
Mitochondriální enzym ABAD a jeho role v rozvoji a léčbě Alzheimerovy nemoci
Mitochondrial enzyme ABAD and its role in the development and treatment of Alzheimer’s disease
The amyloid-ß peptide (Aß) has been associated with Alzheimer’s disease (AD) for some time. The original amyloid cascade hypothesis declared that the insoluble extracellular plaques were responsible for main Aß toxicity. Nowadays, this hypothesis is outdated and soluble intracellular Aß forms and their effects within the cell have come into the centre of attention. There are many intracellular proteins interacting with Aß including the mitochondrial enzyme amyloid-binding alcohol dehydrogenase (ABAD). The interaction between ABAD and Aß impairs mitochondrial functions and ultimately results in cell death. In this review, current findings concerning the enzyme ABAD are summarized. Its role in AD development and its interaction with Aß as a potential therapeutic target are discussed.
Keywords:
Alzheimer’s disease, amyloid-ß peptide, mitochondrial dysfunction, amyloid-binding alcohol dehydrogenase, frentizole
Authors:
Ondřej Benek; Kamil Musílek; Kamil Kuča
Published in:
Čes. slov. Farm., 2012; 61, 144-149
Category:
Review Articles
Došlo 15. května 2012 / Přijato 2. července 2012
Overview
Již delší dobu je známa souvislost mezi peptidem ß-amyloidem (Aß) a Alzheimerovou nemocí (AD). Původní hypotéza považující za hlavní toxickou formu Aß nerozpustné extracelulární plaky je dnes již překonána a zájem se upírá k rozpustným formám Aß a jejich působení uvnitř buňky. Je známo mnoho intracelulárních proteinů interagujících s Aß, mezi nimi též mitochondriální enzym amyloid vázající alkoholdehydrogenasa (ABAD). Vazba Aß na tento enzym poškozuje zatím ne zcela známým mechanismem mitochondrie a v konečném důsledku vede až k zániku buňky. V této práci jsou shrnuty dosavadní poznatky o enzymu ABAD, jeho roli v rozvoji AD a možnostech ovlivnění interakce ABAD-Aß jako potenciálním cíli pro farmakoterapii tohoto závažného onemocnění.
Klíčová slova:
Alzheimerova nemoc, amyloid-ß peptid, mitochondriální dysfunkce, amyloid vázající alkohol dehydrogenasa, frentizol
Úvod
Alzheimerova choroba je nejběžnějším neurodegenerativním onemocněním vyskytujícím se u starší populace. Poprvé bylo onemocnění popsáno na začátku 20. století Aloisem Alzheimerem. Je pro ni charakteristické progresivní poškození kognitivních funkcí a paměti. Ztráta mozkových funkcí vede k naprosté sociální nesamostatnosti a v konečném důsledku ke smrti. Navzdory intenzivnímu výzkumu není dosud známa přesná příčina vzniku AD a mechanismus patologie v raném stadiu onemocnění, a tedy ani neexistuje efektivní terapie.
Hlavními patologickými znaky vyskytujícími se v postižených oblastech mozku jsou přítomnost extracelulárních senilních plaků tvořených peptidem ß-amyloidem (Aß), intracelulárních neurofibrilárních klubek vznikajících agregací fosforylovaného tau-proteinu a ztráta neuronů, především cholinergních1, 2). AD je dále spjata se zánikem synapsí, poškozením mitochondrií a zánětlivou reakcí3–7).
Ačkoliv není známa přesná příčina AD, předpokládá se, že hlavní roli v patologii onemocnění hraje zvýšená tvorba peptidu ß-amyloidu (Aß). Aß vzniká proteolytickým štěpením amyloidového prekurzorového proteinu (APP) enzymy ß - a γ-sekretasou. Při štěpení APP vznikají peptidy o délce převážně 40 a 42 aminokyselin8, 9). Původní hypotéza tzv. amyloidní kaskády předpokládala, že ukládání nerozpustného Aß v extracelulárním prostoru je prvotní příčinou rozvoje onemocnění. Novější data však ukazují, že nositelem toxicity jsou rozpustné formy Aß oligomerů vyskytující se intracelulárně, jejichž kumulace uvnitř buňky časově předchází vzniku senilních plaků10–13). Na základě těchto zjištění byla upravena původní hypotéza a výzkum se zaměřil na působení Aß uvnitř buňky.
Stále více důkazů ukazuje na mitochondrie jako hlavní místo toxicity intracelulárního Aß (obr. 1)14). Již delší dobu je známo, že AD je spojena s oxidativním stresem a poškozením mitochondrií. Byly zjištěny změny v počtu a velikosti mitochondrií15), snížení energetického metabolismu16), zvýšený oxidativní stres17), poruchy homeostázy vápníku18) a změny v mitochondriální DNA19). Dále byla prokázána přítomnost Aß v mitochondriích zvířecího modelu onemocnění i u lidských pacientů a jeho negativní dopad na jejich fungování20, 21). In vitro bylo prokázáno, že Aß působí poškození dýchacího řetězce, snížení membránového potenciálu, únik cytochromu C, zvýšenou tvorbu mitochondriálního permeabilního tranzitního kanálu (mPTP) a zvýšenou tvorbu volných radikálů22–24). Jakou cestou se Aß dostává do mitochondrií, zatím nebylo jednoznačně zodpovězeno. Nejpravděpodobnější se jeví možnost prostupu skrze transportní kanály TOM40 (vnější mitochondriální membrána) a TIM22 (vnitřní mitochondriální membrána)25). Další možností je transport přes tzv. MAMs (membrány asociované s mitochondriemi), které jsou součástí endoplazmatického retikula26, 27).
