
Kam směřuje vývoj nových antituberkulotik?
Where does the development of new antituberculotics aim at?
Several research teams in the Czech and Slovak Republics are oriented on the development of new antitubeculotics. The present article is based mainly on the information from the Chemical Abstracts from the year 2011 and the beginning of the year 2012. It is a selection from almost three thousand reports aiming to help our scientists. The article presents topical information which may be of interest to several pharmaceutical professions.
Keywords:
Mycobacterium tuberculosis, antituberculotics, multiresistant mycobacteria, QSAR, evaluation in vivo, biochemistry of mycobacteria
Authors:
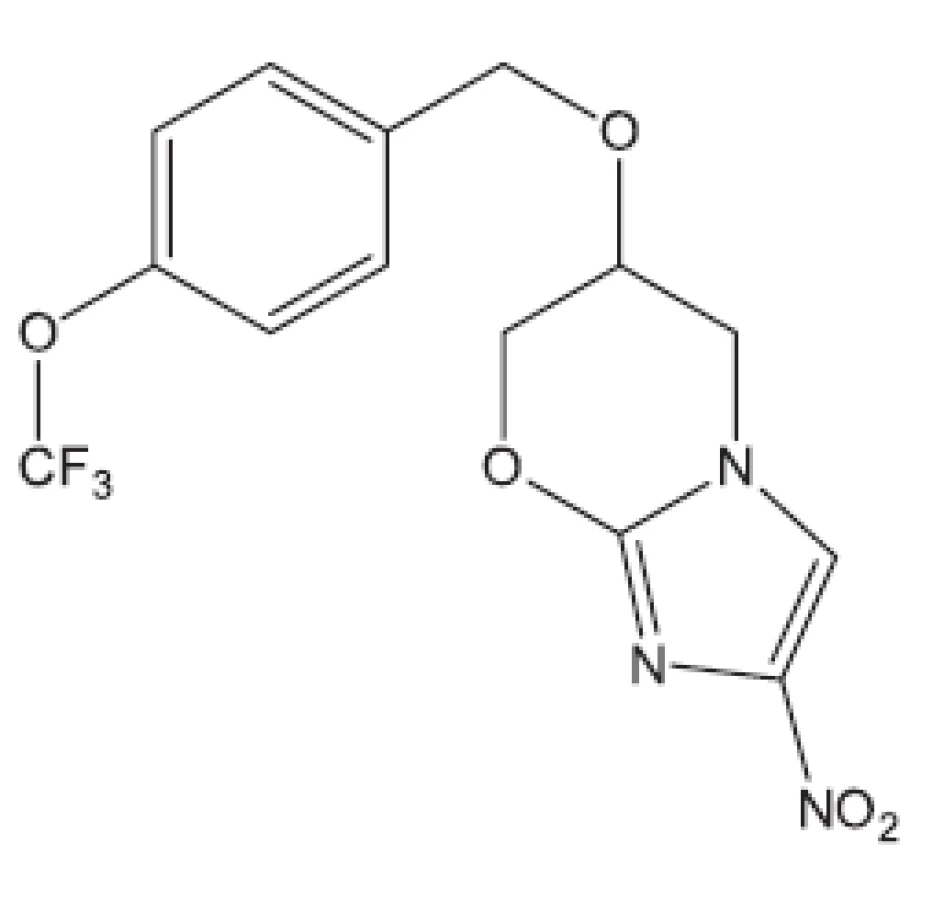
Karel Waisser
Published in:
Čes. slov. Farm., 2012; 61, 150-158
Category:
Review Articles
Došlo 24. května 2012 / Přijato 11. července 2012
Overview
Několik vědeckých skupin v České a Slovenské republice je orientováno na vývoj antituberkulotik. Uveřejněný článek byl připraven převážně z informací z Chemical Abstracts z roku 2011 a počátku roku 2012. Je to výběr z téměř 3000 zpráv pro pomoc našim vědeckým pracovníkům. Článek přináší aktuální informace, které mohou zajímat širokou farmaceutickou veřejnost.
Klíčová slova:
Mycobacterium tuberculosis, antituberkulotika, multirezistentní mykobakterie, QSAR, hodnocení in vivo, biochemie mykobakterií
Úvod
V osmdesátých letech 20. století jsme uveřejnili soubor přehledných sdělení o vývoji antituberkulotik za posledních 15 let. Tehdy ročně vycházelo okolo 20 původních prací. Bylo snadné je sledovat a před sepsáním původních sdělení je utřídit podle struktur antimykobakteriálně účinných látek. Odborné prognózy předpokládaly, že okolo roku 2000 tuberkulóza přestane být v technicky vyspělé části světa hrozbou. V současné době se však tuberkulóza dostává do popředí zájmu farmaceutické chemie. V tabulce 1 je uvedena statistika prací (podle Chem. Abstr.) věnovaných vývoji nových antituberkulotik. Co způsobilo to, že zájem o tento obor patří dnes mezi přední? V roce 2008 bylo celosvětově diagnostikováno 9,4 milionů nových případů onemocnění a 1,9 milionů postižených zemřelo1). V rozvojové části světa byla to nedostatečná lékařská péče. Určitou roli zde hrál i nedostatek finančních prostředků. K příčině potíží lze připočíst i neukázněnost pacientů, opouštějících chemoterapii onemocnění ještě před jeho úplným vyléčením. Tak vznikaly multirezistentní kmeny mykobakterií, vzdorující soudobým léčivům. Pro současnou velkou migraci obyvatelstva se rezistentní kmeny dostávaly do vyspělé části světa. Velký vliv má i choroba HIV. Pacienti se sníženou imunitou velmi často umírají na mykobakteriální onemocnění. Multirezistetní kmeny bakterií, které mají rezistenci vůči nejúčinnějším antituberkulotikům, tj., hydrazidu isonikotinové kyseliny (INH) a rifampicinu, se označují MDR-Tb. Pokud přibývá ještě rezistence vůči fluorovaným chinolonům a jednomu za tří dalších léčiv podávaných injekčně (kanamycinu, kapreomycinu a arrkacinu), kmeny mykobakterií se nazývají extrémně multirezistentní a označují se XDR Tb. Nedávno byl nalezen kmen M. tuberculosis rezistentní vůči všem antituberkulotikům (označení TMR Tb, tj. totálně rezistentní). Počítá se, že na 500 000 případů MDR-Tb přichází 3–5 % XDR-Tb2). Výzkum antituberkulotik proti rezistentním kmenům je hlavním trendem současného vývoje2).
Z uvedeného úvodu lze shrnout, že tímto vážným problémem se dnes zabývají stovky vědeckých pracovišť o pravděpodobně tisících pracovníků. Publikační výstupy lze rozdělit na studie hledající potenciální antituberkulotika účinné in vitro, dále dokonalejší studie zaměřující se na hodnocení in vivo. Za nejperspektivnější oblast lze považovat práce vycházející s poznatků o biochemických pochodech mykobakteriíí. Přehled je věnován výzkumům od roku 2011, protože do roku 2010 byl již vypracován3).
Potenciální antituberkulotika roku 2011
V tomto roce o perspektivních skupinách antimykobakteriálních látek byla sepsána řada přehledných referátů4–20). Některé přehledné články se zabývají biologickou aktivitou určité skupiny látek a o antituberkulózních aktivitách je tam jen málo informací4, 10, 12, 16). Jiné analyzují skupiny látek, které se jako antituberkulotika nedostaly do praxe, avšak vykazují velkou aktivitu12, 13). Nitroskupina je funkční skupinou v mnohých soudobých antituberkulotikách (např. PA-824, OPC-37688) a referáty iniciují syntézy mnohých nových aktivních sloučenin. Řada přehledů je v úvodech prakticky zaměřených článků5, 6). Často úspěšné látky jsou označeny pouze kódem (např. TMC207 je úspěšný derivát chinolinu). Cesta k němu je také popsána v přehledném referátu7). Zajímavý je i přehled věnovaný 1,4-di-N-oxidům chinoxalinu8). Kdysi na Farmaceutické fakultě Univerzity Karlovy byly studovány N-oxidy pyridinu jako potenciální antituberkulotika. Studované látky byly vysoce účinné in vitro, avšak neúčinné in vivo9). Za úspěšné látky jsou považovány fluochinolony19). Otázce léčby tuberkulózy vyvolané rezistentními kmeny ve Velké Británii je věnován přehledový článek20). V mnoha laboratořích v poslední době se hodnotí účinnost i vůči některým multirezistentním mykobakteriálním kmenům.
Potenciální antituberkulotika hodnocená in vitro
Potenciální antituberkulotika hodnocená in vitro tvoří nejpočetnější skupinu studovaných látek. Důvodem je patrně nepříliš obtížná a nákladná pracovní metodika. V České a Slovenské republice je to téměř jediný přístup. Kdysi se provádělo hodnocení in vivo ve výzkumném ústavu Preventivného lekárstva v Bratislavě. Patrně z finančních důvodů byly laboratoře vedené dr. Odlerovou zrušeny. Náklady u studií in vivo jsou daleko vyšší než u studií prováděných in vitro. Protože studie opírající se o hodnocení in vitro jsou příliš početné (v roce 2011 se jedná přibližně o 2000 sdělení), rozsah již přesahuje možnosti našeho přehledného referátu. Zpravidla se touto cestou zprvu ubírá celý výzkum antituberkulózní aktivity látek. Lépe je však aktivitu označit za antimykobakteriální. Pracovníci naší fakulty zpravidla spolupracují s Regionálním zdravotním ústavem v Ostravě. Standardní stanovení se provádí na Mycobacterium tuberculosis, Mycobacterium kansasii (INH rezistentním), Mycobacterium avium (INH rezistentním) a Mycobacterium kansasii klinicky izolovaném, který nevykazuje rezistenci vůči INH. Na vyžádání ústav provádí i hodnocení vůči několika multirezistentním kmenům.
Potenciální antituberkulotika hodnocená in vivo
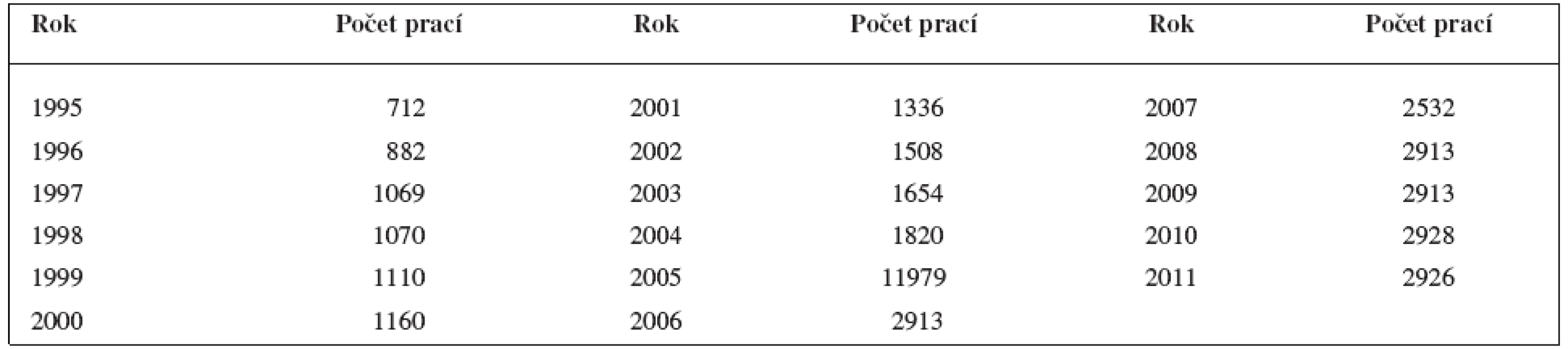
Hodnocení in vitro je náročnější, avšak přináší více informací. Předně pokusná zvířata musí být geneticky podobná, aby v pokusné skupině nebyly mezi zvířaty velké rozdíly. Nejčastěji se pracuje s experimentální tuberkulózou myší nebo morčat. Řada prací vykazuje nesmírně rozsáhlou chemickou část a velmi skromnou biologickou část. Ve skupině pokusných zvířat musí být známa akutní toxicita. Ukážeme si některé příklady. Mezi uvedené práce patří již citovaná studie o protituberkulózních derivátech skořicové kyseliny12). Ve skupině indofenazin-1,2.5-trisubstituovaných pyrazolinových derivátů21) podobných benzofuranu byla analýza prováděna na myších. Na obrázku 1 je uvedena úspěšná struktura skupiny látek, přičemž nejaktivnější deriváty měly R nitroskupinu v poloze o- nebo m-.
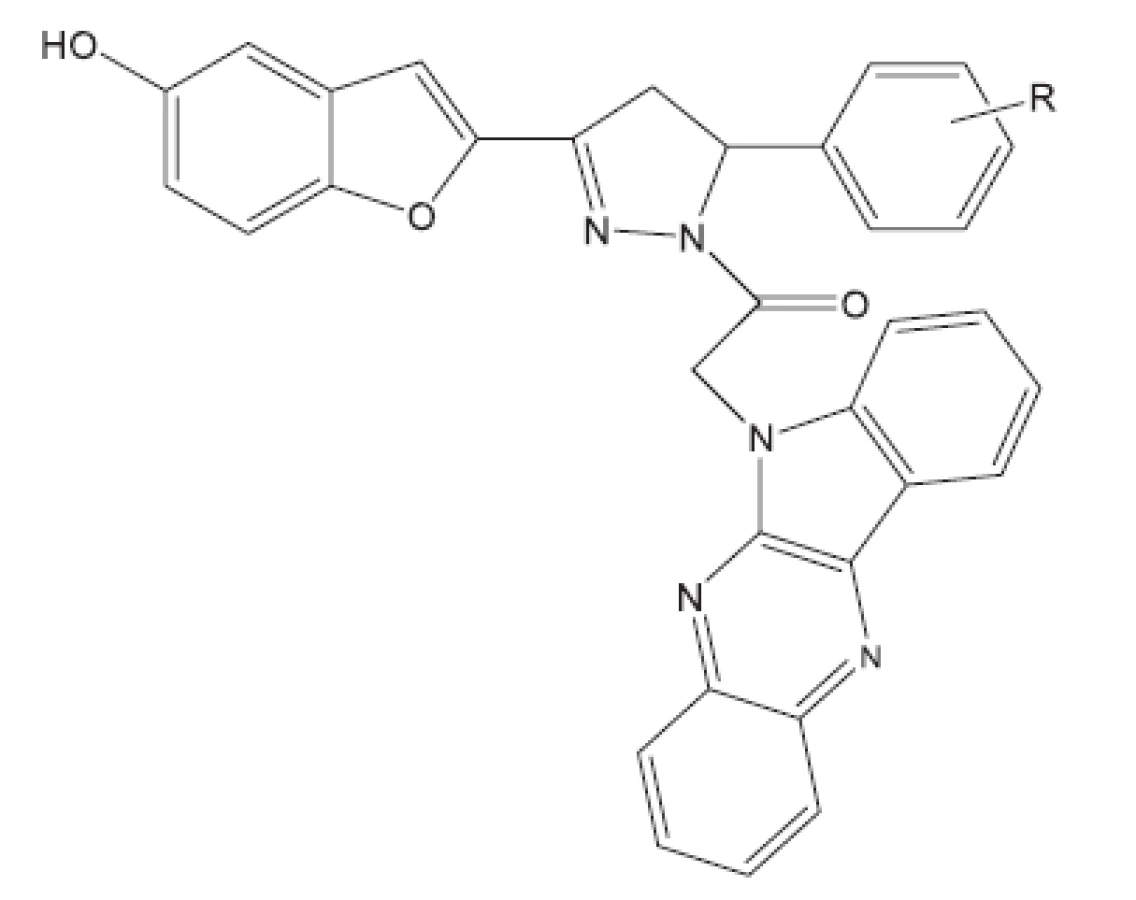
Aryloxy azolyl chalkony22) (obr. 2) byly studovány v roce 2011. Je zajímavé, že mnoho chalkonů bylo před časem studováno na Farmaceutické fakultě Univerzity Komenského v Bratislavě a na Farmaceutické fakultě Univerzity Karlovy v Hradci Králové23–26). Převážně se jednalo o azachalkony, kde jako aren byl převážné pyrazin. Hodnoceny byly pouze in vitro. Pokud nejsou látky hodnoceny in vivo, obtížně se dále prosazují do praxe. Na nových sloučeninách je vidět to, co je pro dnešní vývoj charakteristické. Výzkum se ubírá ke složitějším látkám a hodnocení se uplatňuje také in vivo. Z první skupiny nejaktivnější látka měla R1 2-Cl a R2 4-Cl. Ve druhé skupině nejaktivnější látka obsahovala v těchto polohách fluor (obr. 3).
