
Optimalizace technologických postupů pro přípravu tablet s nízkým obsahem warfarinu metodou přímého lisování
Optimization of technological processes for the preparation of tablets with a low content of warfarin by direct compression
Warfarin is adrug with anarrow therapeutic index. It is commercially available in the form of tablets with immediate release from many generic manufacturers. Though attempts are being made to replace it with new drugs, in the U.S.A.it is still the antithrombotic agent of the first choice. In the past there were cases when after replacement of the original brand preparation with the generic one, the patient suffered from complications such as the loss of control of anticoagulation and increased frequency of patient’s visits to the physician. One of the critical parameters of tablets containing an active substance with anarrow therapeutic index (NTI) is content uniformity, which must comply with the pharmacopoeial requirements as well as the strict criteria of regulatory authorities in the validation of the manufacture of the solid dosage form. Content uniformity is affected by anumber of factors such as density, particle shape and size distribution, electrostatic charge, and concentration of the individual components. Of the technological parameters, it is mainly the intensity and length of mixing, shape of the mixing vessel and the mixer, the size of the charge, or the degree of filling of the mixing device, etc. This paper deals with the influence of the mixing time and concentration of the drug on the content uniformity of warfarin-containing tablets. In mixing the mixtures of solid substances, where the active substance is included in alow concentration, there occurs the so-called mixing-out and segregation of the active substance. For this reason it is necessary to optimize the period of mixing. This study managed to optimize the mixing time of mixtures prepared with the use of the patented technology of the Veterinary and Pharmaceutical University Brno and further to prepare tablets with varying content of warfarin (2–10 mg) from acommon blend, which fulfil the pharmacopoeial requirements as well as the requirements of regulatory authorities for content uniformity.
Keywords:
content uniformity • warfarin • blending time • narrow therapeutic index • common blend
Authors:
Jan Muselík; Aleš Franc; Jana Štarková; Zuzana Matějková
Authors‘ workplace:
Veterinární a farmaceutická univerzita Brno, Farmaceutická fakulta, Ústav technologie léků
Published in:
Čes. slov. Farm., 2014; 63, 217-221
Category:
Original Articles
Overview
Warfarin je lékem súzkým terapeutickým indexem. Komerčně je dostupný ve formě tablet sokamžitým uvolňováním od mnoha generických výrobců. Ačkoliv je snaha jej nahradit novými léky, vUSA je stále antitrombotikem první volby. Vminulosti se vyskytly případy, kdy po převedení pacienta zoriginálního léčivého přípravku na generický přípravek došlo ke komplikacím, jako je ztráta kontroly antikoagulace azvýšení frekvence návštěv pacienta ulékaře. Jedním z kritických parametrů tablet s obsahem léčiva s úzkým terapeutickým indexem (NTI) je obsahová stejnoměrnost, která musí vyhovět lékopisným požadavkům ataké přísným kritériím regulačních autorit při validaci výroby pevné lékové formy. Obsahovou stejnoměrnost ovlivňuje celá řada faktorů, jako je hustota, distribuce velikosti atvar částic, elektrostatický náboj akoncentrace jednotlivých složek. Ztechnologických parametrů se jedná hlavně ointenzitu adélku mísení, tvar mísící nádoby amíchadla, velikost násady, resp. míru zaplněnosti mísícího zařízení apod. Tato práce se zabývá vlivem doby mísení akoncentrace léčiva na obsahovou stejnoměrnost tablet s obsahem warfarinu. Při mísení směsí pevných látek, kde je léčivá látka obsažena vnízké koncentraci, dochází ktzv. „vymíchávání“ asegregaci účinné látky. Z tohoto důvodu je nutné optimalizovat čas mísení. V rámci této práce se podařilo optimalizovat dobu mísení směsi připravených patentovanou technologií VFU Brno adále připravit tablety s různým obsahem warfarinu (2–10 mg) ze společné směsi, které splňují lékopisné požadavky ipožadavky regulačních autorit na obsahovou stejnoměrnost.