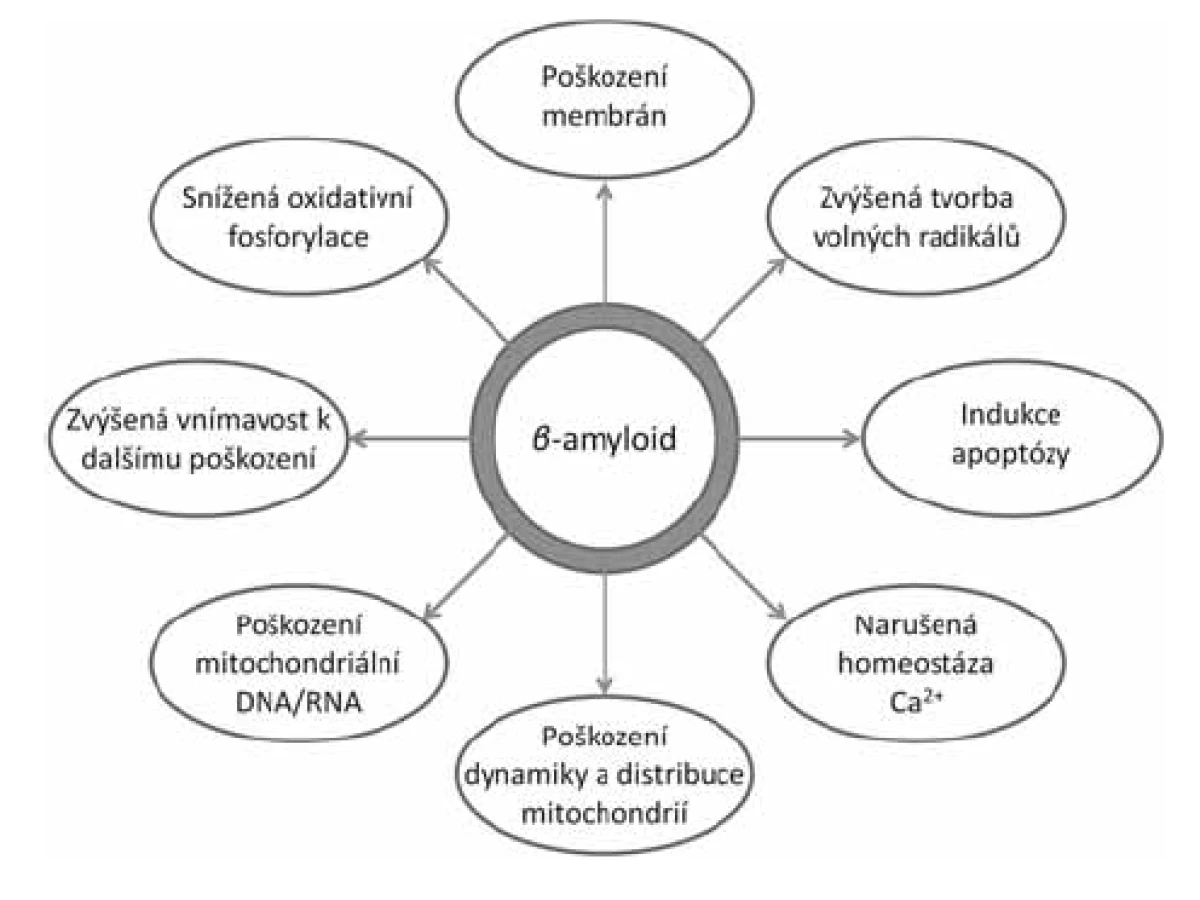
Bylo identifikováno několik mitochondriálních enzymů, které přímo interagují s Aß za ovlivnění jejich fyziologické funkce, např. ABAD, CypD, GAPDH, HtrA228–31). Mezi nimi amyloid vázající alkoholdehydrogenasa (ABAD) se zdá být klíčovou pro rozvoj neurotoxicky mitochondriálního Aß21). V tomto článku jsou shrnuty dosavadní poznatky o tomto enzymu, jeho roli v rozvoji AD a možnostech farmakologického ovlivnění interakce ABAD-Aß jako perspektivním cíli pro terapii AD.
ABAD a jeho fyziologická funkce
Enzym ABAD poprvé popsali v roce 1997 Yan et al.28) a o rok později byl identifikován jako lidská obdoba dříve objevené hovězí hydroxyacyl-CoA dehydrogenasy typu II32). Původně byl označen jako ERAB (s endoplazmatickým retikulem asociovaný amyloid vázající protein) podle kompartmentu, ve kterém byl původně chybně identifikován. Jeho skutečná lokalizace je uvnitř mitochondriální matrix33). Enzym má ještě další alternativní názvy jako SCHAD34), HADH II35), 17ß-HSD1036), MBHD37). Tyto názvy byly odvozeny podle různých substrátů, jejichž přeměnu je ABAD schopen katalyzovat.
ABAD je NAD-dependentní oxidoreduktasa patřící do SDR (dehydrogenasy/reduktasy s krátkým řetězcem) skupiny enzymů. Je to multifunkční enzym katalyzující redukci aldehydů a ketonů a oxidaci alkoholů (obr. 2)38). ABAD je poměrně málo substrátově specifický a experimentálně pro něj bylo popsáno mnoho potenciálních substrátů. Je však otázkou, které z nich enzym skutečně přeměňuje v prostředí organismu27).
