alzheimer’s disease: gamma secretase inhibitors. Drug Discov Today: Therapeutic Strategies 2008; 5, 169–175.
42. Weggen S., Eriksen J. L., Sagi S. A., Pietrzik C. U., Golde T. E., Koo E. H. A beta 42-lowering nonsteroidal anti-inflammatory drugs preserve intramembrane cleavage of the amyloid precursor protein (APP) and ErbB-4 receptor and signaling through the APP intracellular domain. J Biol Chem 2003; 278, 30748–30754.
43. Becker R. E., Greig N. H. Why so few drugs for Alzheimer’s disease? Are methods failing drugs? Curr Alzheimer Res 2010; 7, 642–651.
44. Mintzer J. E., Wilcock G. K., Black S. E., Zavitz K. H., Hendrix S. B. MPC-7869, a selective Abeta42-lowering agent, delays time to clinically significant psychiatric adverse events in Alzheimer’s disease: analysis from a 12-month phase 2 trial. Presented as a poster exhibit at the 10th International Conference on Alzheimer’s Disease and Related Disorders; 2006 Jul 15–20; Madrid, Spain.
45. Aisen P. S., Gauthier S., Vellas B., Briand R., Saumier D., Laurin J., Garceau D. Alzhemed: A potential treatment for Alzheimer’s disease. Curr Alzheimer Res 2007; 4, 473–478.
46. Bilikiewicz A., Gaus W. Colostrinin (a naturally occurring, proline-rich, polypeptide mixture) in the treatment of Alzheimer’s disease. J Alzheimer Dis 2004; 6, 17–26.
47. Leszek J., Inglot A. D., Janusz M., Byczkiewicz F., Kiejna A., Georgiades J., Lisowski J. Colostrinin proline-rich polypeptide complex from ovine colostrum-a long-term study of its efficacy in Alzheimer’s disease. Med Sci Monitor 2002; 8, PI93–PI96.
48. Townsend M., Cleary J. P., Mehta T., Hofmeister J., Lesne S., O’Hare E., Walsh D. M., Selkoe D. J. Orally available compound prevents deficits in memory caused by the Alzheimer amyloid-beta oligomers. Ann Neurol 2006; 60, 668–676.
49. Fassbender K., Simons M., Bergmann C., Stroick M., Lutjohann D., Keller P., Runz H., Kuhl S., Bertsch T., von Bergmann K., Hennerici M., Beyreuther K., Hartmann T. Simvastatin strongly reduces levels of Alzheimer’s disease beta-amyloid peptides A beta 42 and A beta 40 in vitro and in vivo. P Natl Acad Sci USA 2001; 98, 5856–5861.
50. Wolozin B., Kellman W., Ruosseau P., Celesia G. G., Siegel G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors. Arch Neurol 2000; 57, 1439–1443.
51. Li G., Higdon R., Kukull W. A., Peskind E., Moore K. V., Tsuang D., van Belle G., McCormick W., Bowen J. D., Teri L., Schellenberg G. D., Larson E. B. Statin therapy and risk of dementia in the elderly: a community-based prospective cohort study. Neurology 2004; 63, 1624–1628.
52. Jick H., Zornberg G. L., Jick S. S., Seshadri S., Drachman D. A. Statins and the risk of dementia. Lancet 2000; 356, 1627–1631.
53. Rockwood K., Kirkland S., Hogan D. B., MacKnight C., Merry H., Verreault R., Wolfson C., McDowell I. Use of lipid-lowering agents, indication bias, and the risk of dementia in community-dwelling elderly people. Arch Neurol 2002; 59, 223–227.
54. McGuiness B., O`Hare J., Craig D., Bullock R., Malouf R., Passmore P. Statins for the treatment of dementia (Review). Cochrane Database Syst Rev 2010; 4.
55. Yaffe K., Barrett-Connor E., Lin F., Grady D. Serum lipoprotein levels, statin use, and cognitive function in older women. Arch Neurol 2002; 59, 378–384.
56. Rea T. D., Breitner J. C., Psaty B. M., Fitzpatrick A. L., Lopez O. L., Newman A. B., Hazzard W. R., Zandi P. P., Burke G. L., Lyketsos C. G., Bernick C., Kuller L. H. Statin use and the risk of incident dementia: the cardiovascular health study. Arch Neurol 2005; 62, 1047–1051.