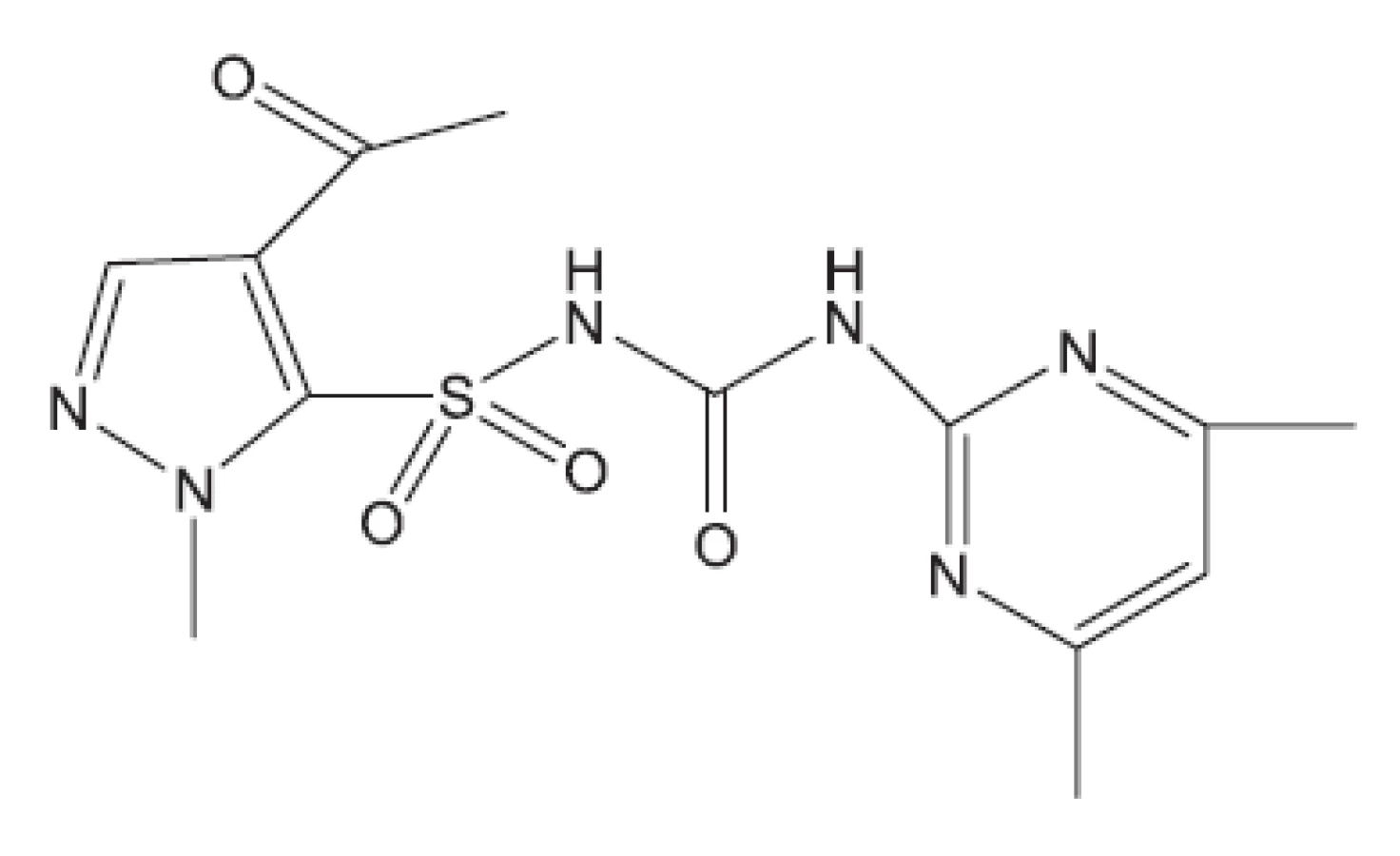
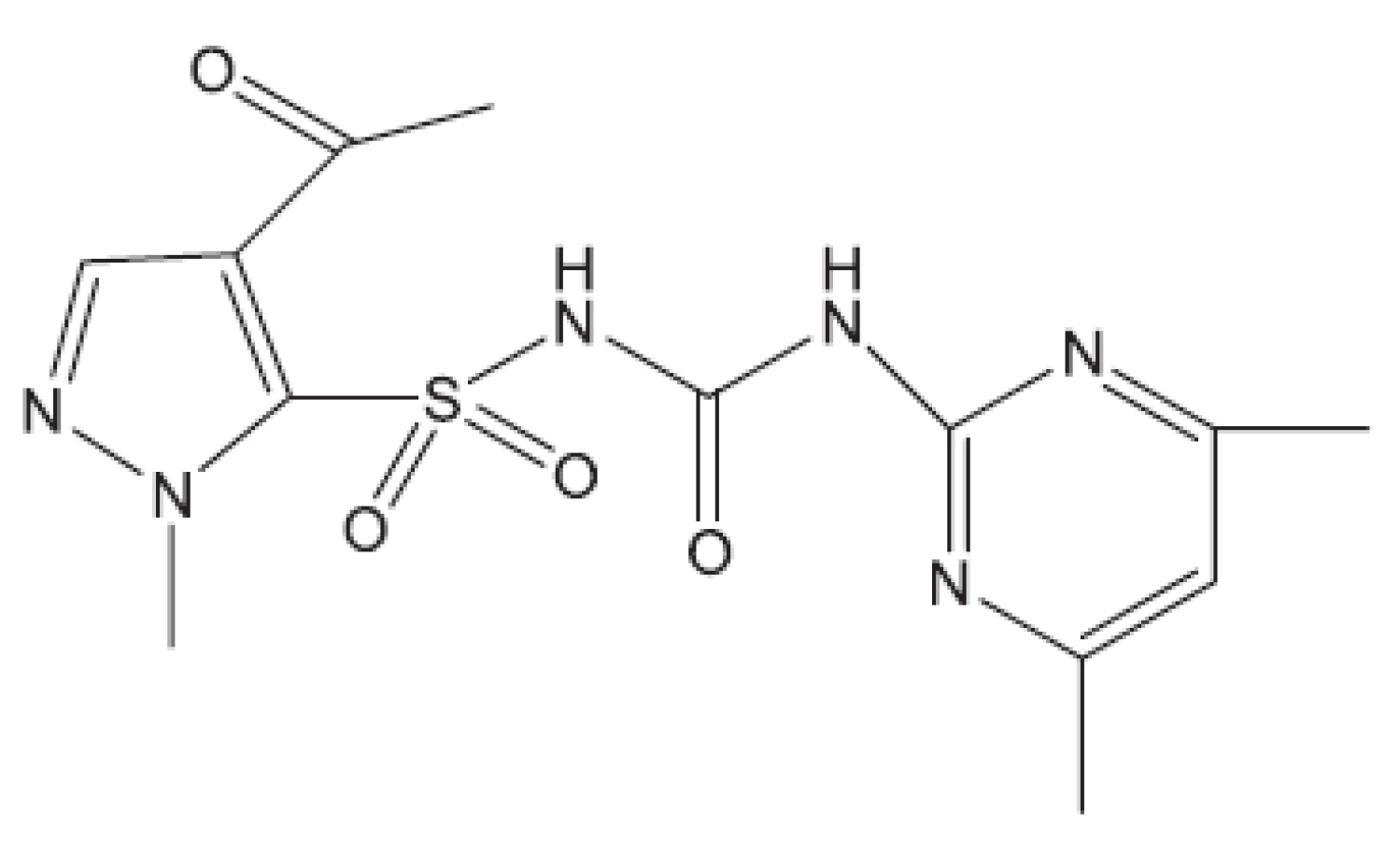
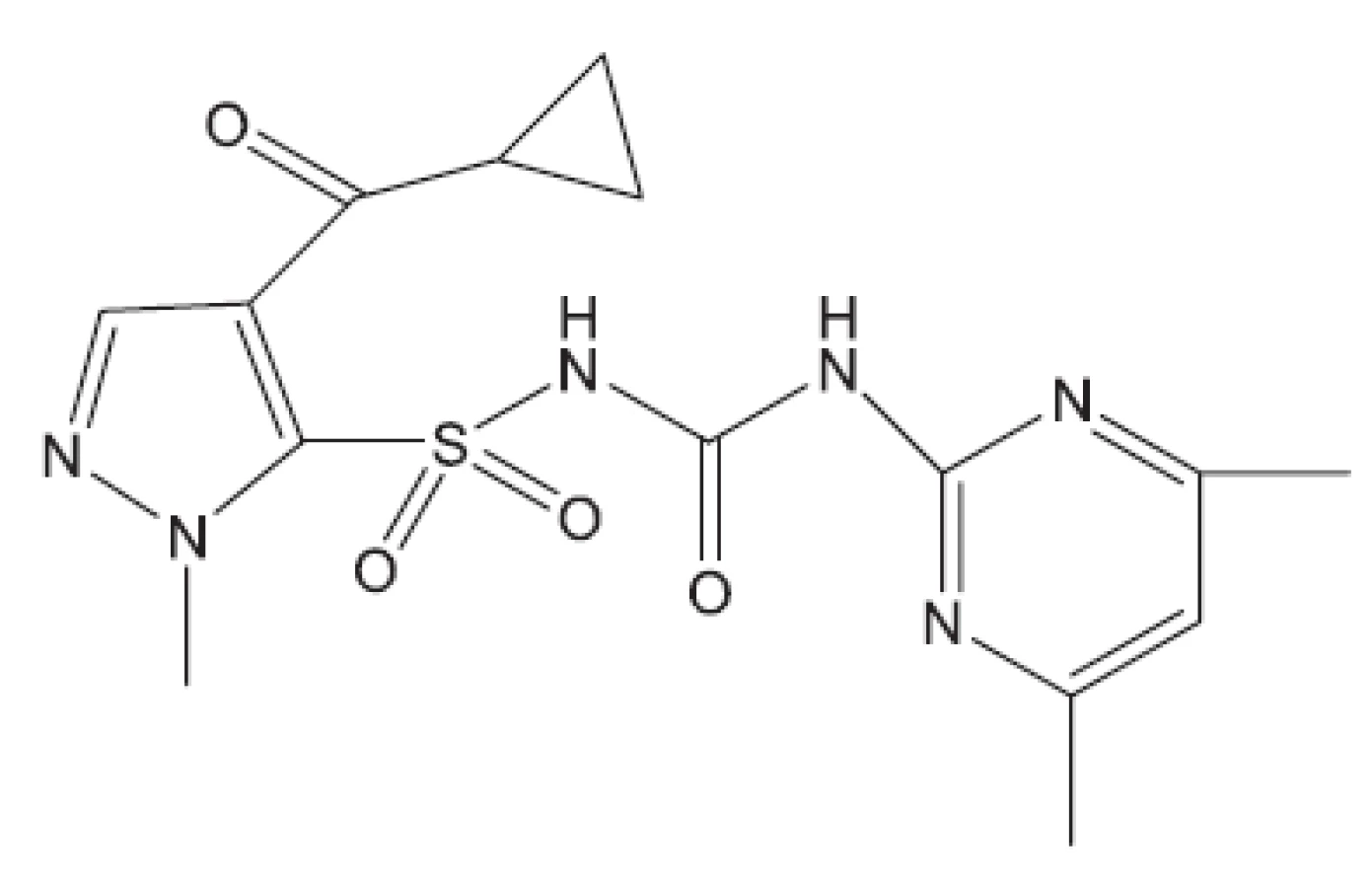
V některých případech jako pokusné zvíře bylo používáno morče28). V citované práci byl in vivo hodnocen rifampin, pyrazinamid a TMC207. S rifampinem a pyrazinamidem se setkáváme v mnoha učebnicích. Zbývá nám jen ukázat strukturu TMC207 (obr. 4). Je to látka vyvinutá proti multirezistentním kmenům. Mechanismus účinku spočívá v inhibici syntézy ATP7).