Klíčová slova:
obsahová stejnoměrnost • warfarin • doba mísení • úzký terapeutický index • společná směs
Úvod
Warfarin je derivátem dikumarolu, který se používá ve formě sodné soli nebo jejího klathrátu s izopropanolem. Patří mezi nejstarší antikoagulancia a v USA bývá stále považován za lék první volby při prevenci trombóz1). Tablety s obsahem sodné soli warfarinu byly poprvé registrovány americkou firmou DuPont Pharma počátkem čtyřicátých let 20. století pod názvem Coumadine. Ochranná lhůta vypršela v roce 1962, avšak z hlediska úzkého terapeutického indexu warfarinu nebylo snadné přistoupit k jeho generické substituci. Důvodem se stala zkušenost z roku 1980 z Boston City Hospital, kde při přechodu na generikum bylo shledáno zvýšené riziko úmrtnosti, zdravotních komplikací, včetně akutního krvácení2). Jaffr a Bragg zde nalézají příčinu v tom, že generikum obsahovalo amorfní formu namísto krystalické soli, což v důsledku vedlo k vyřazení amorfního warfarinu ze substituční léčby v USA3). Nicméně Wittkowská naznačuje, že na vině mohlo být ve skutečnosti kolísání obsahu warfarinu v tabletách, neboť při titraci pacienta na terapeutickou hladinu se dávka snižuje či zvyšuje pouze o 5 až 15 %. Jestliže lékopis v té době připouštěl chybu obsahové stejnoměrnosti v jednotlivých tabletách ± 15 % a RSD do 6 %, pak kolísání dávky již mohlo způsobit citelný farmakologický efekt, a tím i změnu času krevní srážlivosti. Tomuto lékopisnému limitu odpovídaly i tablety generického výrobce4).
Originální výrobce, DuPont Pharma, měl však vnitřní limit obsahové stejnoměrnosti přísnější, a to obsah warfarinu v rozsahu 92,5–107,5 % a RSD do 3 %, a do roku 1996 vlastnil na distribuci warfarinu v USA monopol5). Ve snaze zabránit generické náhradě již v roce 1996 žádal DuPont Pharma úřad FDA i o striktnější limit pro zkoušku bioekvivalence a svůj přísnější limit obsahové stejnoměrnosti hodlal prosadit do USP. Obojí bylo zamítnuto6). Taktéž následné studie Halkina7) a Swansona8) potvrdily dostatečnost stávajících zkoušek bioekvivalence při generických záměnách.
Teprve v září 1997 byla FDA schválena generická substituce Coumadinu tabletami s obsahem sodné soli warfarinu klathrátu od firmy Barr Laboratories (nyní Teva). Přesto dva důležití generičtí výrobci tablet s obsahem warfarinu, Taro-Warfarin (Taro Pharmaceuticals Inc) a Apo-Warfarin (Apotex Inc.) mají ve své specifikaci jak přísnější limity pro bioekvivalenci, tak pro obsahovou stejnoměrnost. Konfidenční interval limitů AUC a cmax 0,8–1,25 je zde počítán pro pravděpodobnost 95 % namísto obvyklých 90 % a limit pro obsahovou stejnoměrnost je obsah v jednotlivých tabletách v intervalu 92,5–107,5 % průměru a hodnota RSD do 3 %9).
V roce 2006 se firma Barr stala majitelem patentu chorvatské firmy Pliva na výrobu tablet s obsahem warfarinu, registrovaných v USA, s vysokou mírou obsahové stejnoměrnosti. V tomto patentu je uvedena technologie výroby tablet, kde jednotlivé obsahy jsou v intervalu 97–103 % průměrné hodnoty obsahu a hodnota RSD do 2 %10). Technologie je zde založena na impregnaci sorbentu vodným roztokem sodné soli warfarinu klatrátu, aniž by po vysušení byla jakkoliv definována krystalová či amorfní struktura warfarinu v lékové formě11).
Z výše uvedeného plyne, že obsahová stejnoměrnost je nadále jednou z nejdůležitějších otázek při formulaci lékových forem s NTI. To se shoduje i s prohlášením Beneta, podle něhož komplikace při léčbě NTI bývají způsobeny především interindividuální variabilitou kvality léků12).
Pokusná část
Příprava směsí a tablet
Složení směsí a základní fyzikálně chemické vlastnosti jednotlivých složek vychází z patentu VFU Brno13) a jsou uvedeny v tabulce 1. Proměnnými zde byly celková délka mísení tabletové směsi, tj. součet času mísení bez lubrikantu a s lubrikantem (12, 15, 20 a 25 min) a obsah warfarinu sodné soli v tabletovině (0,55 a 2,73 %), což odpovídá obsahu v tabletě (1 a 5 mg). V tomto experimentu byly zužitkovány výsledky z předchozích pokusů, kde jsme hodnotili výsledky obsahové stejnoměrnosti tablet vyrobených metodou přímého lisování směsí s obsahem plniv s rozdílnou velikostí distribuce částic a s různou hustotou, k nimž byla v různém stupni mísení přidána kluzná látka14–16).