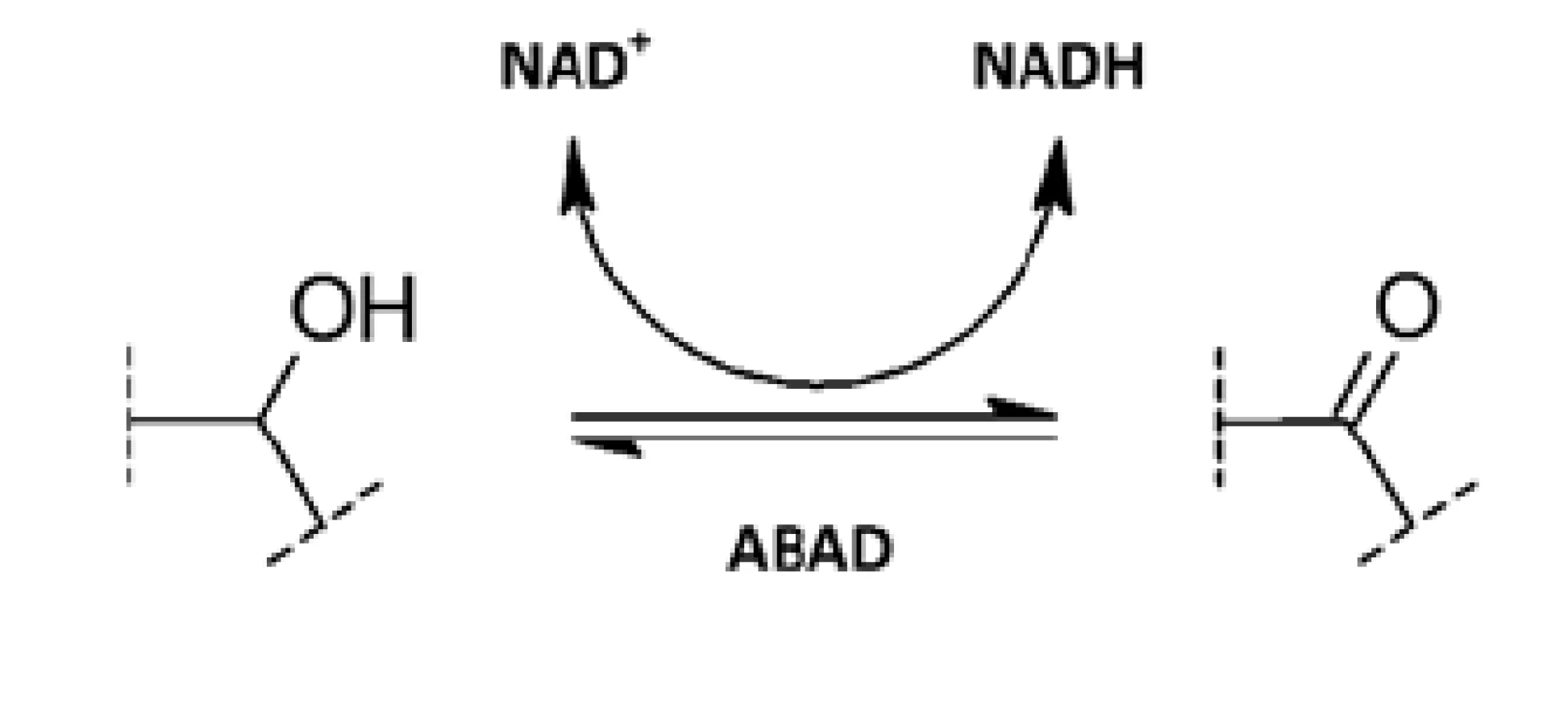
Hlavní fyziologickou funkcí ABAD je pravděpodobně třetí krok ß-oxidace mastných kyselin s krátkým rozvětveným řetězcem, kde vystupuje jako L-3-hydroxyacyl-CoA dehydrogenasa. Tomu by napovídala i lokalizace enzymu uvnitř mitochondriální matrix39). Z hlediska energetického metabolismu se jeví významná též schopnost metabolizovat ketolátky např. ß-hydroxybutyrát, a tím dodávat buňce energii při nedostatku kyslíku40). Účastní se též degradační cesty isoleucinu. Nesmyslná mutace v genu kódujícím ABAD vede k poruše katabolismu isoleucinu, která se klinicky projeví jako progresivní ztráta mentálních a motorických schopností, mentální retardací a epilepsií37).
Dalšími zvažovanými funkcemi jsou oxidace a inaktivace estradiolu a steroidních modulátorů GABAA receptoru. Role ABAD v inaktivaci estradiolu by mohla vysvětlit, proč AD trpí častěji ženy než muži38, 41). ABAD je také pravděpodobně součástí mitochondriální RNAsy P. Tato funkce nesouvisí s enzymatickou aktivitou ABAD. RNAsa P je enzym důležitý pro správnou tvorbu tRNA a následně i syntézu proteinů v mitochondriích42, 43).
Struktura a mechanismus funkce
ABAD se v buňce vyskytuje ve formě homotetrameru. Katalytická triáda sestává z aminokyselin Ser155, Tyr168 a Lys172 a je tedy stejná jako u ostatních enzymů z SDR skupiny39). Kofaktor NAD+/NADH se váže na enzym v blízkosti aminokyselin katalytické triády a vytváří s nimi nevazebné interakce. Vazebné místo pro substrát obsahuje oblast s kladně nabitými zbytky aminokyselin lysinu a histidinu, se kterými interaguje záporně nabitá část substrátů obsahujících v molekule CoA. Proto jsou substráty, které ve své molekule CoA neobsahují, enzymem přeměňovány méně efektivně. Hydrofobní oblast oddělující katalytickou triádu a oblast s kladným nábojem je vhodně uspořádána pro interakci s alifatickým řetězcem mastné kyseliny. Všechny tyto skutečnosti podporují domněnku, že acyl-CoA jsou primárním substrátem ABAD39, 44).
Mechanismus katalýzy lze přiblížit na příkladu redukce ketonu na alkohol. Tyr168 interaguje s karbonylovou skupinou substrátu, čímž zvyšuje elektrofilitu karbonylového uhlíku. Amoniová skupina Lys172 interaguje s tyrosinem za zvýšení jeho acidity. Donorem hydridu pro vlastní redukci je kofaktor NADH. Při přenosu hydridu na aktivovaný karbonyl současně dochází k deprotonaci Tyr168 a proton je přenesen na nově vytvořený hydroxylový anion. Záporný náboj vzniklý na Tyr168 je stabilizován vytvořením vodíkové vazby se Ser15539).
Na rozdíl od ostatních enzymů SDR skupiny obsahuje molekula ABAD navíc dvě sekvence aminokyselin (rezidua 102–107 a 141–146). Právě oblast zahrnující rezidua 102–107 a nacházející se v blízkosti aktivního centra enzymu je místem vazby Aß21, 39, 44).
Interakce ABAD-Aß a její důsledky
Interakce ABAD s Aß byla poprvé prokázána v roce 1997 pomocí kvasinkového dvouhybridního systému28). V následujících letech bylo toto zjištění potvrzeno za použití dalších metod, např. ELISA45), krystalografie21), SPR (surface plasmon resonance)21, 46), ko-imunoprecipitace21, 28) nebo imunocytochemie následované konfokální mikroskopií21).
Vazebným místem Aß je oblast aminokyselinových reziduí 100–110, označovaná též jako Loop-D. Bodové mutace v oblasti Loop-D (konkrétně rezidua 98–101 a 108–110) zabránily vazbě Aß na enzym21). Ostatní enzymy SDR skupiny tuto oblast ve své molekule neobsahují39). To by vysvětlovalo, proč pouze ABAD a nikoliv ostatní SDR enzymy interaguje s Aß.