57. Shepherd J., Blauw G. J., Murphy M. B., Bollen E. L., Buckley B. M., Cobbe S. M., Ford I., Gaw A., Hyland M., Jukema J. W., Kamper A. M., Macfarlane P. W., Meinders A. E., Norrie J., Packard C. J., Perry I. J., Stott D. J., Sweeney B. J., Twomey C., Westendorp R. G.; PROSPER study group. PROspective Study of Pravastatin in the Elderly at Risk. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet 2002; 360, 1623–1630.
58. Zandi P. P., Sparks D. L., Khachaturian A. S., Tschanz J., Norton M., Steinberg M., Welsh-Bohmer K. A., Breitner J. C.; Cache County Study investigators. Do statins reduce risk of incident dementia and Alzheimer disease? The Cache County Study. Arch Gen Psychiatry 2005; 62, 217–224.
59. Wang Q., Yan J., Chen X., Li J., Yang Y., Weng J., Deng C., Yenari M. A. Statins: multiple neuroprotective mechanisms in neurodegenerative diseases. Exp Neurol 2011; 230, 27–34.
60. Craft S. Insulin resistance syndrome and Alzheimer’s disease: age - and obesity-related effects on memory, amyloid, and inflammation. Neurobiol Aging 2005; 26, S65–S69.
61. Pedersen W. A., McMillan P. J., Kulstad J. J., Leverenz J. B., Craft S., Haynatzki G. R. Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp Neurol 2006; 199, 265–273.
62. Watson G. S., Cholerton B. A., Reger M. A., Baker L. D., Plymate S. R., Asthana S., Fishel M. A., Kulstad J. J., Green P. S., Cook D. G., Kahn S. E., Keeling M. L., Craft S. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am J Geriat Psychiat 2005; 13, 950–958.
63. Geldmacher D. S., Frolich L., Doody R. S., Erkinjuntti T., Vellas B., Jones R. W., Banerjee S., Lin P., Sano M. Realistic expectations for treatment success in Alzheimer’s disease. J Nutr Health Aging 2006; 10, 417–429.
64. Craft S. Insulin resistance syndrome and Alzheimer disease: pathophysiologic mechanisms and therapeutic implications. Alz Dis Assoc Dis 2006; 20, 298 –301.
65. Gupta A., Bisht B., Dey C. S. Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and Alzheimer’s-like changes. Neuropharmacology 2011; 60, 910–920.
66. Cuajungco M. P., Frederickson C. J., Bush A. I. Amyloid-beta metal interaction and metal chelation. Sub-Cell Biochem 2005; 38, 235–254.
67. Cherny R. A., Atwood C. S., Xilinas M. E., Gray D. N., Jones W. D., McLean C. A., Barnham K. J., Volitakis I., Fraser F. W., Kim Y., Huang X., Goldstein L. E., Moir R. D., Lim J. T., Beyreuther K., Zheng H., Tanzi R. E., Masters C. L., Bush A. I. Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 2001; 30, 665–676.
68. Ritchie C. W., Bush A. I., Mackinnon A., Macfarlane S., Mastwyk M., MacGregor L., Kiers L., Cherny R., Li Q. X., Tammer A., Carrington D., Mavros C., Volitakis I., Xilinas M., Ames D., Davis S., Beyreuther K., Tanzi R. E., Masters C. L. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol 2003; 60, 1685–1691.
69. Dedeoglu A., Cormier K., Payton S., Tseitlin K. A., Kremsky J. N., Lai L., Li X., Moir R. D., Tanzi R. E., Bush A. I., Kowall N. W., Rogers J. T., Huang X. Preliminary studies of a novel bifunctional metal chelator targeting Alzheimer’s amyloidogenesis. Exp Gerontol 2004; 39, 1641–1649.
70. Lee J. Y., Friedman J. E., Angel I., Kozak A., Koh J. Y. The lipophilic metal chelator DP-109 reduces amyloid pathology in brains of human beta-amyloid precursor protein transgenic mice. Neurobiol Aging 2004; 25, 1315–1321.
71. Reznichenko L., Amit T., Zheng H., Avramovich-Tirosh Y., Youdim M. B., Weinreb O., Mandel, S. Reduction of iron-regulated amyloid precursor protein and beta-amyloid peptide by (-)-epigallocatechin-3-gallate in cell cultures: implications for iron chelation in Alzheimer’s disease. J Neurochem 2006; 97, 527–536.
72. Fisher A., Pittel Z., Haring R., Bar-Ner N., Kliger-Spatz M., Natan N., Egozi I., Sonego H., Marcovitch I., Brandeis R. M1 muscarinic agonists can modulate some of the hallmarks in Alzheimer’s disease: implications in future therapy. J Mol Neurosci 2003; 20, 349–356.