V této práci byly jednotlivé komponenty tabletoviny (složení 1 a 2 – tab. 1) přesítovány sítem o velikosti oka 250 μm a míseny 10 minut bez přídavku stearanu hořečnatého, který byl přidán následně a mísení pokračovalo dalších 2, 5, 10 nebo 15 minut (technologie A, B, C nebo D). Každou z těchto technologií byly připraveny dvě šarže s obsahem warfarinu sodné soli 5 mg. Technologie B byla dále použita k přípravě dvou šarží tablet s obsahem sodné soli warfarinu 1 mg.
Pro porovnání vlivu koncentrace warfarinu v tabletovině byla připravena také jedna šarže směsi postupem popsaným výše (technologie B) s obsahem warfarinu sodné soli klathrátu 2 % (složení 3 – viz tab. 1). Tento postup odpovídá patentované technologii VFU Brno13). Z této společné směsi byly připraveny tablety s různou cílovou hmotností (108 mg, 163 mg, 271 mg a 542 mg) s teoretickým obsahem warfarinu sodné soli 2, 3, 5 a 10 mg.
Mísení všech výše uvedených složení probíhalo v homogenizačním zařízení Turbula (T2C, Švýcarsko) rychlostí 40 otáček za minutu a tablety byly připraveny pomocí excentrického lisu (Korsch EK0, Německo).
Stanovení obsahu warfarinu v tabletových směsích a tabletách
Připravená tabletová směs byla vysypána do nádoby tvaru válce o průměru 25 cm. Následně byla hladina vyrovnána horizontálními pohyby. Poté byl povrch rozdělen do deseti částí a z každé části byl lžičkou vykrojen vzorek o hmotnosti přibližně přesně 500 mg. Přesná hmotnost vzorků byla zaznamenána.
Pro stanovení obsahu warfarinu v tabletách bylo během lisování v pravidelných časových intervalech odebráno z každé šarže deset tablet.
Odebrané a zvážené tabletové směsi nebo tablety byly vloženy do odměrných baněk a ponechány po dobu 12 hodin v rozpouštědle (voda : methanol v poměru 9 : 1; v/v). Poté byla směs doplněna rozpouštědlem po rysku a nerozpuštěné pomocné látky byly odstraněny centrifugací (10 minut, 5000 ot. min-1). Takto připravené vzorky byly analyzovány pomocí HPLC a obsah warfarinu byl stanoven na základě kalibrační křivky.
Chromatografická analýza
Pro stanovení obsahu warfarinu byl použit kapalinový chromatograf YL 9100 (Young Lin Instrument, Jižní Korea) s kvarternární pumpou, automatickým dávkovačem a detektorem s diodovým polem. Chromatografické separace byly provedeny na koloně BDS HYPERSIL C18 (150 × 4,6 mm; velikost částic 5 μm). Složení mobilní fáze bylo 64 % methanolu a 36 % kyseliny mravenčí (0,04 M). Průtok mobilní fáze byl 1,4 ml min-1, teplota kolony byla 25 °C. Spektra byla snímána při 280 nm. Vždy bylo dávkováno 10 μl vzorku. Celková délka analýzy byla 6 minut.
Měření velikosti částic
Účinná látka i pomocné látky byly změřeny na zařízení Malvern Mastersaizer 5000 v kapalné fázi. Pro měření účinné látky, sodné soli warfarinu kltahrátu, bylo jako média použito cyklohexanu, pro ostatní pomocné látky bylo použito izopropanolu.
Statistické zpracování výsledků
Výsledky stanovení vzorků směsí a tablet z každé šarže (počet vzorků n = 10) byly přepočteny relativně vůči teoretickému obsahu warfarinu sodné soli. Z těchto hodnot byl pro každou připravenou šarži vypočítán průměrný obsah, směrodatná odchylka, relativní směrodatná odchylka a Cpk indexy pro limity dle Ph.Eur. 2009 čl. 2.9.6 (85–115 %).