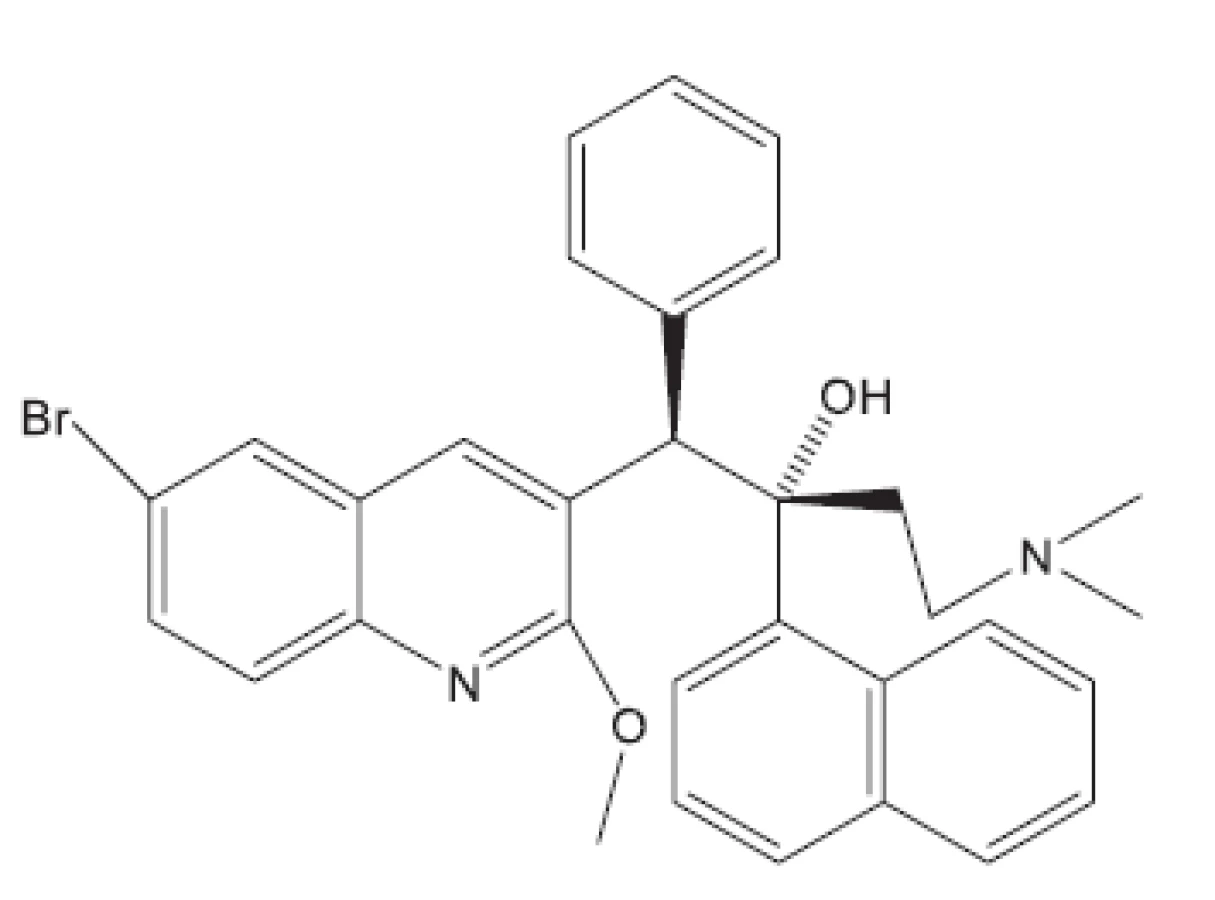
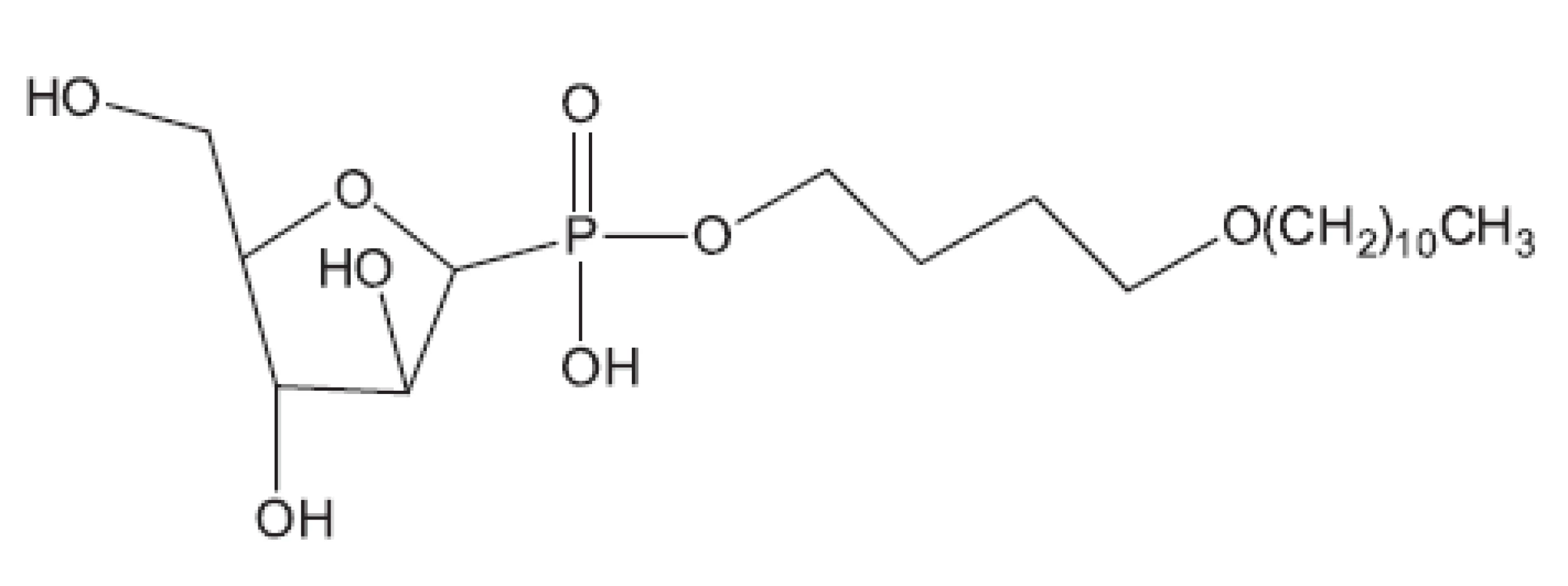
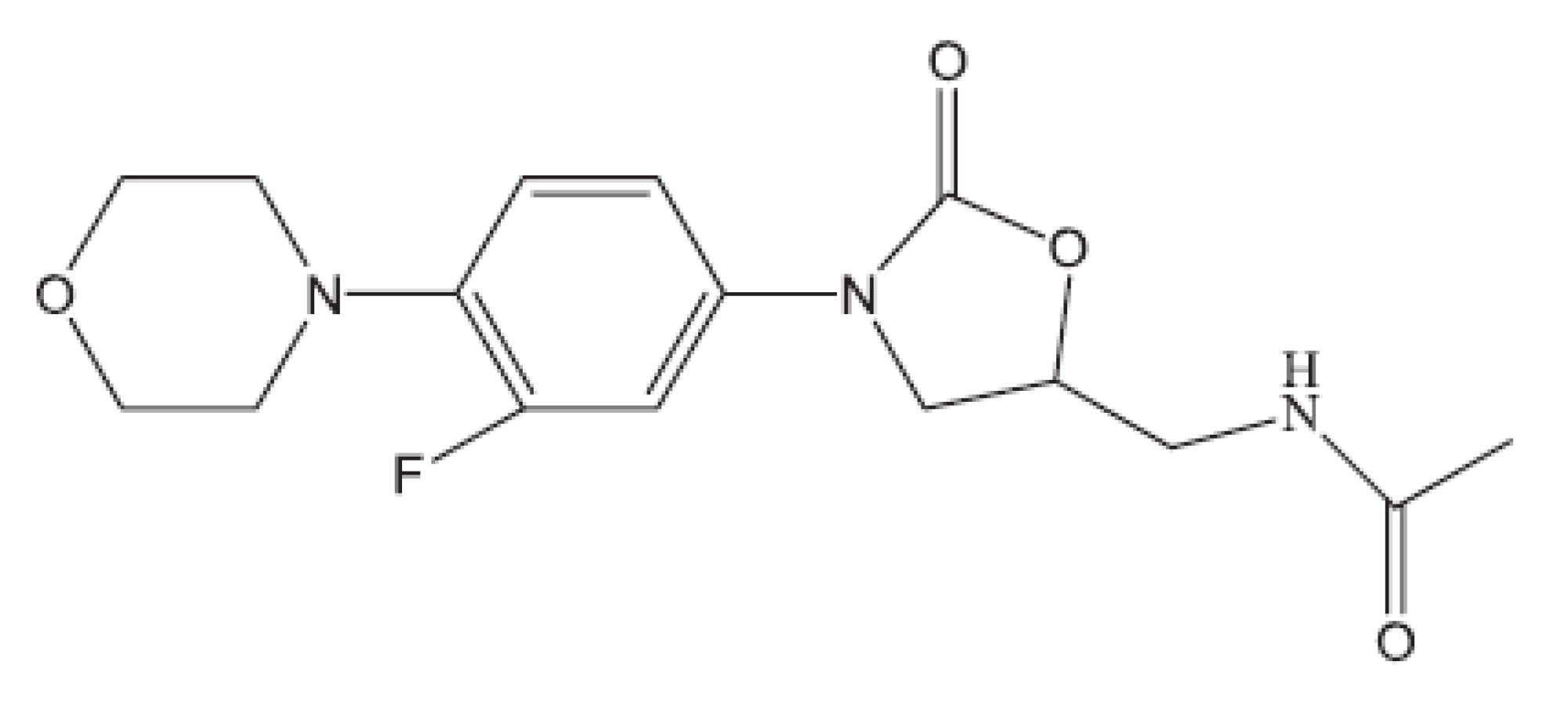
V mnohých studiích in vivo není uveden druh zvířat, pouze je uvedeno pokusná zvířata („animals“). Možná, že je použito více druhů zvířat tak, jak pracuje současný farmakologický výzkum. Další studie hodnocené in vivo uvádíme bez nákresů struktur, protože jejich struktury vyplývají z názvu článku29–36). Pozastavíme se u práce vedoucí k celé skupině in vivo aktivních sloučenin37); jako předloha byla použita látka označovaná SQ609 (obr. 5), která je již používána v praxi proti multirezistentním kmenům.
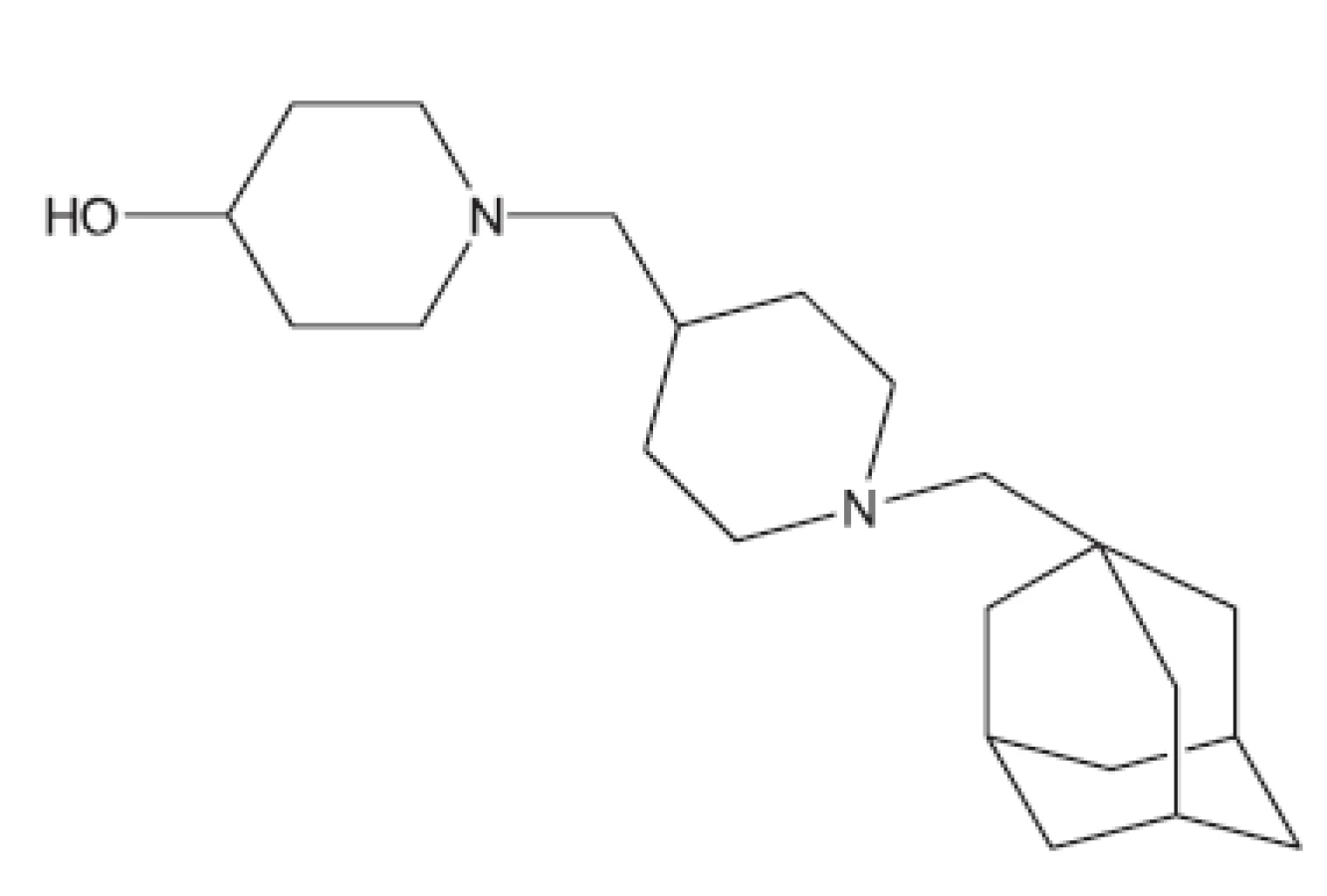
Zajímavými sloučeninami účinnými in vivo jsou kalixareny. Jsou to makrocyklické sloučeniny obsahující v makrocyklu čtyři až sedm cyklických substrukturních jednotek38).
Hodnocení in vivo je některých případech spojeno s klinickým výzkumem34).