Vazba Aß na enzym způsobí sterické změny v oblasti vazebného místa pro kofaktor NAD+, který se pak nemůže na enzym vázat, a tím je inhibována jeho funkce. Naopak enzym s navázaným kofaktorem s Aß neinteraguje. Ačkoliv se tedy Aß a NAD+ vážou na různá místa enzymu, jejich současné navázání se vylučuje21, 46).
Aß inhibuje funkci enzymu (Ki = 1,2–1,6 μM) pro substrát acetoacetyl-CoA47, 48). Zajímavé však je, že vazba Aß na ABAD je popsána již při koncentracích v řádu desítek nM28, 46). Možné vysvětlení předpokládá, že pro inhibici enzymu nestačí navázání monomerní formy Aß, ale je jí dosaženo až agregovanou oligomerní formou amyloidu27).
Byly provedeny pokusy na živých buňkách, které ve zvýšené míře exprimovaly v různých kombinacích ABAD, enzymaticky inaktivní ABAD, Aß nebo APP. Při těchto pokusech bylo zjištěno, že zvýšenou toxicitu vykazoval Aß (resp. APP) pouze v těch buňkách, které současně exprimovaly i funkční ABAD. Samotný Aß/APP nebo v kombinaci s inaktivní formou ABAD měl podstatně nižší toxické účinky. Z toho lze usuzovat, že toxicita Aß nespočívá v inhibici enzymu, ale ve změně jeho vlastností (např. funkce nebo lokalizace). Nositelem toxicity je tedy katalyticky aktivní enzym po navázání Aß, čemuž by odpovídalo i výše zmíněné zjištění, že vazba Aß na enzym probíhá již v o dva řády nižších koncentracích, než jaká je potřebná k jeho inhibici47). Při jiném experimentu potvrzujícím tuto domněnku vedlo podání protilátek proti ABAD ke snížení toxicity Aß28).
Proteomické studie odhalily dva proteiny, jejichž exprese se zvyšuje v důsledku interakce ABAD-Aß. Peroxiredoxin II je antioxidačně působící enzym a jeho zvýšená exprese působí protektivně proti toxickým účinkům Aß49). Druhý z proteinů, endofilin I (EP-I), působí naopak cytotoxicky, když aktivuje proapoptotickou signální kaskádu50, 51).
Mechanismus toxicity komplexu ABAD-Aß
Mechanismus, kterým Aß po navázání na ABAD rozvíjí svoji neurotoxicitu, není dosud znám, avšak existuje několik teorií. Nejčastěji zmiňovaná teorie předpokládá, že ABAD po navázání Aß produkuje toxické aldehydy HNE (4-hydroxynonenal) a MDA (malondialdehyd), které za normálních okolností naopak detoxikuje. Tato teorie je podpořena výsledky experimentů na buněčných liniích. V nich pouze buňky, které exprimovaly katalyticky aktivní ABAD a současně Aß, vykazovaly zvýšenou tvorbu HNE a MDA. Naopak buňky exprimující pouze ABAD vykazovaly zvýšenou odolnost proti toxickým účinkům těchto aldehydů. K tomu může docházet buď na základě změny funkce enzymu, anebo změny distribuce enzymu, jenž následně dostane možnost metabolizovat substráty z jiných kompartmentů47, 52, 53).
Jiná teorie předpokládá, že ABAD po interakci s Aß ve zvýšené míře metabolizuje hormon estradiol, ať už vinou změny distribuce enzymu, nebo v důsledku jeho zvýšené exprese a následně i aktivity36). Bylo prokázáno, že estradiol působí protektivně vůči toxickým účinkům Aß a jeho preventivní podávání snižuje riziko onemocnění AD54).
Další možností je poškození funkce mitochondriální RNAsy P, které je ABAD součástí. To by narušilo syntézu mitochondriálních proteinů, včetně součástí dýchacího řetězce, s následným poškozením energetického metabolismu buňky42, 43).
V neposlední řadě může být toxicita interakce ABAD-Aß zprostředkována proteinem EP-I, jehož exprese je v důsledku této interakce zvýšená. EP-I aktivuje enzym JNK (c-Jun N-terminální kinasa), který je součástí proapoptotické signální kaskády, což může v konečném důsledku vést až k zániku buňky. Zvýšená aktivace JNK je spojována s patologií AD již delší dobu a protein Ep-I se tak zdá být možným propojením mezi ní a působením Aß50, 51).
Ovlivnění interakce ABAD-Aß jako potenciální cíl terapie AD
Pro ověření, zda je Loop-D skutečně místem vazby Aß a zda její přerušení sníží toxicitu, byl syntetizován tzv. ABAD-návnadový peptid (ABAD-decoy peptide; ABAD-DP), jehož aminokyselinová sekvence (rezidua 92–120) oblast Loop-D zahrnuje. Předpokládalo se, že ABAD-DP vyváže Aß, a tím ochrání vlastní enzym. Peptid blokoval vazbu ABAD-Aß s Ki v řádu jednotek μM. Kd pro komplex Aß s ABAD-DP byla podobná jako pro ABAD-Aß. Při pokusech na buněčných liniích, které byly vystaveny působení Aß, snížilo podání ABAD-DP tvorbu volných radikálů, uvolňování cytochromu C z mitochondrií a zvýšilo životaschopnost buněk21). Systémová léčba pomocí ABAD-DP (obohaceného o aminokyselinové sekvence umožňující přechod přes membrány a vstup do mitochondrií) u zvířecího modelu AD zlepšila kognitivní schopnosti v porovnání s neléčenou skupinou55). Na základě výše uvedených údajů se jeví interakce ABAD-Aß jako perspektivní cíl pro farmakoterapeutický zásah.
ABAD-DP je jako peptid svým charakterem nevhodný pro běžnou léčbu. Peptidy jsou málo dostupné po perorálním podání a v organismu mají nízkou stabilitu. Je proto snaha nalézt nízkomolekulární látky schopné inhibovat interakci ABAD-Aß. První výsledky v této oblasti publikoval Xie et al.45).