Výsledky a diskuze
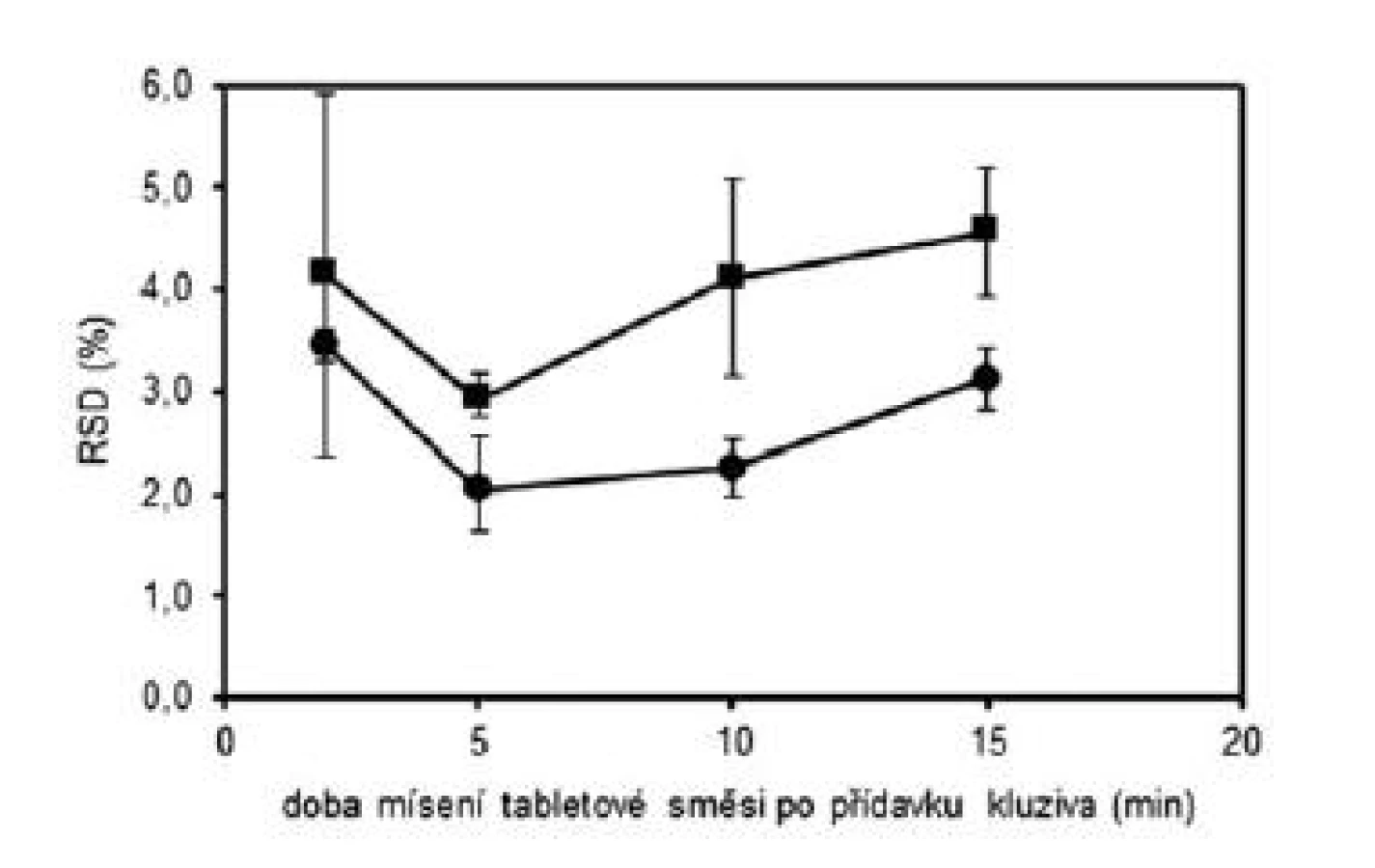
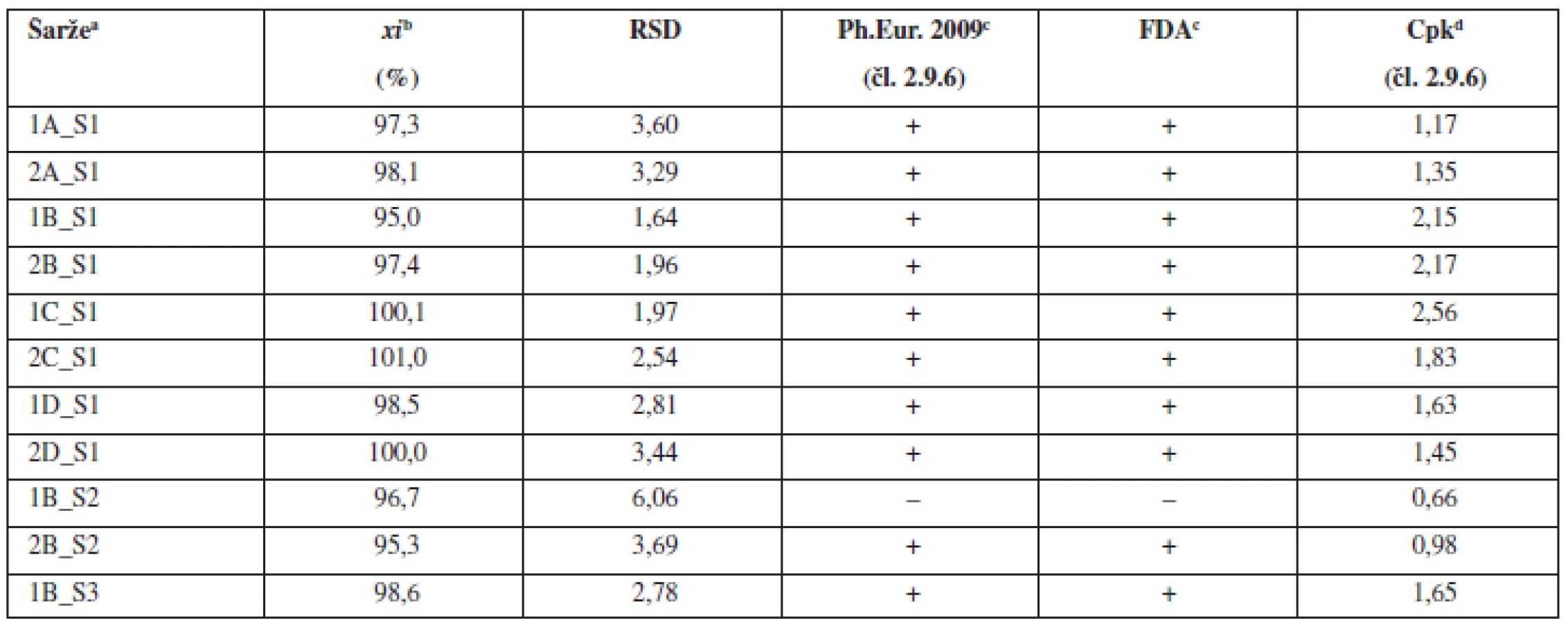
V první části experimentu byly připraveny tabletové směsi a tablety dle tabulky 1 (složení 1) s obsahem warfarinu sodné soli klatrátu 2,73 %. Byly zvoleny celkem čtyři technologie mísení, které se lišily délkou mísení po přídavku kluziva do tabletové směsi. Doby „domísení“ směsi byly zvoleny 2 minuty (technologie A), 5 minut (technologie B), 10 minut (technologie C) a 15 minut (technologie D). Z takto připravených směsí byly následně nalisovány tablety s obsahem warfarinu sodné soli 5 mg. Každou technologií (A–D) byly připraveny dvě šarže. Takto připravené směsi byly dále hodnoceny dle požadavků Ph.Eur. (čl. 2.9.6), požadavků FDA (nalezené obsahy v intervalu 90–110 % průměrné hodnoty, RSD do 5 %) a výpočtu indexu způsobilosti (Cpk). Tablety byly hodnoceny dle požadavků Ph.Eur 2009 (čl. 2.9.6 a čl. 2.9.40) a výpočtu indexu způsobilosti (Cpk). Výsledné hodnoty průměrných obsahů a RSD tabletových směsí uvádí tabulka 2 a tablet tabulka 3. Všechny takto připravené směsi a tablety