Význam metody QSAR
Zkratka QSAR byla vytvořena z anglického názvu „quantitative structure-activity relationships“. Zdomácněla snad ve všech jazycích. Významné začátky přístupu lze zařadit do šedesátých let 20. století, i když její kořeny sahají do ještě dřívějšího období38). V této době cílem QSAR studií bylo omezit počty syntetizovaných látek, protože tehdy chemické syntézy léčiv tvořily největší část nákladů. Od konterganové aféry se však bilance nákladů změnila a největší jejich část zaujímá nyní farmakologie. Význam metod QSAR však nepoklesl. Umožňují chápat logiku závislostí biologické aktivity na chemické struktuře a často pracovníky přivedou k aktivnějším sloučeninám. Často QSAR studie pomáhají ve studované skupině vyhledat nejúčinnější látky. Například na našem pracovišti nás QSAR studie41) přivedly do oblasti vysoce účinných antimykobakteriálních látek42). QSAR studii jsme uveřejnili teprve za rok po informaci o vysoce účinných látkách. Studie není dosud ukončena, protože sloučeniny jsou aktivní i proti multirezistentním kmenům. Poslední poznatek jsme dosud nepublikovali a látky jsou dále studovány na katedře biochemických věd naší fakulty. QSAR přístupy byly dále zdokonalovány. Tak vznikaly více-dimenzionální přístupy (dvou-dimenzionální 2D, troj-dimenzionální 3D). Používají větší počet deskriptorů. Jako příklad uvádíme studii ve skupině 1-cyklopropyl-6-fluor-8-methoxy-3-karboxy-4-chinolonů. Byl sledován vliv obměn substituentů v poloze 5 a 7 pomocí dvou-dimenzionálního přístupu na aktivu vůči M. tuberculosis H37Rv43). Při QSAR studii byly používány strukturní, topologické, elektrotopologické a termodynamické deskriptory. Fluorované chinolony jsou úspěšně používány při tuberkulóze vyvolané MDR-Tb bakteriemi. Cílem další studie bylo hledání obecných vztahů mezi strukturou a aktivitou. QSAR studie se zaměřila na 20 prací používajících hodnocení vztahů mezi strukturou a antimykobakteriální aktivitou metodou QSAR (z toho tři z našeho pracoviště). Autoři vybrali 3224 deskriptorů, které postupně omezovali, až bylo dosaženo dobré obecné predikce44). Dále se omezíme jen na citace dvou-dimenzionálního QSAR45-48) a troj-dimenzionálního QSAR48–51) ve studiích protituberkulózní aktivity. Deskriptory by měly být na sobě nezávislé, jinak je prognóza obtížná. Pokus používat jako deskriptor hodnoty z 13C NMR jsou v přehledném referátu Verna a Hansche52). Metody QSAR jsou přínosem k výzkumu, avšak nejsou jediným racionálním přístupem. Dosti rozšířené jsou práce používající SAR (structure-activity-relationships). Jako příklad kombinace více přístupů uvádíme práci kolektivu kolem prof. L. Denga53).
Poznatky biochemie mykobakterií