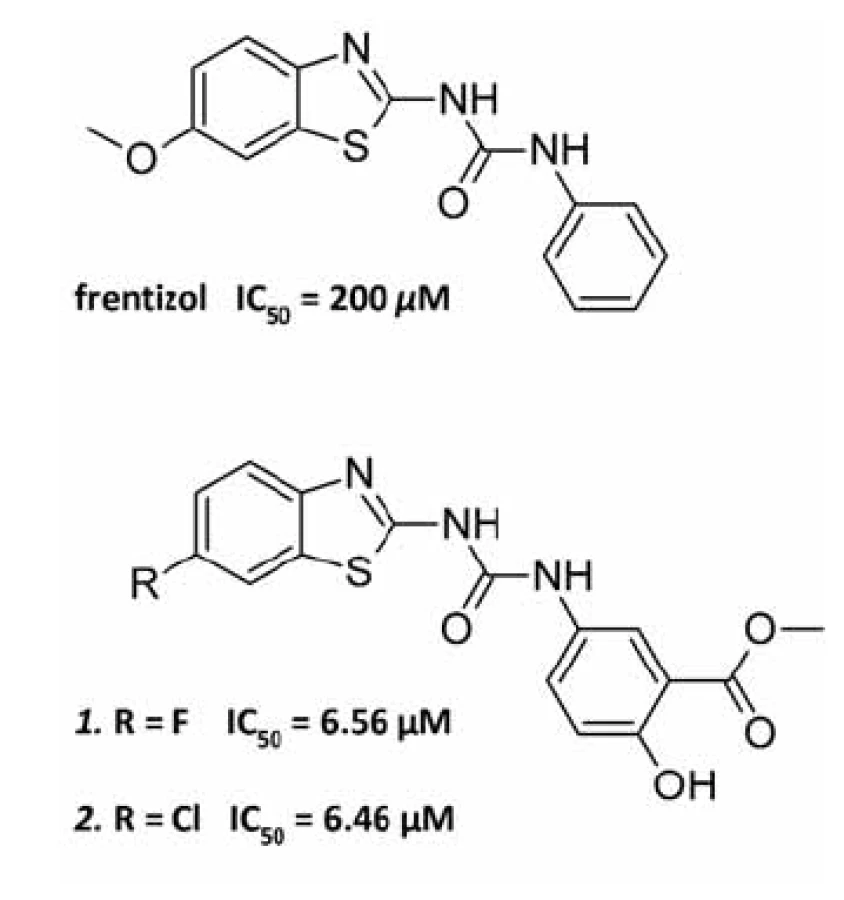
Byl proveden screening skupiny 50 látek, o nichž se vědělo, že interagují buď s Aß, nebo s ABAD, anebo působí neuroprotektivně, na schopnost inhibovat interakci ABAD-Aß. Aktivitu vykázaly tři sloučeniny: Aß vážící barviva Kongo červeň a thioflavin-T a neuroprotektivní látka resveratrol. Jako nejvhodnější kandidát pro další zkoumání byl vybrán thioflavin-T pro svou nízkou toxicitu a dobrý průnik přes biologické membrány. Při následném screeningu analogických struktur byla objevena účinnější látka frentizol, známé imunosupresivum a antivirotikum používané například k léčbě revmatické artritidy. Na základě předlohové molekuly frentizolu byla připravena a otestována série jeho strukturních analogů, z nichž dva nejúčinnější vykazovaly 30× vyšší inhibiční aktivitu vůči ABAD ve srovnání s frentizolem (obr. 3)45). Zatím však nebylo prokázáno, zda jsou inhibiční vlastnosti těchto látek specifické pro vazbu ABAD-Aß a zda jsou schopné snižovat toxicitu Aß.
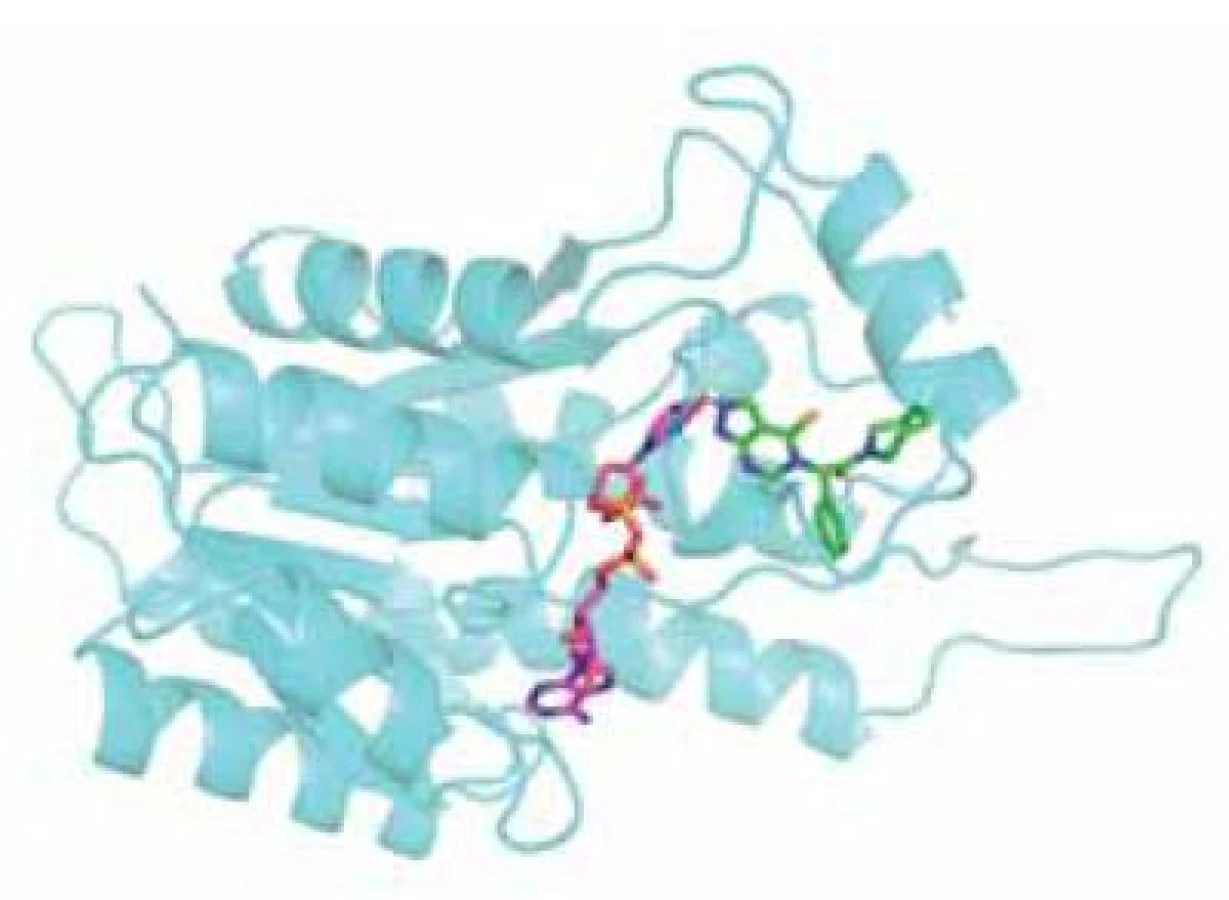
Jinou možností jak zabránit ß-amyloidem indukované neurotoxicitě je inhibovat katalytickou funkci ABAD, která se pro ni jeví jako nezbytná. Tento přístup je však méně specifický pro vlastní onemocnění a bude jím ovlivněna i fyziologická funkce enzymu, což by se mohlo projevit v podobě nežádoucích účinků. Za tímto účelem byla testována sloučenina AG18051 (obr. 4)44, 56).