vyhověly požadavkům Ph.Eur. na obsahovou stejnoměrnost. Připravené směsi vyhověly i požadavkům FDA a u všech směsí byl index Cpk ≥ 1,0. U tablet nalisovaných z těchto směsí byly již patrné rozdíly v závislosti na použité technologii. Kritérium Cpk ≥ 1,0 splnily obě šarže pouze pro technologii B. Dále byl hodnocen vliv délky mísení na obsahovou stejnoměrnost vyjádřenou jako RSD. Na obrázku 1 lze pozorovat počáteční pokles hodnot RSD do 5. minuty, kde má RSD minimální hodnotu, při delším mísení směsí hodnoty RSD opět stoupají. Vlivem mísení dochází nejdříve k homogenizaci směsi, která dosáhne maxima a v další fázi mísení dochází již k postupné segregaci směsi a zhoršování uniformity. Stejné závěry byly zjištěny i u tablet připravených z těchto směsí. Z obrázku 1 je patrné, že tablety vykazovaly vyšší hodnoty RSD než směsi, z nichž byly tablety připravené. To lze vysvětlit tím, že následné skladování a zpracování směsi do konečných tablet může způsobovat její rozdružování. Na základě těchto výsledků byl zvolen optimální čas „domísení“ směsí po přídavku kluziva 5 minut (technologie B).