Mykobakterie se biochemickými pochody a svoji strukturou odlišují od běžných bakterií. Právě proto tuberkulóza vyvolávaná Mycobacterium tuberculosis (a ještě několika dalšími mykobakteriemi, jako M. bovis, M. africanum, M. cansasii) byla v historii lidstva nesmírně nebezpečnou chorobou. Většina antibakteriálních chemoterapeutik nepůsobila na původce tuberkulózy. Mykobakterie chrání silná buněčná stěna, která je vytvořena ze tří vrstev. První vrstvu tvoří polymer vytvořený z mykolových kyselin. Mykolové kyseliny obsahují pouze mykobakterie a korynebakterie (obr. 6). Jejich obecný vzorec odpovídá 2-alkyl-3alkyl-3-hydroxypropanové kyselině. Alkyly jsou zastoupeny uhlovodíkovým, a to i rozvětvenými řetězci. Mykolové kyseliny mykobakterií obsahují 75–90 atomů uhlíku, u korynebakterií 32–36 atomů uhlíku.
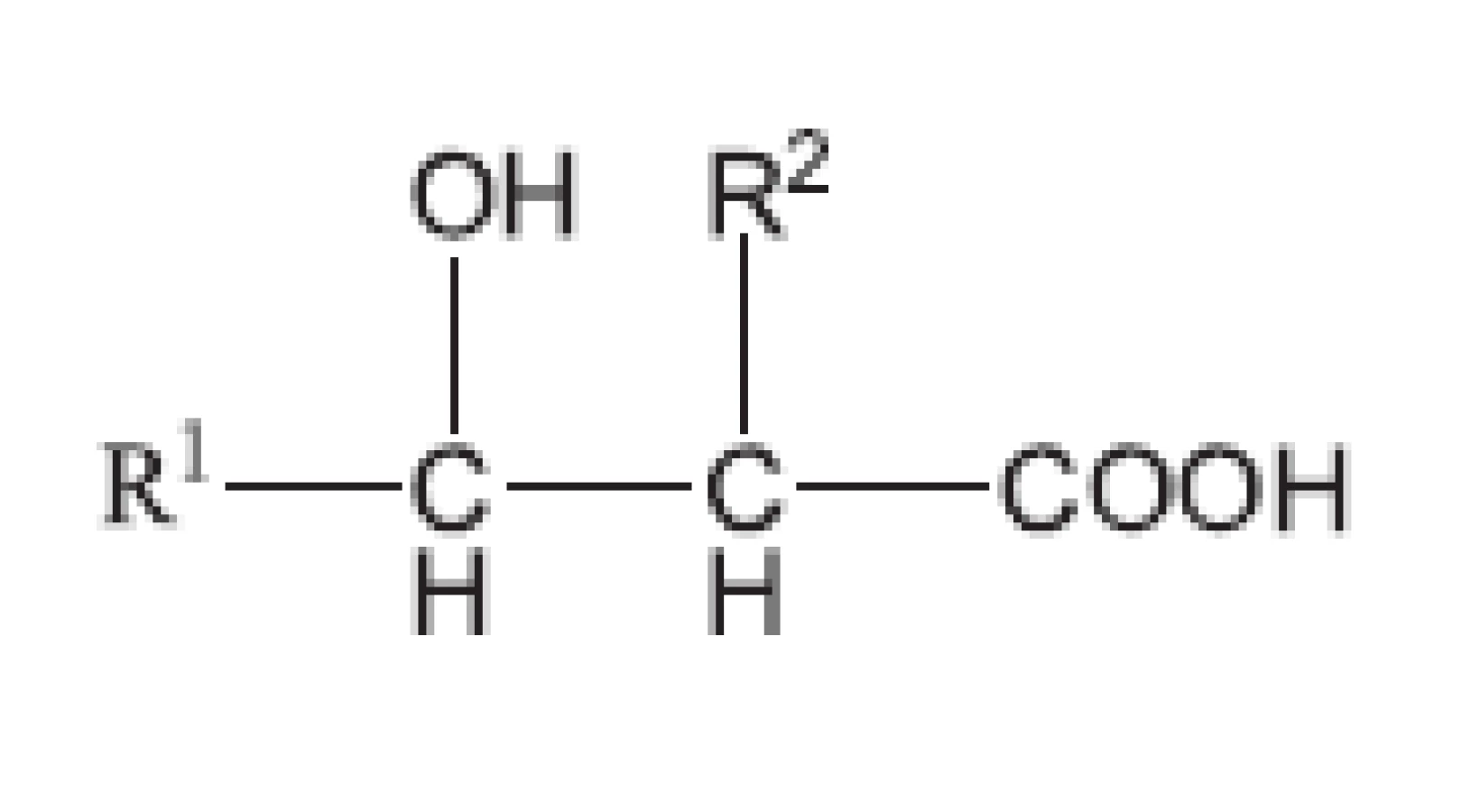
Karboxylové skupiny a hydroxyly umožňují vytvářet velmi pevná esterová spojení. Druhou vrstvu vytváří arabinogalaktan. Opět je to vysokomolekulární látka, ve které jsou monosacharidové jednotky spojeny glykosidickou vazbou. Třetí vrstvu tvoří peptidoglykan. Má však poněkud odlišné složení od peptidoglykanu běžných bakterií. Kyselina N-acetylmuramová je nahrazena N-glykolylmuramovou. V peptidových řetězcích však, podobně jako peptidoglykan běžných bakterií, obsahuje také D-aminokyseliny. Všechny tři vrstvy jsou spojeny chemickými vazbami, např. vrstva mykolových kyselin je esterovou vazbou připoutána k arabinogalaktanu. Proto jsou všechny tři vrstvy společně označovány jako mykolyl-arabinogalaktanyl-peptidoglykanový komplex (mAGP). K tomuto komplexu je dále nekovalentně připojen lipoarabinomanan. Je tedy zřejmé, že antituberkulotika, útočící na takto složitý útvar, musí mít odlišnou strukturu od běžných antibakteriálních chemoterapeutik (obr. 7).
Klasická antituberkulotika této skupiny byla objevena náhodou, bez znalosti biochemie mykobakterií. Příklady uvádíme: Peptidoglykan ohrožuje cykloserin, který inhibicí alanin racemasy brání tvorbě D-alaninu. Syntézu arabinogalaktanu ohrožuje ethambutol. Do syntézy mykolových kyselin zasahuje INH (hydrazid isonikotinové kyseliny) a thioamidy substituované isonikotinové kyseliny (ehtioamid, prothioamid), thiosemikarbazon 4 acetaminobenzaldehydu (thiacetazon), 4,4’-bis-(isopentyloxy)thiokartbonalid (isoxyl). Tyto vzorce opět neuvádíme, protože jsou v běžných učebnicích. Proti multirezistentním kmenům bylo nutné vytvořit nové struktury. Ukážeme54) si některá léčiva vyvinutá před rokem 2011 (obr. 8 až 15).
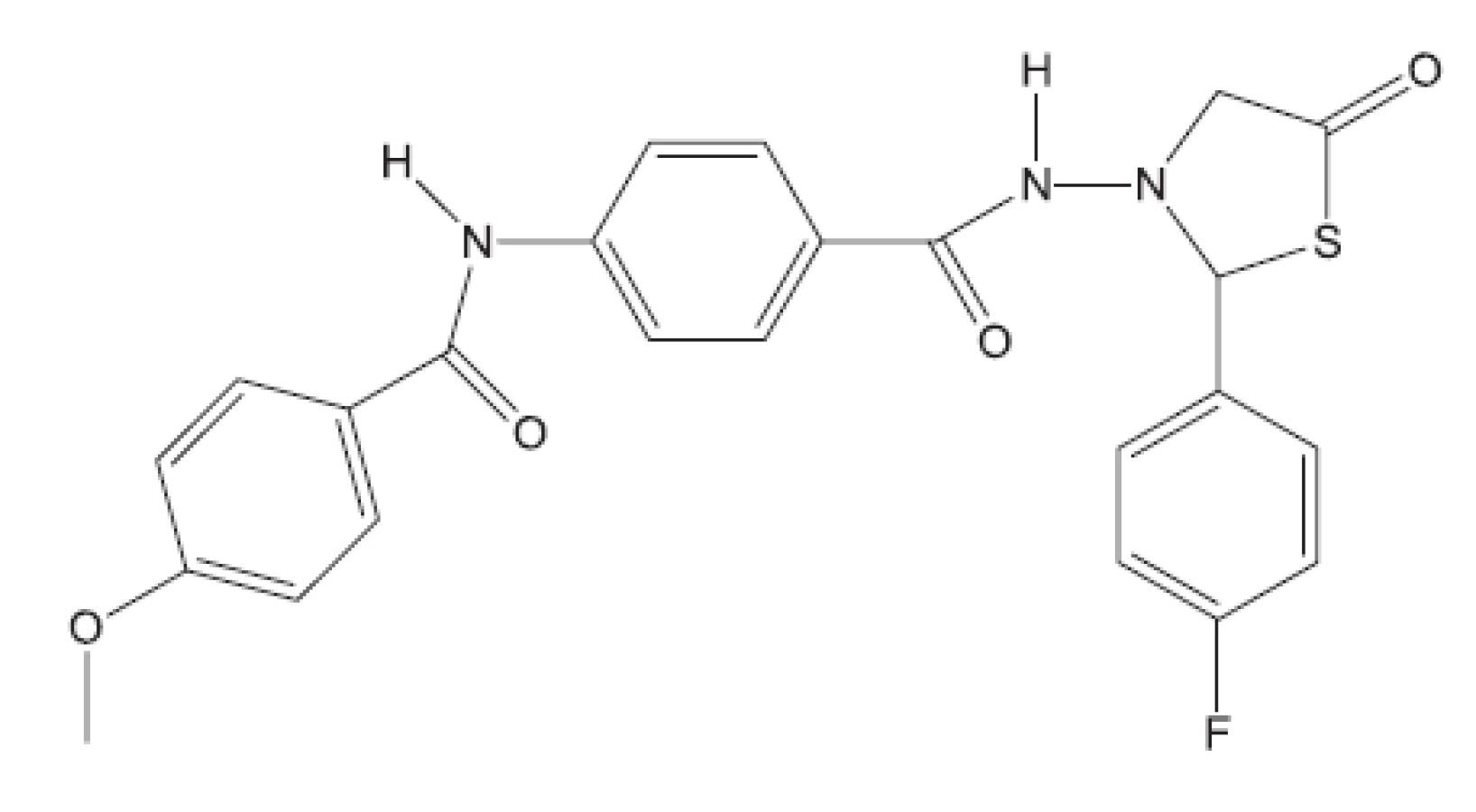
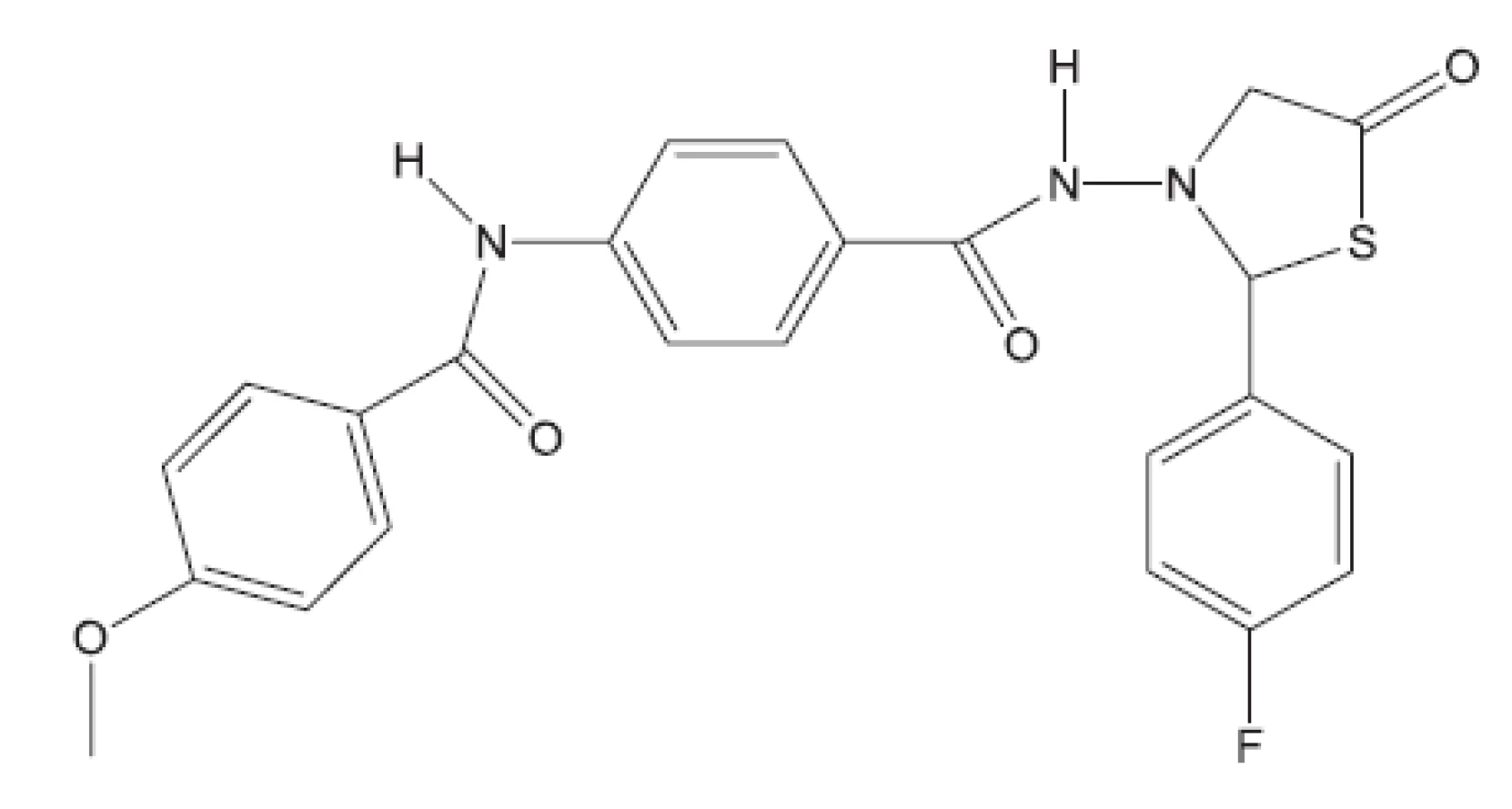
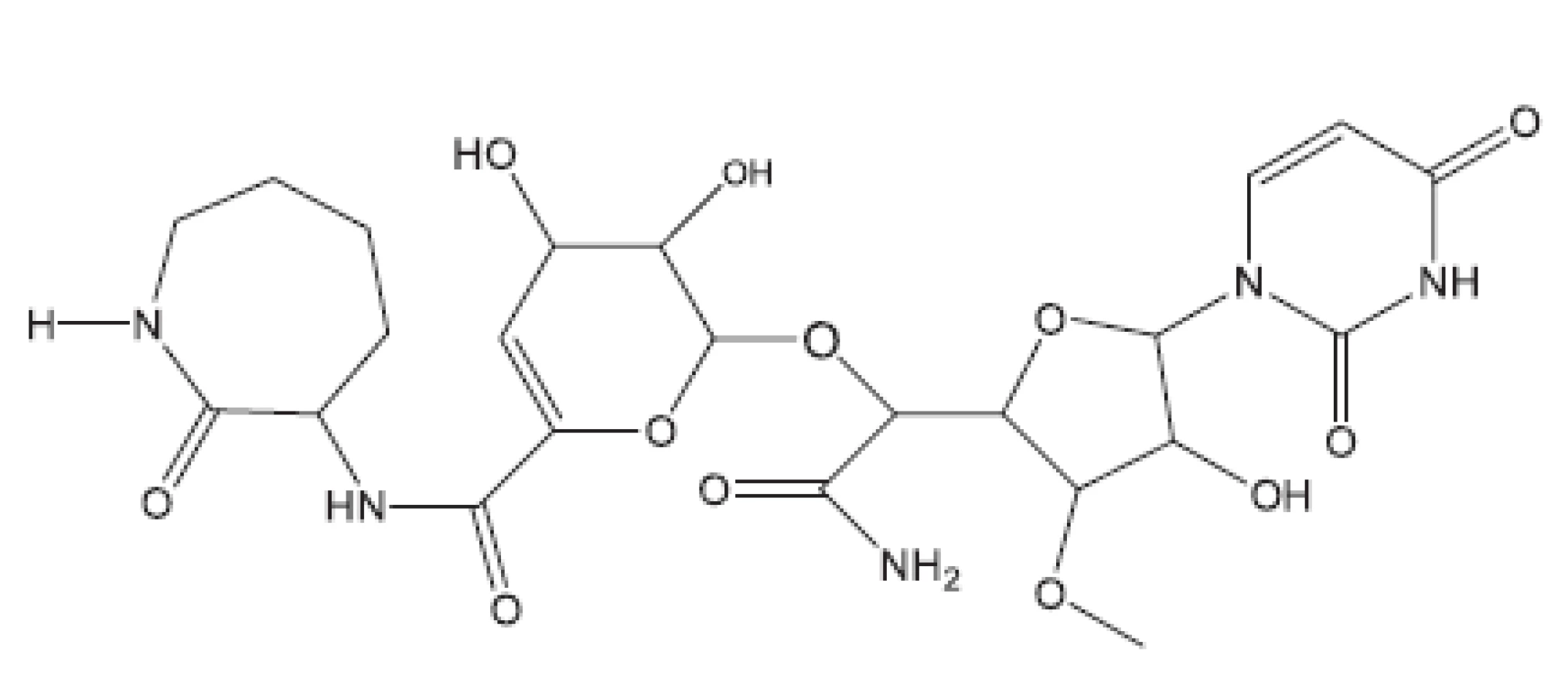
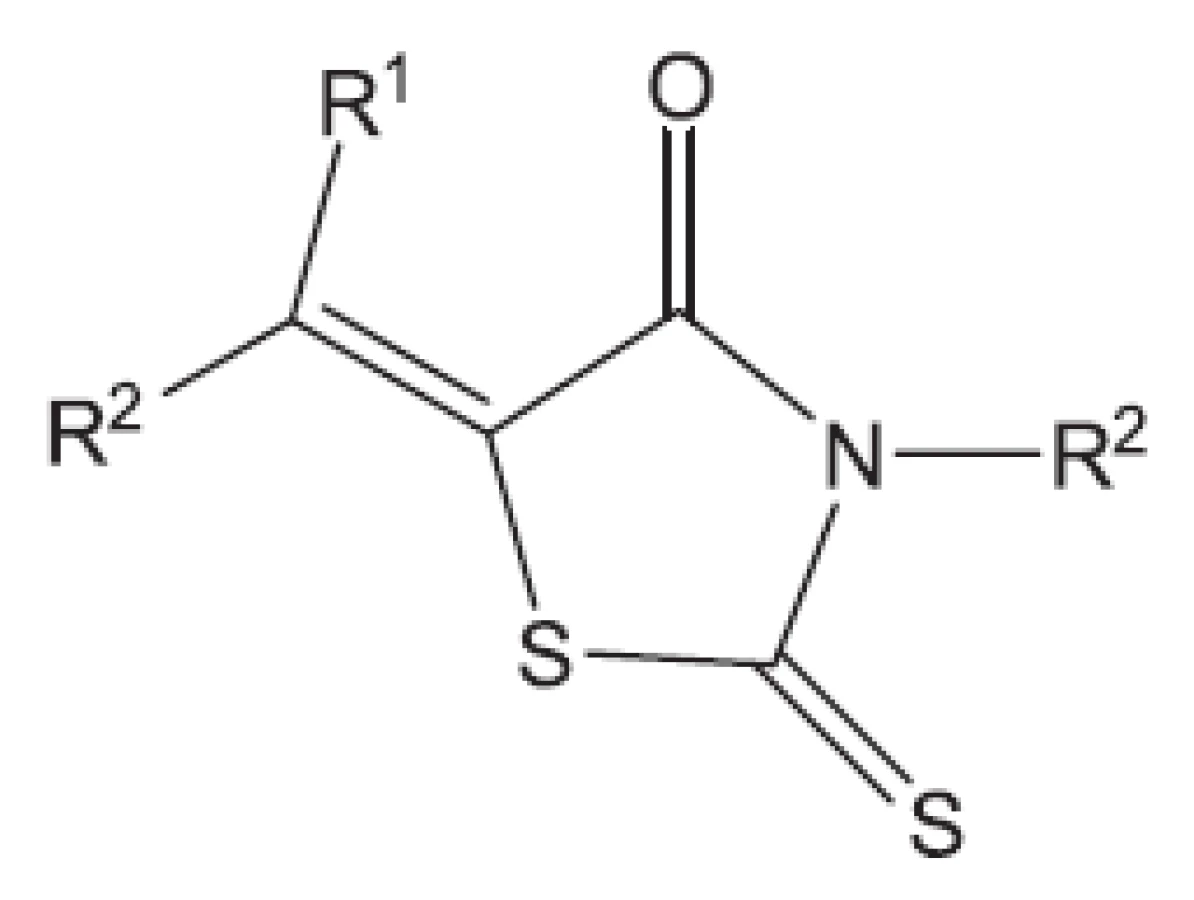
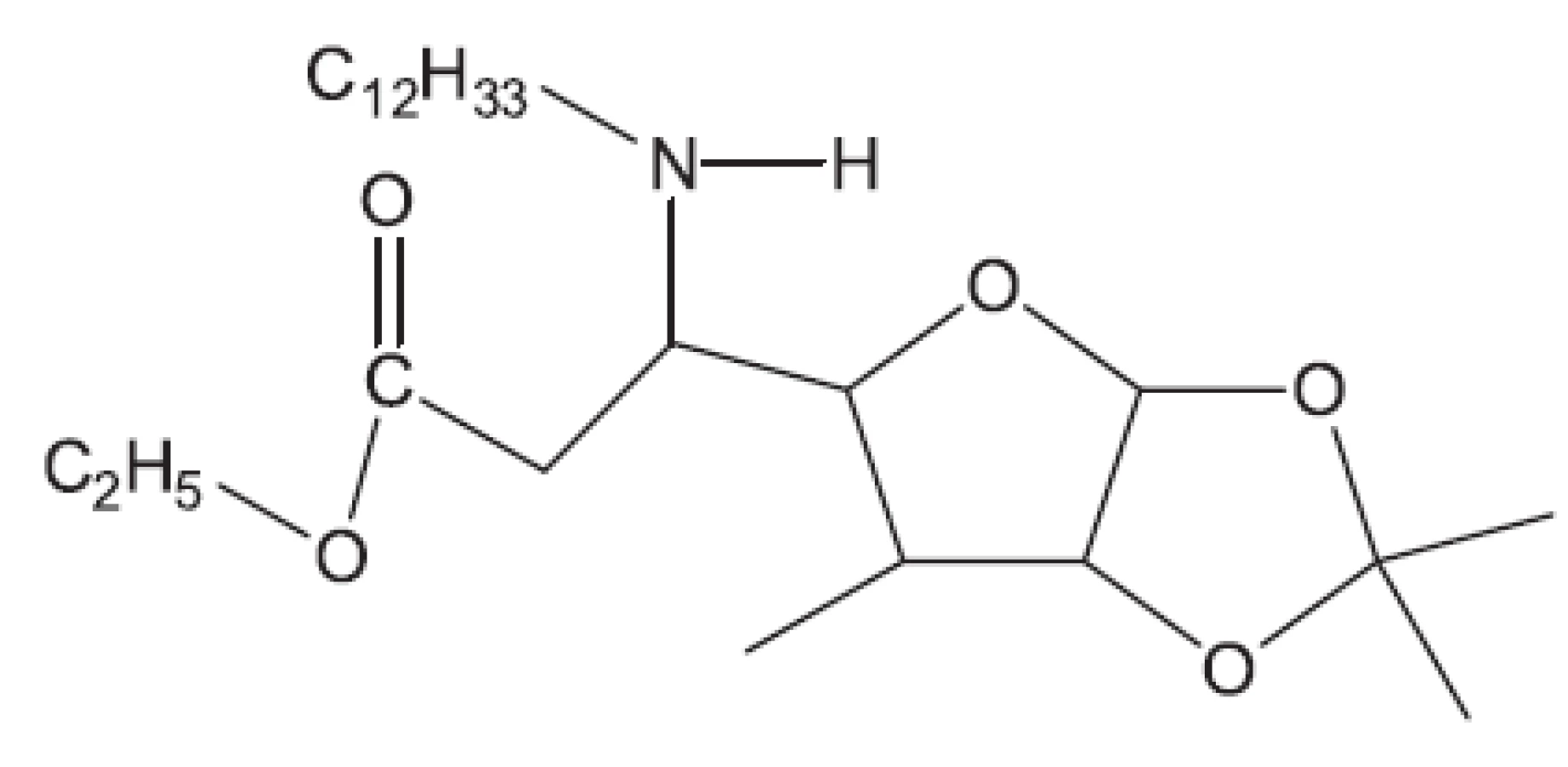
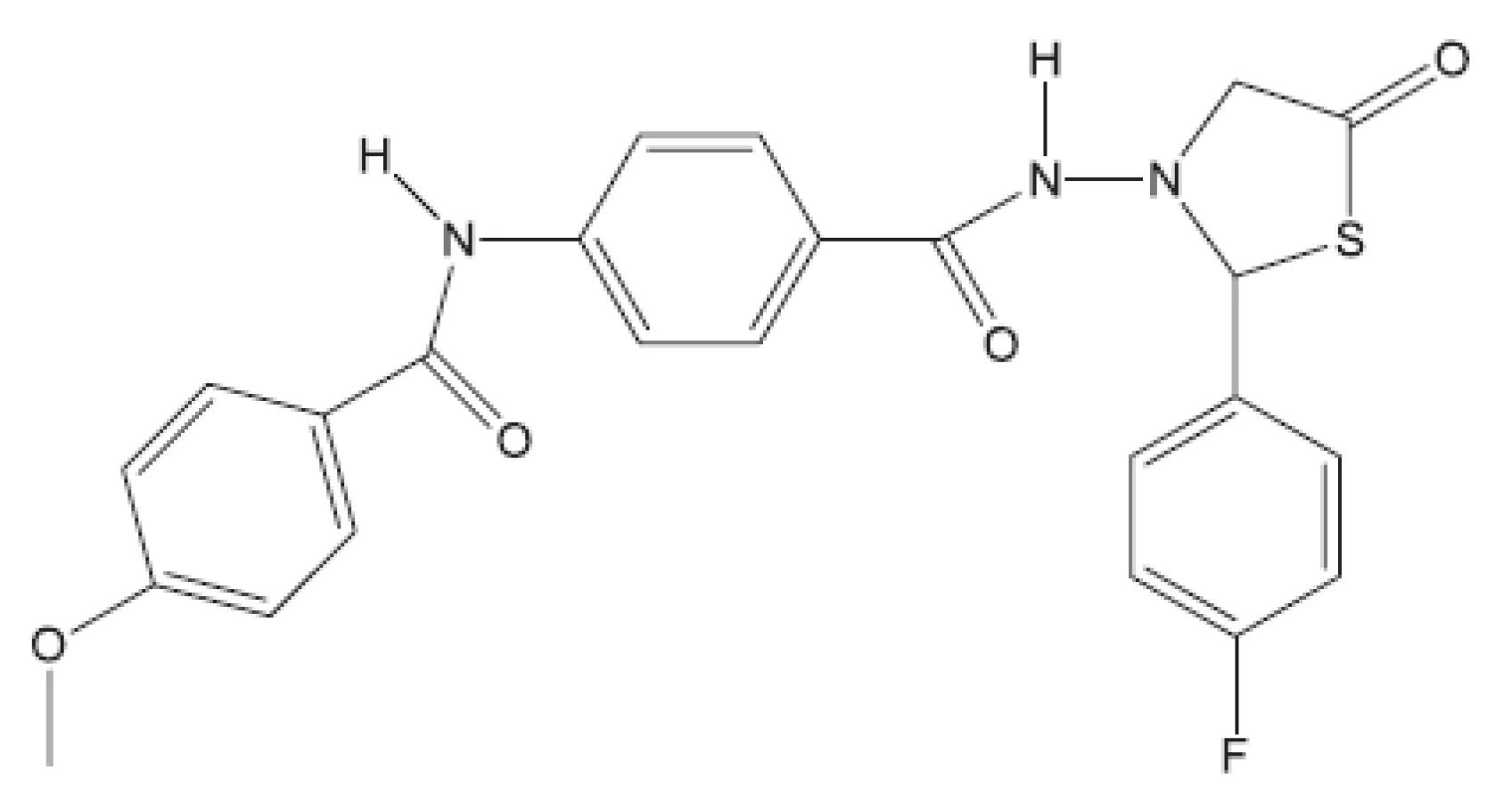
Z uvedených příkladů vyplývá, že útoky na buněčné stěny mohou přinést slibné výsledky. Hodně úspěšných klasických antituberkulotik bylo cíleno také tímto směrem. Řada dalších antituberkulotik působí inhibici biosyntézy některých bílkovin, inhibici biosyntézy některých aminokyselin, zasahuje do respiračního řetězce a inhibici biosyntézy, syntézy případně štěpení DNA, nebo RNA. Opět se vraťme před rok 2011.