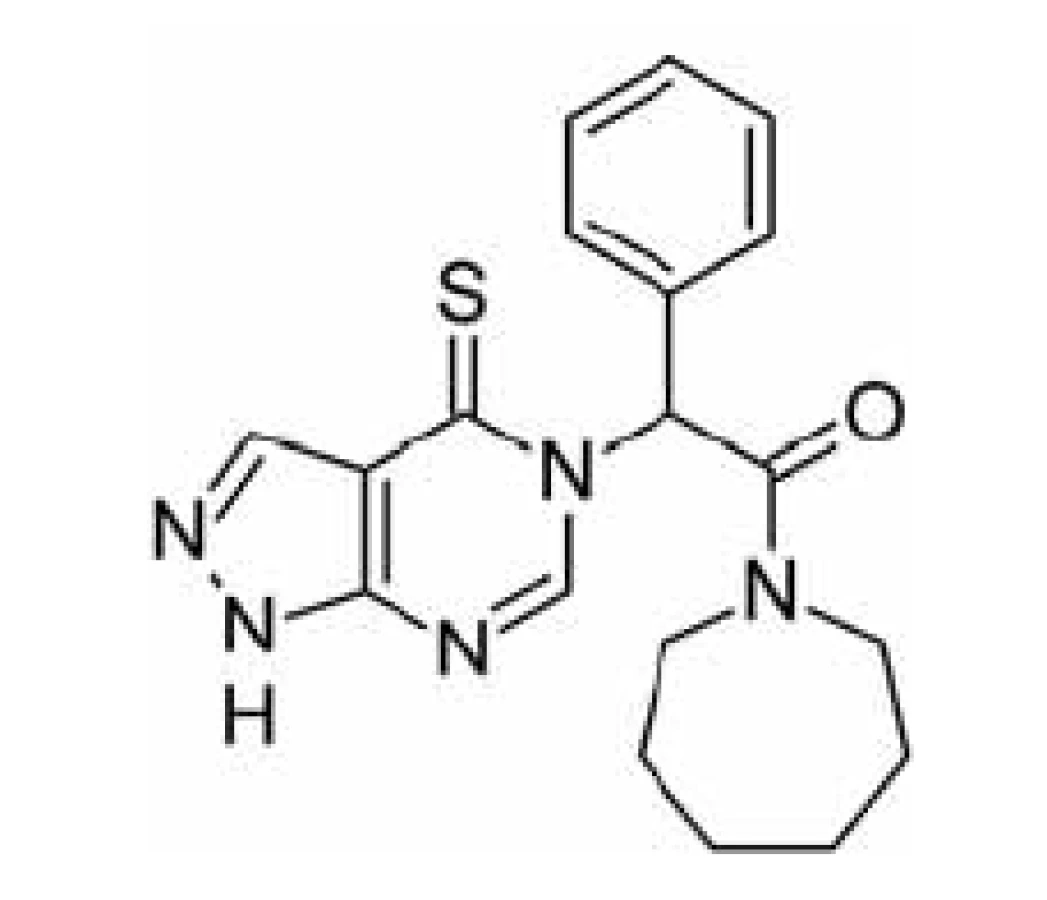
AG18051 je specifický inhibitor ABAD, který nemá strukturní podobnost se žádným ze známých substrátů. Váže se na aktivní místo enzymu, kde se orientuje do oblasti vazebného místa pro substrát a vytváří kovalentní vazbu s kofaktorem NAD+ (obr. 5). Na živých buňkách bylo potvrzeno, že inhibice ABAD pomocí AG18051 snižuje toxicitu Aß. Bylo též prokázáno, že AG18051 částečně brání vazbě Aß na ABAD. Vzhledem k mechanismu inhibice odpovídá toto zjištění výše zmíněné skutečnosti, že současná vazba kofaktoru a Aß na enzym se vylučuje. Schopnost inhibovat vazbu ABAD-Aß tak pravděpodobně přispívá k neuroprotektivním účinkům AG1805144, 56).
Závěr
ABAD je dosud nejlépe prozkoumaným intracelulárním proteinem interagujícím s Aß. Přímá vazba Aß na tento mitochondriální enzym byla jednoznačně prokázána pomocí různých metod. Interakce ABAD-Aß vede k poškození mitochondrií typickému pro AD. Inhibice této interakce tedy představuje perspektivní cíl pro terapii AD, což bylo potvrzeno za použití ABAD-DP. První pokusy o nalezení nízkomolekulárního inhibitoru vedly k syntéze série derivátů frentizolu. Pro další vývoj léčiv tohoto typu je však nezbytné hlouběji porozumět podstatě interakce ABAD-Aß a zjistit přesný mechanismus jejího toxického působení.
Střet zájmů: žádný.
O. Benek
Univerzita obrany, Fakulta vojenského zdravotnictví, Katedra toxikologie a Centrum pokročilých studií, Hradec Králové
doc. PharmDr. Kamil Musílek, Ph.D.