Obsahová stejnoměrnost se obvykle snižuje s klesajícím množstvím léčiva v tabletě. Proto byla pro další experiment použita tabletová směs s obsahem warfarinu sodné soli klatrátu 0,55 %, což odpovídá obsahu warfarinu sodné soli v tabletě 1 mg (při hmotnosti tablety 200 mg). Pro mísení byla použita technologie B, která vykazovala v předchozí části experimentu nejlepší obsahovou stejnoměrnost v porovnání s ostatními technologiemi mísení. Tímto postupem byly připraveny dvě šarže směsí a z nich byly následně nalisovány dvě šarže tablet. Výsledky hodnocení obsahové stejnoměrnosti dle požadavků Ph.Eur. připravených směsí (tab. 2) a tablet (tab. 3) ukazují, že vždy jedna z připravených šarží lékopisným požadavkům nevyhověla. Výsledky potvrzují komplikovanost procesu mísení a dosažení požadované obsahové stejnoměrnosti směsí a tablet u přípravků s nízkým obsahem léčiva.
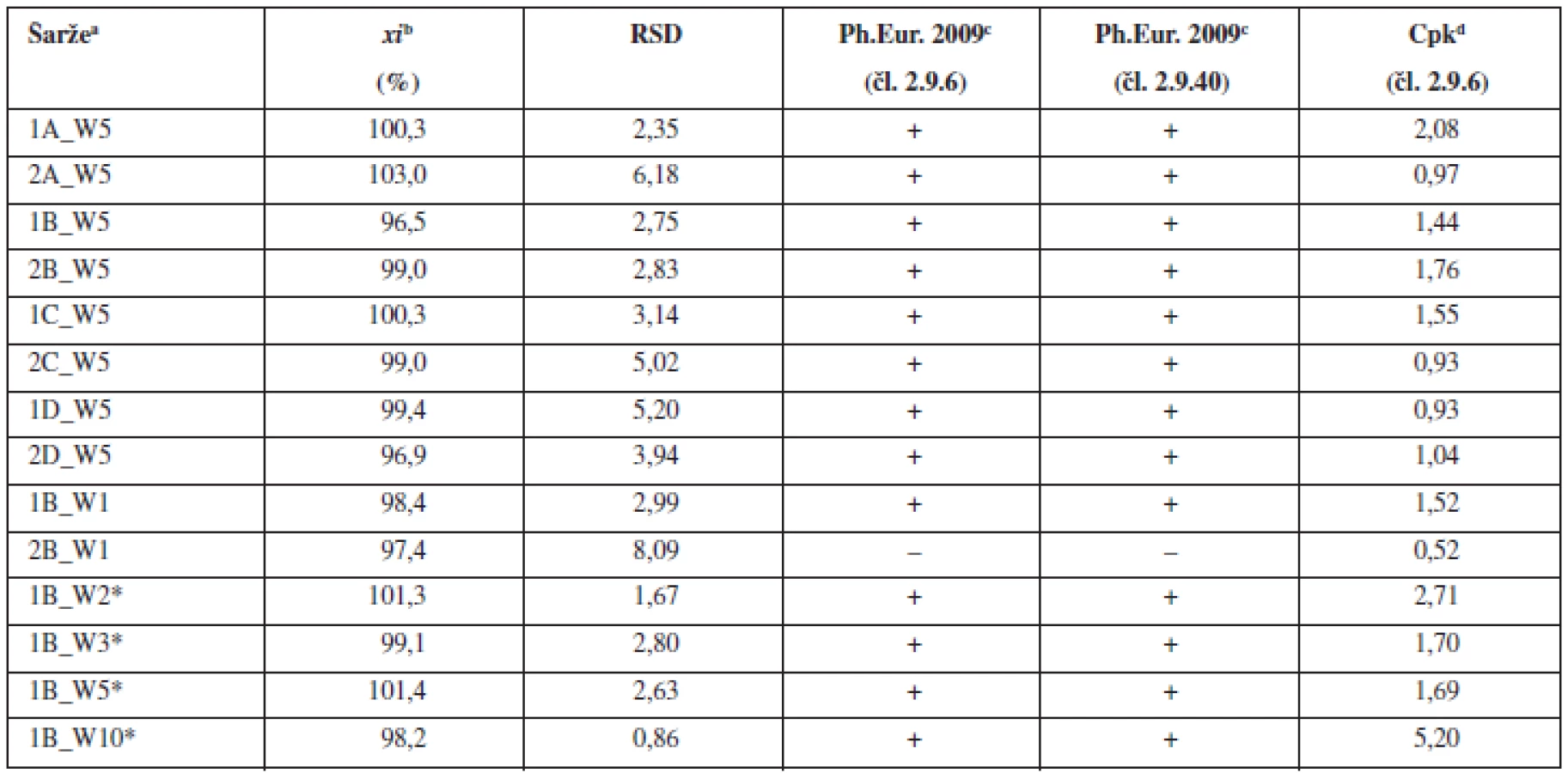
Na základě těchto výsledků, kdy tablety s nízkým obsahem léčiva (1 mg) nevyhověly hodnoceným kritériím, byla pro porovnání ve třetí části experimentu připravena jedna šarže dle patentované technologie VFU Brno s obsahem warfarinu sodné soli klatrátu 2,0 %. Z jedné společné směsi byly nalisovány tablety s různou hmotností a obsahem warfarinu sodné soli 2, 3, 5 a 10 mg. Jak směs, tak z ní připravené tablety o různých obsazích warfarinu sodné soli, vyhověly všem hodnoceným kritériím (tab. 2 a tab. 3). Použití společné směsi pro přípravu tablet s různým obsahem warfarinu sodné soli vede k přípravě tablet, které svými parametry vyhovují nejen lékopisným požadavkům, ale i hodnoceným požadavkům pro procesní validaci (Cpk index). Použití společné směsi se na základě těchto výsledků jeví jako vhodný postup k přípravě tablet s nízkým obsahem léčivé látky metodou přímého lisování. Tímto postupem se podařilo připravit tablety s obsahem warfarinu v celém rozsahu komerčně dostupných přípravků v Evropě.