Inhibitory biosyntézy rozvětvených aminokyselin se svým účinkem podobají některým herbicidům. Z látek vyvinutých proti tuberkulóze si uvedeme K12147 (obr. 16), K13010 (obr. 17) a K13030 (obr. 18).
Proteosyntézu inhibují streptomycin, kanamycin, kapreomycin, viomycin klaritromycin, linezolid (obr. 19), RU 66252 (obr. 20), Rbx7644 (obr. 21), Rbx8700 (obr. 22), DA2867 (obr. 23) a pleuromutilin (obr. 24). Mechanismus účinku inhibitorů proteosyntézy spočívá ve vazbách na ribozomální podjednotky.
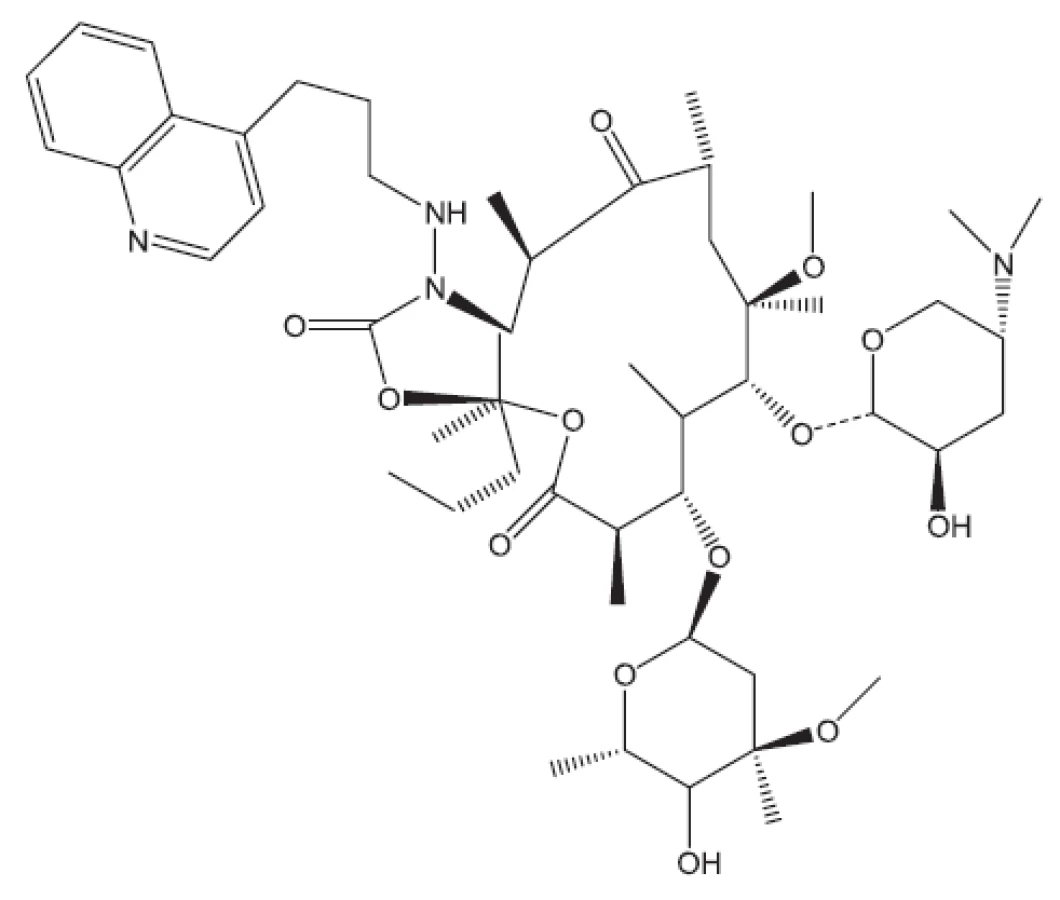
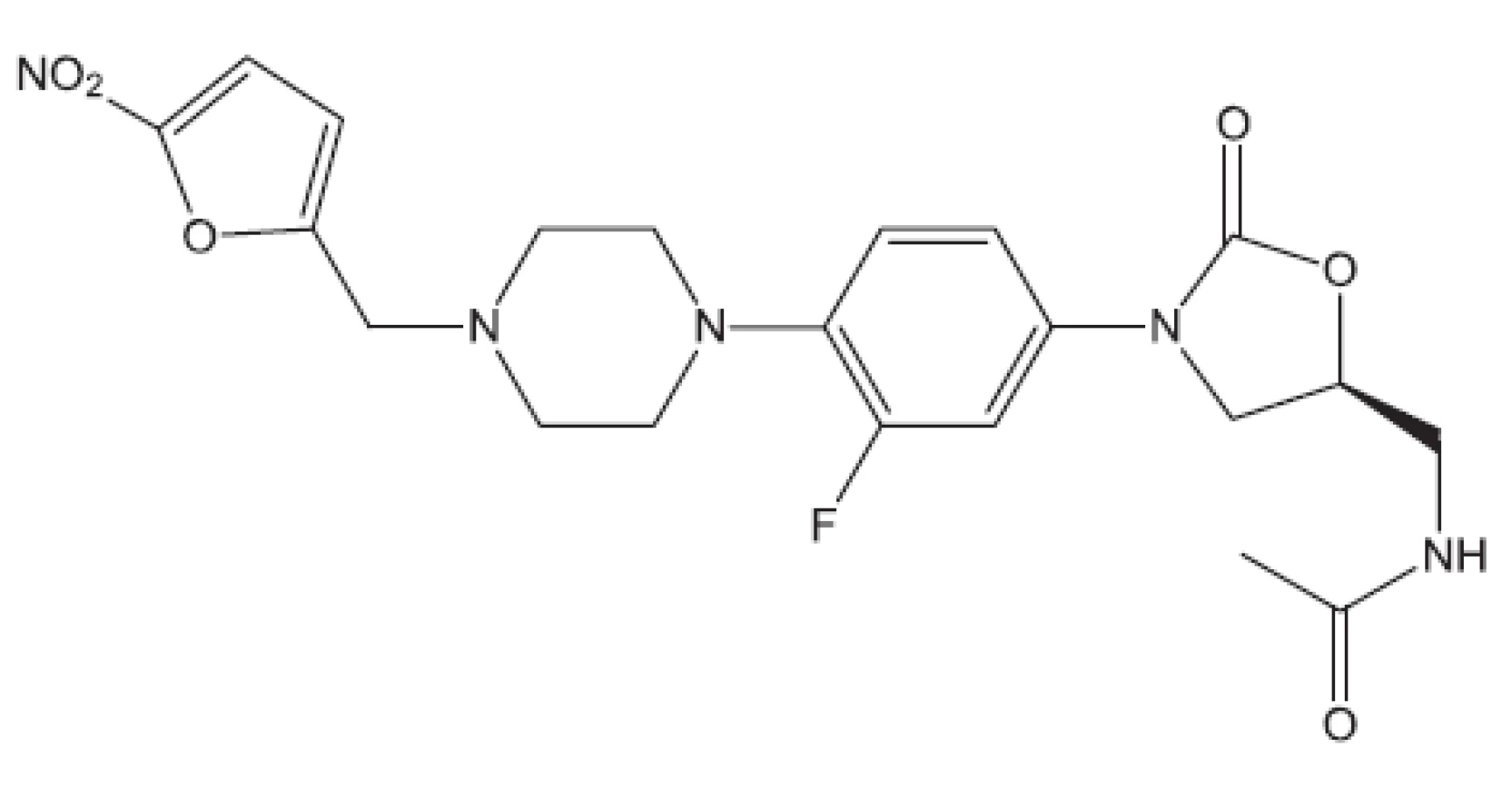
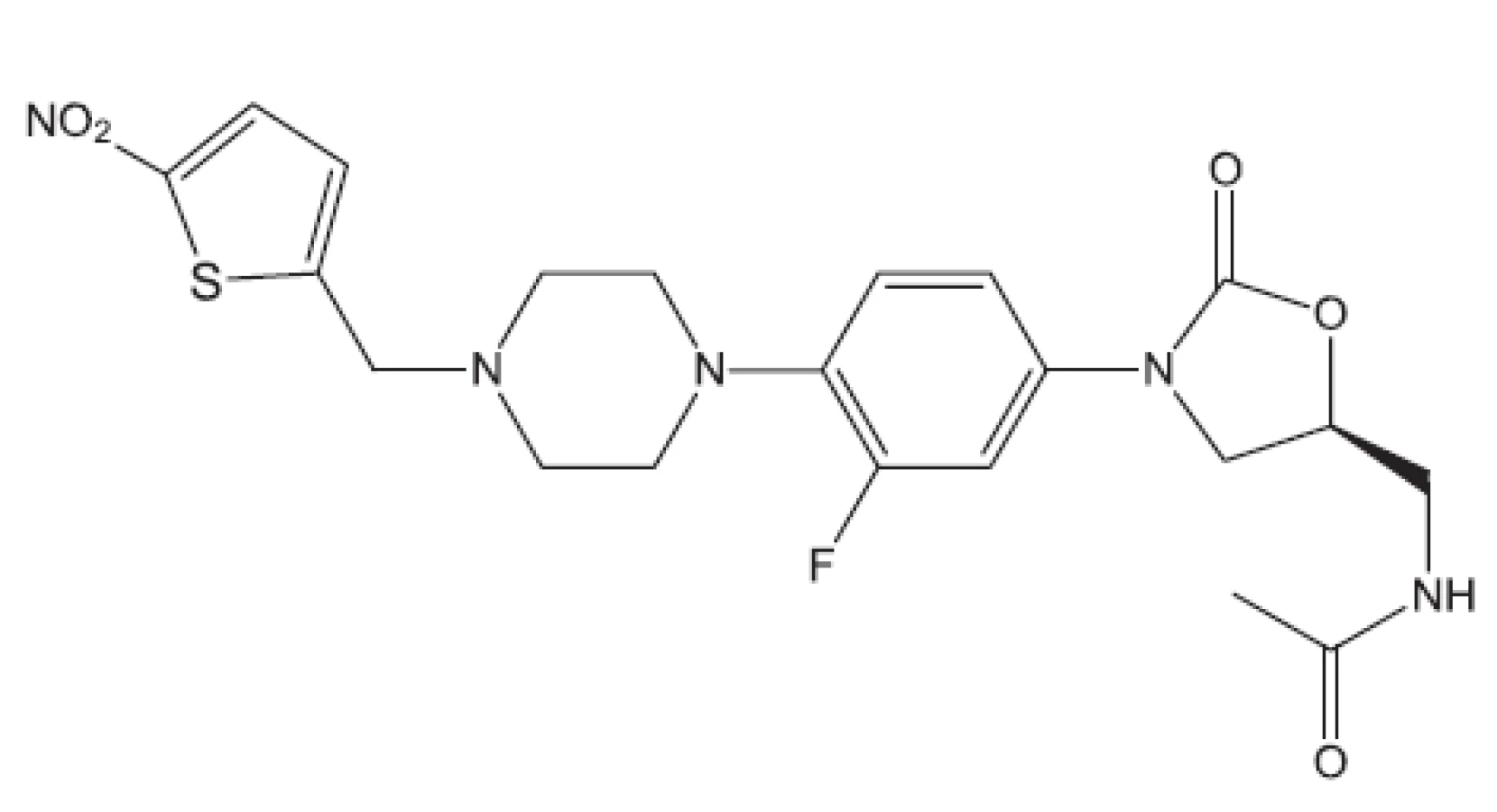
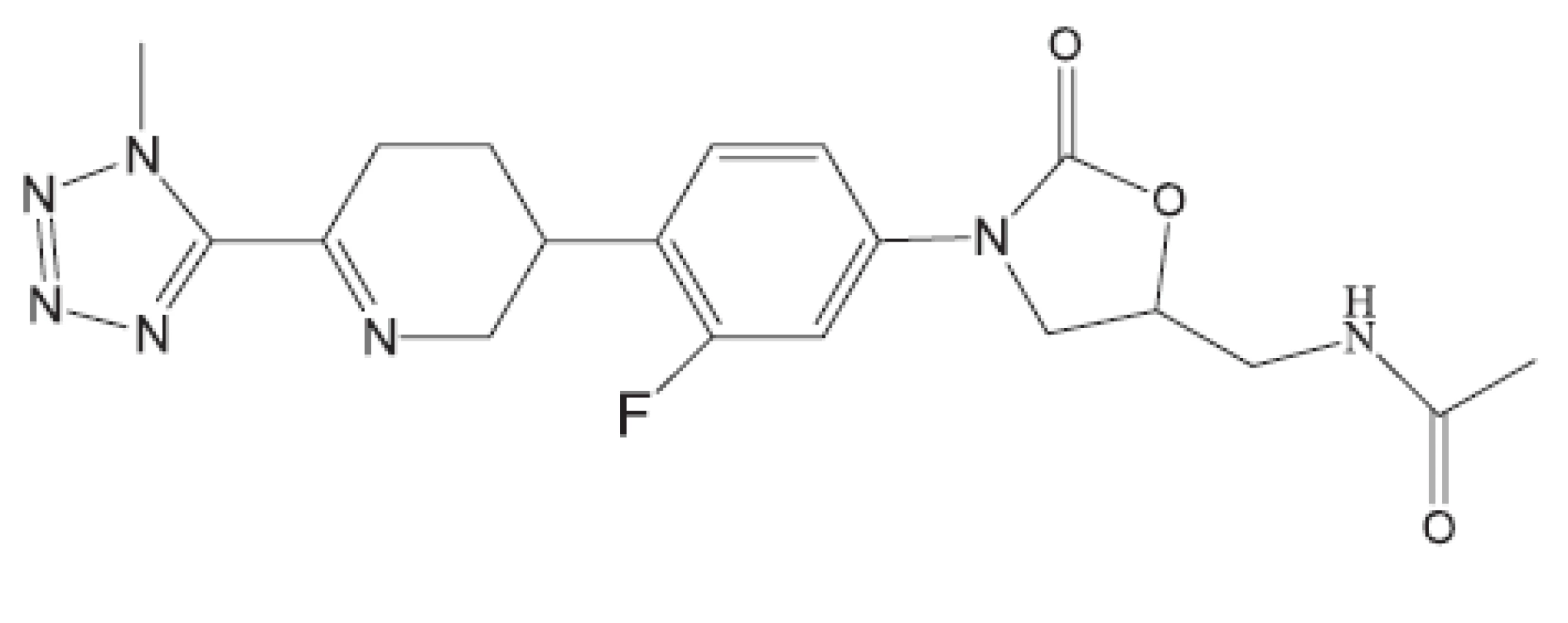
Uvedené příklady nejsou vyčerpávající. Setkáváme se s inhibitory buněčného dělení, inhibitory některých kinas, s látkami zasahujícími do respiračního řetězce, inhibitory některých reduktas, inhibitory buněčné replikace (fluorované chinolony), inhibitory biosyntézy ATP. Lze říci, že již před rokem 2011 byla připravena řada protituberkulózních látek pro případné zásahy proti multirezistentním kmenům, z nichž některé se dostaly až na klinické testy. Svět se tedy připravuje na útoky multirezistentních bakterií. Vraťme se však opět do roku 2011.