Univerzita Hradec Králové, Přírodovědecká fakulta, Katedra chemie
Rokitanského 62, 500 03 Hradec Králové, Česká republika
e-mail: kamil.musilek@gmail.com
K. Kuča
Fakultní nemocnice, Hradec Králové
Sources
1. Price D. L., Sisodia S. S.:Mutant genes in familial Alzheimer’s disease and transgenic models. Annu. Rev. Neurosci. 1998; 21, 479–505.
2. Selkoe D. J. Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature. 1999; 399(Suppl), A23–31.
3. Reddy P. H., Mani G., Park B. S., et al. Differential loss of synaptic proteins in Alzheimer’s disease: implications for synaptic dysfunction. J. Alzheimers Dis. 2005; 7(2), 103–117; discussion 173–180.
4. Golde T. E. Inflammation takes on Alzheimer disease. Nat. Med. 2002; 8(9), 936–938.
5. Reddy P. H., McWeeney S., Park B. S. et al. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Hum. Mol. Genet. 2004; 13(12), 1225–1240.
6. Reddy P. H., Beal M. F. Are mitochondria critical in the pathogenesis of Alzheimer’s disease? Brain Res. Brain Res. Rev. 2005; 49(3), 618–632.
7. Reddy P. H., McWeeney S. Mapping cellular transcriptosomes in autopsied Alzheimer’s disease subjects and relevant animal models. Neurobiol. Aging. 2006; 27(8), 1060–1077.
8. Keller J. N. Age-related neuropathology, cognitive decline, and Alzheimer’s disease. Ageing Res. Rev. 2006; 5(1), 1–13.
9. Selkoe D. J. Alzheimer’s disease: genes, proteins, and therapy. Physiol. Rev. 2001; 81(2), 741–766.
10. Hardy J. A., Higgins G. A. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992; 256(5054), 184–185.
11. Dahlgren K. N., Manelli A. M., Stine W. B. Jr., Baker L. K., Krafft G. A., LaDu M. J. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J. Biol. Chem. 2002; 277(35), 32046–32053.
12. Gouras G. K., Tsai J., Naslund J. et al. Intraneuronal Abeta42 accumulation in human brain. Am. J. Pathol. 2000; 156(1), 15–20.
13. Wirths O., Multhaup G., Bayer T. A. A modified beta-amyloid hypothesis: intraneuronal accumulation of the beta-amyloid peptide-the first step of a fatal cascade. J. Neurochem. 2004; 91(3), 513–520.
14. Tillement L., Lecanu L., Papadopoulos V. Alzheimer’s disease: effects of ß-amyloid on mitochondria. Mitochondrion. 2011; 11(1), 13–21.
15. Hirai K., Aliev G., Nunomura A. et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001; 21(9), 3017–3023.
16. Bubber P., Haroutunian V., Fisch G., Blass J. P., Gibson G. E. Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann. Neurol. 2005; 57(5), 695–703.
17. Butterfield D. A., Drake J., Pocernich C., Castegna A. Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid beta-peptide. Trends Mol Med. 2001; 7(12), 548–554.
18. LaFerla F. M. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 2002; 3(11), 862–872.
19. Manczak M., Park B. S., Jung Y., Reddy P. H. Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: implications for early mitochondrial dysfunction and oxidative damage. Neuromolecular Med. 2004; 5(2), 147–162.
20. Fernández-Vizarra P., Fernández A. P., Castro-Blanco S. et al. Intra - and extracellular Abeta and PHF in clinically evaluated cases of Alzheimer’s disease. Histol. Histopathol. 2004; 19(3), 823–844.
21. Lustbader J. W., Cirilli M., Lin C. et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science. 2004; 304(5669), 448–452.
22. Kim H. S., Lee J. H., Lee J. P. et al. Amyloid beta peptide induces cytochrome C release from isolated mitochondria. Neuroreport. 2002; 13(15), 1989–1993.
23. Aleardi A. M., Benard G., Augereau O. et al. Gradual alteration of mitochondrial structure and function by beta-amyloids: importance of membrane viscosity changes, energy deprivation, reactive oxygen species production, and cytochrome c release. J. Bioenerg. Biomembr. 2005; 37(4), 207–225.
24. Moreira P. I., Santos M. S., Moreno A., Rego A. C., Oliveira C. Effect of amyloid beta-peptide on permeability transition pore: a comparative study. J. Neurosci. Res. 2002; 69(2), 257–267.
25. Hansson Petersen C. A., Alikhani N., Behbahani H. et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008; 105(35), 13145–13150.
26. Area-Gomez E., de Groof A. J. C., Boldogh I., et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 2009; 175(5), 1810–1816.
27. Muirhead K. E. A., Borger E., Aitken L., Conway S. J., Gunn-Moore F. J. The consequences of mitochondrial amyloid beta-peptide in Alzheimer’s disease. Biochem. J. 2010; 426(3), 255–270.
28. Yan S. D., Fu J., Soto C. et al. An intracellular protein that binds amyloid-beta peptide and mediates neurotoxicity in Alzheimer’s disease. Nature. 1997; 389(6652), 689–695.
29. Du H., Guo L., Fang F. et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008; 14(10), 1097–1105.
30. Verdier Y., Földi I., Sergeant N. et al. Characterization of the interaction between Abeta 1-42 and glyceraldehyde phosphodehydrogenase. J. Pept. Sci. 2008; 14(6), 755–762.
31. Park H. J., Seong Y. M., Choi J. Y., Kang S., Rhim H. Alzheimer’s disease-associated amyloid beta interacts with the human serine protease HtrA2/Omi. Neurosci. Lett. 2004; 357(1), 63–67.
32. He X. Y., Schulz H., Yang S. Y. A human brain L-3-hydroxyacyl-coenzyme A dehydrogenase is identical to an amyloid beta-peptide-binding protein involved in Alzheimer’s disease. J. Biol. Chem. 1998; 273(17), 10741–10746.
33. He X. Y., Merz G., Yang Y. Z., Mehta P., Schulz H., Yang S. Y. Characterization and localization of human type10 17beta-hydroxysteroid dehydrogenase. Eur. J. Biochem. 2001; 268(18), 4899–4907.
34. He X. Y., Yang Y. Z., Schulz H., Yang S. Y. Intrinsic alcohol dehydrogenase and hydroxysteroid dehydrogenase activities of human mitochondrial short-chain L-3-hydroxyacyl-CoA dehydrogenase. Biochem. J. 2000; 345(Pt 1), 139–143.