73. Fisher A., Brandeis R., Bar-Ner R. H., Kliger-Spatz M., Natan N., Sonego H., Marcovitch I., Pittel Z. AF150(S) and AF267B: M1 muscarinic agonists as innovative therapies for Alzheimer’s disease. J Mol Neurosci 2002; 19, 145–153.
74. Caccamo A., Fisher A., Laferla F. M. M1 Agonists as a potential disease-modifying therapy for Alzheimer’s disease. Curr Alzheimer Res 2009; 6, 112–117.
75. Caccamo A., Oddo S., Billings L. M., Green K. N., Martinez-Coria H., Fisher A., LaFerla F. M. M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron 2006; 49, 671–682.
76. Hock C., Maddalena A., Raschig A., Müller-Spahn F., Eschweiler G., Hager K., Heuser I., Hampel H., Müller-Thomsen T., Oertel W., Wienrich M., Signorell A., Gonzalez-Agosti C., Nitsch R. M. Treatment with the selective muscarinic m1 agonist talsaclidine decreases cerebrospinal fluid levels of A beta (42) in patients with Alzheimer’s disease. Amyloid 2003; 10, 1–6.
77. Bodick N. C., Offen W. W., Levey A. I., Cutler N. R., Gauthier S. G., Satlin A., Shannon H. E., Tollefson G. D., Rasmussen K., Bymaster F. P., Hurley D. J., Potter W. Z., Paul S. M. Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease. Arch Neurol 1997; 54, 465–473.
78. Fang L., Jumpertz S., Zhang Y., Appenroth D., Fleck C., Mohr K., Tränkle C., Decker M. Hybrid molecules from xanomeline and tacrine: enhanced tacrine actions on cholinesterases and muscarinic M1 receptors. J Med Chem 2010; 53, 2094–2103.
79. Mulugeta E., Karlsson E., Islam A., Kalaria R., Mangat H., Winblad B., Adem A. Loss of muscarinic M4 receptors in hippocampus of Alzheimer patients. Brain Res 2003; 960, 259–262.
80. Heinrich JN, Butera JA, Carrick T, Kramer A, Kowal D, Lock T, Marquis KL, Pausch MH, Popiolek M, Sun SC, Tseng E, Uveges AJ, Mayer SC. Pharmacological comparison of muscarinic ligands: historical versus more recent muscarinic M1-preferring receptor agonists. Eur J Pharmacol 2009; 605, 53–56.
81. Neeper M., Schmidt A. M., Brett J., Yan S. D., Wang F., Pan Y. C., Elliston K., Stern D., Shaw A. Cloning and expression of a cell-surface receptor for advanced glycosylation end-products of proteins. J Biol Chem 1992; 267, 14998–15004.
82. Mackic J. B., Stins M., McComb J. G., Calero M., Ghiso J., Kim K. S., Yan S. D., Stern D., Schmidt A. M., Frangione B., Zlokovic B. V. Human blood-brain barrier receptors for Alzheimer’s amyloid-h 1–40. Asymmetrical binding, endocytosis, and transcytosis at the apical side of brain microvascular endothelial cell monolayer. J Clin Invest 1998; 102, 734–743.
83. Deane R., Du Yan S., Submamaryan R. K., LaRue B., Jovanovic S., Hogg E., Welch D., Manness L., Lin C., Yu J., Zhu H., Ghiso J., Frangione B., Stern A., Schmidt A. M., Armstrong D. L., Arnold B., Liliensiek B., Nawroth P., Hofman F., Kindy M., Stern D., Zlokovic B. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med 2003; 9, 907–913.
84. Matsuoka Y., Saito M., LaFrancois J., Saito M., Gaynor K., Olm V., Wang L., Casey E., Lu Y., Shiratori C., Lemere C., Duff K. Novel therapeutic approach for the treatment of Alzheimer’s disease by peripheral administration of agents with an affinity to betaamyloid. J Neurosci 2003; 23, 29–33.
85. Bachmeier C., Beaulieu-Abdelahad D., Mullan M., Paris D. Selective dihydropyiridine compounds facilitate the clearance of β-amyloid across the blood-brain barrier. Eur J Pharmacol 2011; 659, 124–129.
Labels
Pharmacy Clinical pharmacologyArticle was published in
Czech and Slovak Pharmacy

2012 Issue 1-2
Most read in this issue
- Možnosti inovace individuální přípravy léčivých přípravků v lékárnách v České republice
- Kompatibilita fosforečnanů se solemi vápníku v parenterální výživě
- Metabolomika vo výskume fytoterapeutík
- Zvažované farmakoterapeutické přístupy léčby Alzheimerovy choroby