Byly hledány modifikace odvozené od riifampicinu, tedy léčiva, která se váží na RNA polymerasu31). Rifampicin a INH patřily mezi nejvýznamnějšího antituberkulotika. Velké úsilí bylo zaměřeno na substituční modifikace ethionamidu, tedy inhibitoru syntézy arabinogalaktanu65). Na obrázku 5 je uvedena struktura SQ60937). Pozornost na blokování biosyntézy arabinanů byla zaměřena i na benzothizinony55). Práce však vznikla již v roce 2009. Několik dalších studií bylo také orientováno na inhibici buněčné stěny mykobakterií57–58). Pokus byl o inhibici alanin-racemasy59), tedy zábranu vzniku D-aminokyselin (což je mechanismus podobný cykloserinu, který vede k poškození peptidoglykanu). Setkáváme se také se zásahy do enzymů za účasti dalších aminokyselin. Pozornost byla věnována methionin aminopeptidase60–63). Methionin aminopeptidasa je metallo-proteasa, která odstraňuje N-koncový methionin při syntéze proteinu. Ve studii byla nalezena skupina 2,3-dichlor-1,4-naftochinonů jako vhodný cíl pro další výzkum inhibitorů. Dalším místem zásahu byl thymin. Útok byl veden na inhibici thymidin-monofosfat kinasy65). Dále můžeme uvést některé další kinasy66) nebo gyrasy67) a některé syntetasy68). Je velmi obtížné zpracovat celý rozsah biochemických prací zaměřených na zásahy do biochemických pochodů mykobakterií. Pouze za rok 2011 a počátek roku 2012 jich bylo uveřejněno 84.
Kam kráčí vývoj nových antituberkulotik
Na závěr se budeme snažit odpovědět na otázku, která je v nadpisu celého článku. Rozhodně nejdůležitější studie vycházejí z poznatků o biochemických pochodech mykobakterií. Například v některých citacích22–26) byla zmínka o azachalkonech jako potenciálních antituberkulotikách. V práci z letošního o roku je objasněn mechanismus účinku jako inhibice protein tyrosin fosfatasy69). Nechceme tím říci, že ostatní přístupy jsou nepřínosné. Hodnocení in vitro může iniciovat k dalšímu výzkumu. Nicméně však pracoviště opírající se pouze o in vitro hodnocení jsou v nevýhodě proti pracovním skupinám používajícím dokonalejší přístupy. Při hodnocení in vivo je nutné předem stanovit toxicitu látek vůči používaným zvířatům. Je to další krok k poznání účinku látek. Epidemiologická situace je v Evropské unii poměrně příznivá90). Výskyt choroby se pohybuje pod hladinou 50 případů(100 000 obyvatel. V České republice je ročně hlášeno 900 nových onemocnění a recidiv. Světem však obchází hrozba multirezistentních kmenů M. tuberculosis. Je tedy otázkou, jaký bude další vývoj s pohledem na velkou současnou migraci obyvatelstva. V některých asijských zemích byly nalezeny i totálně rezistentní kmeny odolné vůči všem léčivům (TDR-Tb). Hledání nových potenciálních antituberkulotik se v Čechách a na Slovensku zabývá několik vědeckých týmů. Bohužel jsou však odkázány pouze na postupy hodnocení in vitro. Proti multirezistentním kmenům M. tuberculosis (MDR Tb) jsou v České republice registrovány pouze některé fluorované chinolony a kanamycin. U extrémně multirezistentních kmenů M. tuberculosis (XDR-Tb) je léčba značně obtížná. Naštěstí jejich výskyt je poměrně nevelký. Dalším problémem je špatná komunikace našich vědeckých pracovišť s farmaceutickými výrobními koncerny. I když u nás byla nalezena řada sloučenin s významnou aktivitou, a to i proti rezistentním kmenům, do praxe se nedostala žádná. Vývoj nového léčiva je dnes spojen s tak nesmírnými náklady, že od rozpadu výrobního podniku SPOFA v českém a slovenském regionu jej žádná z farmaceutických firem nedokáže realizovat.
Střet zájmů: žádný.
prof. RNDr. Karel Waisser, DrSc.
Univerzita Karlova v Praze, Farmaceutická fakulta v Hradci Králové
Heyrovského 1203, 500 05 Hradec Králové, Česká republika
e-mail: waisser@faf.cuni.cz
Sources
1. World Health Orgamization. Global tuberculosis kontrol, a short update to the 2009 report. WHO/HTM/TB/2009.426. Geneva: WHO 2009.
2. Da Silva P. E. A., Polomino J. C. Molecular basis and mechanisms of drug resistance in Mycobacterium tuberculosis clasical and new drugs. J. Antimicob. Chemother. 2011; 66, 1417–1430.
3. Yew W. W., Lange C., Leung C. C. Treatment of tuberculosis: update 2010. Eur. Res. J., 2011; 31(2), 441–462.
4. Srikanth L., Raj V. V, Raghunandan N., Venkateshwerlu L. Recent advances and potential pharmacological activities of benzimidazole derivatives. Der Pharma Chemica. 2011; 3(2), 177–193.
5. Lee S. H., Kim S., Yun M. H., Lee Y. S., Cho S. N., Oh T., Kim P. Synthesis and antitubercular activcity of monocyclic nitroimidazole. Insight from acenazole. Bioorg, Med. Chem. Letter 2011; 21(5), 1515–1518.
6. Thomson A. M., Sutherland H. S., Palmer B. D., Kmentova I., Blasser A., Franzblau S. G., Wan B., Wang Y., Ma Z., Denny W. A. Synthesis and structure-activity relationships of varied ether linker analogues of antitubercular drug (6S)-2-nitro-6-((4-(trifluoromethoxy)benzyl)oxy)-6,7-dihydroxy-5H-imidazo-[2,1-b] [1,3]oxazine(PA-824). J. Med. Chem. 2011; 54(19), 6561–6585.
7. Guillemont J., Mayer C., Poncelet A., Bourdrez X., Andries K. Diarylquinolines, synthesis partway and quantitative structure-activity relationship studies leading ti the discovery of TMC 207. Future Med. Chem. 2011; 3(11), 1345–1360.
8. Vicente E., Villar R., Pérez-Silanes S., Aldana I., Goldeman R., Monge A. Quinoxaline 1,4-dioxide and the potential for treating tuberculosis. Infections Disordes – Drug Targets 2011; 11, 196–294.
9. Čeladník M., Košťálová Z., Jíška S., Waisser K., Kubala E., Palát K. Antituberkulotika. XVII. Funkční deriváty kyseliny 4‑halogenpikolinové a jejich N-oxirů. Českoslov. Farm. 1975; 25(5), 181–185.
10. Swarnalatha G., Prasanthi G., Sirisha M., Chetty C. M. 1,4-Dihyropyridine: A multifunctional molecule – A review, International J. ChemTech. Research 2011; 3(1), 75–89.
11. Suddigut N., Ahsan W., Alam M. S., Ali R., Jain S., Azad B., Akhtan J. Triazoles as potential bioactive agents. Int. J. Pharmaceutical Sciences Review and Research 2011; 8(1), 161–169.
12. De P., Yoya G. K., Constaqnt P., Bedo-Belval F., Duran H., Saffon N., Daffé M., Balta M. Design synthesis, and biological evaluation of new cinnamic derivatives as antitubercular agents. J. Med. Chem. 2011; 54(5), 1449–1461.
13. Imramovský A., Pauk K., Pejchal V., Hanousek J. Salicylanilide and their derivates as perspective anti-tuberculosis drugs: Synthetic routes and biological evaluation, Mini-Reviews in Org. Chemistry. 2011; 8(2), 211–220.
14. Krátký M., Vinšová J. Salicylanilides ester prodrugs as potential antimicrobial agents. – a reviews. Curr. Pharmacol. Design 2011; 17(2), 3494–3505.
15. Mayer A. M. S., Rodigues A. D., Berlinck R. G.-S., Fusetano N. Marine pharmacology in 2007-2008: Marine compounds with antibacterial, anticoagulant, antifungal, anti-inflamatory, antimalarial, antiprotozoal, antituberculosis, and antiviral activities: affecting the immune and nervous system and other miscellaneous mechanism of action. Comparative Biochem. and Physiology, Part C Toxicology and Pharmacology 2011; 153(2), 191–222.
16. Kharb R., Sharma P., C., Yar M. S. Pharmacological signifikance or triazole scaffold. J. Enzyme Inhibition and Medicinal. Chem. 2011; 26(1), 1–21.
17. Lamichhane O. Novel target in M. tuberculosis: search for new drugs. Trends in Molecular Medicine 2011; 17(1), 25–33.
18. Chen T. Ch., Lu P. L., Lin Ch. Y., Lin W. R., Chen Y. H. Fluoroquinolones are associated with delayed treatment and resistance in tuberculosis a systematic review and meta-analysis. Int. J. Infect. Diseases 2011; 15(3), e211-e216.
19. Biava M., Porretta G. C., Poce G., Battilicchio C., Alfonso S., de Logu A., Manetti F., Botta M. Developing pyrrole – derivated antimycobacterial agents: a rational lead optimalization approach. Chem. Med. Chem. Minireviews. 2011; 6, 593–599.
20. Study A., Goodmen A., José R. J., Loyse A., OęDonoghue M., Kon O. M., Dedicioot M. J., Harrrison T. S., John L., Lipman M., Cooke G. S. Multidrug-resistant tuberculosis (MDR-TB) treatment the UK: a study of injectable use and toxicity in practice. J. Antimicrob. Chemother. 2011; 66, 1815–1820.
21. Manna K., Agrawal Y. K. Potent in vitro and in vivo antitubercular activity of certain newly synthesized indophenazine 1,3,5-trisubstituted pyrazoline derivatives baaring bezofuran, Med. Chem. Res, 2011; 20(3), 300–306.
22. Marrapu V., Chaturvedi V., Singh Shu, Singh Shy, Sinha S. Novel aryloxy azolyl chalcones with potent activity against Mycobacterium tuberculosis H37Rv. Eur. J. Med. Chem. 2011; 46(9), 4302–4310.
23. Opletalová V. Chalkony a jejich heterocyklická analoga jako potenciální terapeutika bakteriálních onemocnění. Česk. Slov. Farm. 2000; 49(6), 278–284.
24. Opletalová V., Pour M., Kuneš J., Buchta V., Silva L., Kráľová K., Chlupáčková M., Meltrová D., Peterka M., Posledníková M. Synthesis and biological evaluation of (E)-3-(nitrophenyl)-1-(pyrazin-2-yl)prop-2-en-1-ones. Collect. Czech. Chem. Commun. 2006; 71(1), 44–58.
25. Chlupáčová M., Opletalová V., Kuneš J., Silva L., Buchta V., Dušková J., Kráľová K. Synthesis and biological evaluation of same ring-substituted (E)-3-aryl-1-pyrazin-2-ylprop-2-en-1-ones. Folia Pharm. Univ. Carol. 2005; 33, 31–43.
26. Opletalová V., Hartl J., Patel A., Plát K. Jr., Buchta V. Ring substituted 3-phenyl)-1-(2-pyrazinyl)-2-propen-1-ones as potential photosynthesis-inhibiting, antitungal and antimycobacterial agents. Farmaco 2002; 57(2), 135–144.
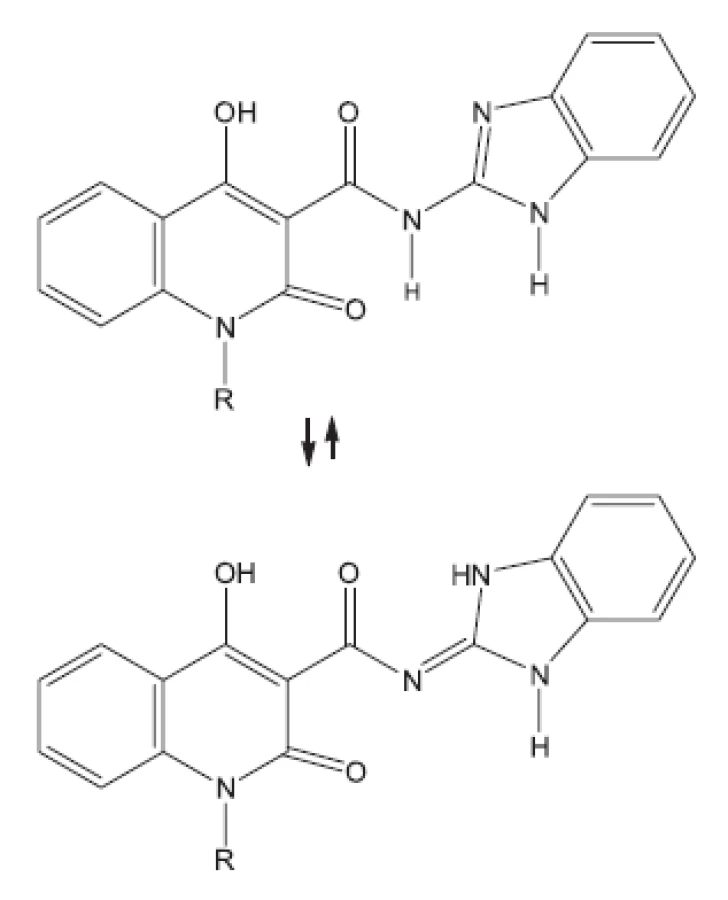
27. Ukrainete I. V., Grinevich L. A., Tkach A. A., Gorokhova O. V., Kravchenko V. N., Sim G. 4-Hydroxy-2-quinolones. 191. Synthesis, tauromerisism and biological activity of benzimidazol-2-ylamides of 1R-4-hydroxy-2-oxo-1.2-dihydroquinoline-3-cyrboxyic acids. Chemistry of Heterocyclic Compounds 2011; 46(11), 1364–1370.