35. Furuta S., Kobayashi A., Miyazawa S., Hashimoto T. Cloning and expression of cDNA for a newly identified isozyme of bovine liver 3-hydroxyacyl-CoA dehydrogenase and its import into mitochondria. Biochim. Biophys. Acta. 1997; 1350(3), 317–324.
36. He X. Y., Wen G. Y., Merz G. et al. Abundant type 10 17 beta-hydroxysteroid dehydrogenase in the hippocampus of mouse Alzheimer’s disease model. Brain Res. Mol. Brain Res. 2002; 99(1), 46–53.
37. Ofman R., Ruiter J. P. N., Feenstra M. et al. 2-Methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency is caused by mutations in the HADH2 gene. Am. J. Hum. Genet. 2003; 72(5), 1300–1307.
38. He X. Y., Merz G., Mehta P., Schulz H., Yang S. Y. Human brain short chain L-3-hydroxyacyl coenzyme A dehydrogenase is a single-domain multifunctional enzyme. Characterization of a novel 17beta-hydroxysteroid dehydrogenase. J. Biol. Chem. 1999; 274(21), 15014–15019.
39. Powell A. J., Read J. A., Banfield M. J. et al. Recognition of structurally diverse substrates by type II 3-hydroxyacyl-CoA dehydrogenase (HADH II)/amyloid-beta binding alcohol dehydrogenase (ABAD). J. Mol. Biol. 2000; 303(2), 311–327.
40. Du Yan S., Zhu Y., Stern E. D. et al. Amyloid beta-peptide-binding alcohol dehydrogenase is a component of the cellular response to nutritional stress. J. Biol. Chem. 2000; 275(35), 27100–27109.
41. He X. Y., Wegiel J., Yang Y. Z., Pullarkat R., Schulz H., Yang S. Y. Type 10 17beta-hydroxysteroid dehydrogenase catalyzing the oxidation of steroid modulators of gamma-aminobutyric acid type A receptors. Mol. Cell. Endocrinol. 2005; 229(1–2), 111–117.
42. Holzmann J., Frank P., Löffler E., Bennett K. L., Gerner C., Rossmanith W. RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell. 2008; 135(3), 462–474.
43. Yang S. Y., He X. Y., Miller D. Hydroxysteroid (17ß) dehydrogenase X in human health and disease. Mol. Cell. Endocrinol. 2011; 343(1–2), 1–6.
44. Kissinger C. R., Rejto P. A., Pelletier L. A. et al. Crystal structure of human ABAD/HSD10 with a bound inhibitor: implications for design of Alzheimer’s disease therapeutics. J. Mol. Biol. 2004; 342(3), 943–952.
45. Xie Y., Deng S., Chen Z., Yan S., Landry D. W. Identification of small-molecule inhibitors of the Abeta-ABAD interaction. Bioorg. Med. Chem. Lett. 2006; 16(17), 4657–4660.
46. Yan Y., Liu Y., Sorci M. et al. Surface plasmon resonance and nuclear magnetic resonance studies of ABAD-Abeta interaction. Biochemistry. 2007; 46(7), 1724–1731.
47. Yan S. D., Shi Y., Zhu A. et al. Role of ERAB/L-3-hydroxyacyl-coenzyme A dehydrogenase type II activity in Abeta-induced cytotoxicity. J. Biol. Chem. 1999; 274(4), 2145–2156.
48. Oppermann U. C., Salim S., Tjernberg L. O., Terenius L., Jörnvall H. Binding of amyloid beta-peptide to mitochondrial hydroxyacyl-CoA dehydrogenase (ERAB): regulation of an SDR enzyme activity with implications for apoptosis in Alzheimer’s disease. FEBS Lett. 1999; 451(3), 238–242.
49. Yao J., Taylor M., Davey F. et al. Interaction of amyloid binding alcohol dehydrogenase/Abeta mediates up-regulation of peroxiredoxin II in the brains of Alzheimer’s disease patients and a transgenic Alzheimer’s disease mouse model. Mol. Cell. Neurosci. 2007; 35(2), 377–382.
50. Ren Y., Xu H. W., Davey F. et al. Endophilin I expression is increased in the brains of Alzheimer disease patients. J. Biol. Chem. 2008; 283(9), 5685–5691.
51. Ramjaun A. R., Angers A., Legendre-Guillemin V., Tong X. K., McPherson P. S. Endophilin regulates JNK activation through its interaction with the germinal center kinase-like kinase. J. Biol. Chem. 2001; 276(31), 28913–28919.
52. Sayre L. M., Zelasko D. A., Harris P. L., Perry G., Salomon R. G., Smith M. A. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J. Neurochem. 1997; 68(5), 2092–2097.
53. Murakami Y., Ohsawa I., Kasahara T., Ohta S. Cytoprotective role of mitochondrial amyloid beta peptide-binding alcohol dehydrogenase against a cytotoxic aldehyde. Neurobiol. Aging. 2009; 30(2), 325–329.
54. Dye R. V., Miller K. J., Singer E. J., Levine A. J. Hormone replacement therapy and risk for neurodegenerative diseases. Int. J. Alzheimers Dis. 2012; 2012, 258454.
55. Yao J., Du H., Yan S. et al. Inhibition of Amyloid-beta (A beta) peptide-binding alcohol dehydrogenase-A beta interaction reduces a beta accumulation and improves mitochondrial function in a mouse model of Alzheimer’s disease. J. Neurosci. 2011; 31(6), 2313–2320.
56. Lim Y. A., Grimm A., Giese M. et al. Inhibition of the mitochondrial enzyme ABAD restores the amyloid-ß-mediated deregulation of estradiol. PLoS ONE. 2011; 6(12), e28887.
Labels
Pharmacy Clinical pharmacologyArticle was published in
Czech and Slovak Pharmacy

2012 Issue 4
Most read in this issue
- Lidokainový gel pro použití na kůži magistraliter připravený
-
Standardní receptura pro přípravu léčivých přípravků v lékárnách V*
Sbírka Dermatologische Magistralrezepturen der Schweiz -
Naše léčivé přípravky v polovině 19. století
II. část – galenické přípravky -
Naše léčivé přípravky v polovině 19. století
I. část – úvod a chemické přípravky