28. Shang S., Shanley C. A., Caraway M. L., Orme E. A., Henao-Tamayo M., Hascall-Dove L., Ackart D., Lenaerts A., Basaraba R. J., Orme I. M., Orodway D. J. Activities TMC 207, Rifampin, and Pyrazinamide against Mycobacterium tuberculosis infection in guinea pigs. Antimicrob. Agents Chemotherapy 2011; 55(1), 124–131.
29. Moraski G. C., Markley L. D., Hipskind P. A., Boshoff H., Cho S., Franzblau S. G., Miller M. J. Advent of imidazolo[1, 2-a] pyridine-3-carboamides with potent multi - and extended drug resistant antituberculosis activity, Med. Chem. Letters 2011; 2(6), 466–470.
30. Cherian J., Choi I. , Nayyar A., Manjunatha U. H., Mukherjee T., Lee Y. S., Boshoff H. I., Singh R., Ha Y. H., Goodwin M., Lakshminarayana S. B., Nioyomrattanakit P., Jiricek J., Ravindran S., Dick T., Keller T. H., Dartois V., Barry C. E. Structure-activity relationships antitubercular nitroimidazoles. 3. Extraploration of linker and lipophilic tail of ((S)-2-nitro-6,7-dinydro-5H-imidazo-[2,1-b][1,3]oxazin-6-yl)-(4-trifluormethoxybenzyl)amine (6-amino PA-824). J. Med. Chem. 2011; 54(16), 5639–5659.
31. Jiri Y., Gill S. K., Kiirchhoff P. D., Wan B., Franzblau S. G., Garcia G. A. Showalter H. D. H. Synthesis and structure activity relationships of novel substituted 8-amino,8-thio, and 1,8-pyrazole congeners of antitubercular rifamycin S and rifampin. Bioorg. Med. Chem. Letters 2011; 21(20), 6094–6099.
32. Halouska S., Fenton R. J., Barletta R. G., Powers R. Prediction the in vivo mechanism of action for drug leads using NMR metabolomics. Chem. Biol. 2012; 7(1), 166–171.
33. Flipo M., Desreses M., Lecat-Guillet N., Dirié B., Carette X., Leroux F., Piveteau C., Demirkaya F., Lens Z., Rucktooa P., Villeret V., Christophe T., Jeon H. K., Locht C., Brodin P., Déprez B., Baulard A. R. Ethionamide boosters: Synthesis, biological aktivity, and structrure-activity relationships of a series of 1,2,4-oxadiazole EhR inmhibitors. J. Med. Chem. 2011; 54(8), 2994–3010.
34. Escribano J., Rivero-Hernández C., Rivera H., Barros D., Castro-Pichel J., Pérez-Herrán E., Mendoza-Losana A., Angulo-Barturen I., Ferrer-Bazaga S., Jiménerz-Navarro E., Ballell L. 4 Subsituted thioquinolines and thiazoloquinolines: Potent, selective, and Tween-80 in vitro dependent families of antitubercular agents with moderate in vivo activity. Chem. Med. Chem. 2011; 6(12), 2252–2263.
35. Singh N., Pandey S. K., Arand N., Dwivedi R., Singh S., Sinha S. K., Chaturvedi V., Jaiswal N., Srivastava A. K., Shah P., Siddiqui M. I., Tripathi R. P. Synthesis, molecular modeling and bio-evaluation of cycloalkyl fused 2-aminopyrimidines as antitubercular and antidiabetic agents Bioorg. Med. Chem. Letters 2011; 21(16), 4404–4408.
36. Amaral L., Martins M., Viveiros M. Thioridazine: Alternative and potentially effective therapy of the XDR-Tb patient. Letters in Drug Design and Discovery 2011; 8, 130–132.
37. Bogatcheva E., Hanrahan C., Nikonenko B., de Santos G., Reddy V., Chen P., Barbosa F., Einck L., Nacy C., Protopopova M. Identification of SQ609 as a lead compound from a library od dipiperidines. Bioorg. Med. Chem. Letters 2011; 21(18), 5353–5357.
38. De Groote M. A., Gilliland J. C., Wells C. I., Brooks E. J., Woolhiser L. K., Gruppo V., Poloquin Ch. A., Orme I. M., Lenaerts A. J. Comparative studies evaluating mause model used for efficacy testing of experimental drugs against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2011; 55(3), 1237–1247.
39. Goodworth K. J., Hervé A. C., Stavropoulos E., Hervé G., Casades I., Hill A. M., Weingarten G. G., Tascon R. E., Colston M. J., Hailes H. C. Synthesis and in vivo biological activity of large-ringed calixarenes against Mycobacterium tuberculosis. Tetrahedron 2011; 67(2), 373–382.
40 . Meermann B., Bockx M., Laenen A., Van Looveren Ch., Cuyckens F., Vanhaecke F. Speciation analysis of bromine-containg drug metabilites in feces samples from a human in vivo study by means of HPLC/ICP-MS comnined with on-line isotope dilution. Anal. Bioanal.. Chem. 2012; 402, 439–448.
41. Petrlíková E., Waisser K., Doležal R., Holý P., Gregor J., Kuneš J., Kaustová J. Antimycobacterial 3-phenyl-4-thioxo-2H-1,3-benzoxazine-2(3H)-ones and 3-phenyl-2H-1,3-benzo xazine-2,4(3H)-dithiones substitutoted on phenyl and benzoxane moiety in position 6. Chem. Papers 2011; 65(3), 352–366.
42. Petrlíková E., Waisser K., Divišová H., Husáková P., Vrabcová P., Kuneš J., Kolář K., Stolaříková J. Highly active antimycobacterial derivatives of benzoxazine. Bioorg. Med.. Chem. 2010; 18(23), 8178–8187.
43. Prajapati K., Singh S., Pathak A. K., Mehla P. QSAR analysis on some 8-methoxy quinoline derivatives as H37Rv (MTB) inhibitors. Int. J. Chem. Tech. Research 2011; 3(1), 408–422.
44. Kovalishyn V., Andres de Sousa J., Ventura C., Leitao R. E., Martins F. QSAR modeling of antitubercular activity of diverse organic compounds. Chemometric and Inteligent Laboratory Systems 2011; 107(1), 69–74.
45. Sharma M. C., Sjarma S. 2D QSAR study of 7-methyljuglone derivatives . An approach to design anti-tubercular agents. J. Pharmacology and Toxicology 2011; 6(6), 499–504.
46. Subramaniam R., Rao G., Pai S. P. N. 2D QSAR studies of some novel quinazolinone derivatives as antitubercular agents. J. Comput. Met. Mol. Design 2011; 1(3), 69–82.
47. Sawant R. L., Wadekar J. B., Lanke P. QSAR analysis of structurally similar antitubercular isatin analogues. Latin American J. Pharmacy 2011; 30(4), 773–780.
48. Khuni R. C., Khedkar V. M., Chawda R. S., Chauhan N. A., Parikh A. R., Coutinho E. C. Synhthesis, antitubercular evaluation and 3D-QSAR study of N - phenyl-3.(4-fluorphenyl)-4-substituted pyrazole derivatives. Bioorg. Med. Chem. Letters 2012; 22(1), 666–678.
49. Kumar U. Ch., Shaik M. 3-D QSAR CoMSTA models of arylamides for prediction of enoyl acyl carrier protein reductase inhibitory activity. J. Pharmaceutical Sciences and Technology 2011; 3(1), 536–542.
50. Puratchikody A., Natarajan R., Jayapal M., Doble M. Synthesis, in vitro antitubercular actrivity and 3D-QSAR of novel quinoxaline derivatives. Chemical Biology and Drug Design 2011; 78(6), 988–998.
51. Khunt R. C., Khedkar V. M., Chawda R. S., Chauhan N. A., Parikh A. R., Coutinho E. C. Synthesis, antitubercular evaluation and 3D-QSAR study of N-phenyl-3-(4-fluorophenyl)-4-substituted pyrazole derivatives. Bioorg. Med. Chem. Letters 2012; 22(1), 666–678.
52. Verna R. P., Hansch C. Use of 13C NMR chemical shift as QSAR/QSPR descriptor. Chem. Reviews 2011; 111(4), 28665–2899.
53. Deng L., Diao J., Chen P., Pujari V., Yao Y., Cheng G., Crick D. C., Prasad B. V. V., Song Y. Inhibition of 1-deoxy-d-xylulose-5-phosphate reductoisomerase by lipophilic phosphonates: SAR, QSAR and crystallographic studies. J. Med. Chem. 2011; 54(13), 4721–4734.
54. Petrlíková E. Protituberkulózní látky a jejich další antimykobakobakteriální obdoby. Dizerační práce Univerzita Karlova v Praze, Farmaceutickáí fakulta v Hradci Králové, Hradec Králové 2010.
55. Makarov V., Manina G., Mikusova K., Möllmann U., Ryabova O., Saint-Joanis B., Dhar N., Pasca M. R., Buroni S., Lucarelli A. P., Milano A., De Rossi E., Balanova M., Bobovska A., Dianiskova P., Kordulakova J., Sala C., Fullam E., Schneider P., McKinney J. D., Brodin P., Christophe T., Waddell S., Butcher P., Albrethsen J., Rosenkrants I., Brosch R., Nandi V., Bharath S., Gaonkar S., Shandil R. K., Balasubramanian V., Balganesh T., Tyagi S., Grossei J., Riccardi G., Cole S. T. Benzothiazinones kill Mycobacterium tuberculosis by blockings arabinan synthesis. Science 2009; 324 801–804.
56. Barry C. S., Backus K. M., Barry C. E., Davis B. G. ESI-MS assai of M. tuberculosis cell wall antigen 85 enzymes permits substrate profillig and design of mechanism-based inhibitors. J. Am. Chem. Soc. 2011; 133(34), 13232–13235.
57. Dutta N. K., Mehra S., Kaushal D. A Mycobacterium tuberculosis sigma factor network responds to cell-envelope damage by promising anti-mycobacterial thioridazine. PloS One 2010; 5(4), e10069.
58. Li Y., Zhou Y., Ma Y., Li X. Design and synthesis of novel cell wall inhibitors of Mycobactrerium tuberculosis GlmM and GmU. Carbohydrate Research 2011; 346(13), 1714–1720.
59. Anthony K. G., Strych U., Yeug K. R., Shoen C. S., Perez O., Krause K. L., Cynymon M. H., Arristoff P. A., Koski R. A. New classes of alanine racemase inhibitors identifies by high-throughput screening show antibacterial actuivity against Mycobacterium tuberculosis. PloS One 2011; 6(5), e20374.
60. Olaley O., Raghunand T. R., Bhat S., He J., Tyagi S., Lamichhane G., Gu P., Zhou J., Zhang Y., Grosset J., Bishai W. R., Liu J. O. Methionine aminopeptidases from Mycobacterium tuberculosis as novel antimycobacterial targets. Chem Biol. 2010; 17(1), 86–97.
61. Lu J. P., Yuan X. H., Ye Q. Z. Structural analysis of inhibition of Mycobacterium tuberculosis metionine aminopeptidase by bengamide derivatives. Eur. J. Med. Chem. 2012; 47(1), 479–484.
62. Olaleye O., Raghunand T. R., Bhat S., Chong C., Gu P., Zhou J., Zhang Y., Bishai W. R., Liu J. O. Characterization of clioquinol and analogues as novel inhibitor of aminopeptidases from Mycobacterium tuberculosis.Tuberculosis 2011; 91, 561–565.
63. Olaleye O. A.. Bishai W. R., Liu J. O. Targeting the role of N‑terminal methionine processing enzyme in Mycobacterium tuberculosis. Tuberculosis 2009; 89, 555–559.
64. Frecer V., Seneci P., Miertus S. Computer-assisted combinatorial design of bicyclic thymidine analogs as inhibitors of Mycobacterium tuberculosis thymidine monophosphate kinase. J. Comput. Aided.Mol. Des. 2011; 25(1), 31–49.
65. Onajole O. K., Govender P., van Helden P. D., Kruger H. G., Maguire G. E. M., Wild I., Govender T. Synthesis and evaluation of S109 analogues as potential anti-tuberculosis candidates. Eur. J. Med. Chem. 2010; 45(5), 2075–2079.
66. Saidenberg D. M., Passerselli A. W., Rodrigues A. V., Basso L. A., Santos D. S., Palma M. S. Shikimate kinase (EC 2.7.1.71) from Mycobacterium tuberculosis: Kinetic and structutural dynamics of potential molecular target for drug development . Current Medicinal Chemistry 2011; 18(9), 1259–1318.
67. Chopra S., Matsuyoma K., Tran T., Malerich J. P., Wan B., Franzblau S. G., Lun S., Guo H., Maiga C. M., Bishai W. R., Madrid P. B. Evaluation of gyrase B as a drug target in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2012; 67(2), 415 –421.
68. Jatana N., Jangid S., Khare G., Tyagi A. K., Latha N. Molecular modeling studies of fatty acyl-CoA synthetase (FadD 13) from Mycobacterium tuberculosis – a potential target for the development of antitubercular drugs. J. Mol. Model. 2011; 17(1), 301–313.
69. Chiaradia L. D., Martins P. G. A., Cordeiro M. N. S., Guido R. V. C., Ecco G., Andricopulo A. D., Yunes R. A., Vernal J., Nunes R. J., Terenzi H. Synthesis, biological evaluation, and molecular modeling of chalcone derivatives as potent inhibitors of Mycobacterium tuberculosis protein tyrosine phosphatases (PtpA and Ptp B) J. Med. Chem. 2012; 55(1), 390–402.
70. Bártů V. Tuberkulóza ve světle 21. století. Medical Tribune 2010; 4, D2 tematická příloha. ISSN 1214-8911.
Labels
Pharmacy Clinical pharmacologyArticle was published in
Czech and Slovak Pharmacy

2012 Issue 4
Most read in this issue
- Lidokainový gel pro použití na kůži magistraliter připravený
-
Standardní receptura pro přípravu léčivých přípravků v lékárnách V*
Sbírka Dermatologische Magistralrezepturen der Schweiz -
Naše léčivé přípravky v polovině 19. století
II. část – galenické přípravky -
Naše léčivé přípravky v polovině 19. století
I. část – úvod a chemické přípravky