Závěr
Tato práce navazuje na již dříve publikované výsledky autorů zaměřené na popis procesu mísení směsí s nízkým obsahem léčivé látky určených pro přímé lisování tablet. V této práci byla optimalizována doba mísení směsi s obsahem nízké koncentrace warfarinu po přídavku kluzné látky. Na základě obsahové stejnoměrnosti směsí a tablet vyjádřené jako RSD byla stanovena optimální délka mísení směsi po přídavku kluziva 5 minut. Dále bylo zjištěno, že při použití velmi nízkých koncentrací léčivé látky (obsah v tabletě 1 mg) produkty nevyhoví lékopisným požadavkům ani v případě použití optimalizovaného složení a technologie mísení. Řešením tohoto problému je příprava společné směsi, ze které jsou následně lisovány tablety s různou hmotností, a tím také s různým obsahem warfarinu. Tímto postupem se podařilo metodou přímého lisování připravit tablety s různým obsahem warfarinu (2–10 mg), které vyhověly všem hodnoceným kritériím pro obsahovou stejnoměrnost.
Seznam zkratek
- FDA Americký úřad pro potraviny a léčiva
- NTI úzký terapeutický index
- Ph.Eur. Evropský lékopis
- RSD relativní směrodatná odchylka
- USP Lékopis USA
Výsledky práce byly získány za finančního přispění IGA VFU v rámci řešení projektu č. 55/2014/FaF.
Střet zájmů: žádný.
Došlo 8. září 2014 / Přijato 16. září 2014
J. Muselík • PharmDr. Aleš Franc, Ph.D. (✉) • J. Štarková • Z. Matějková
Veterinární a farmaceutická univerzita Brno, Farmaceutická fakulta,
Ústav technologie léků
Palackého tř. 1/3, 612 42 Brno
e-mail: franca@vfu.cz
Sources
1. Mant J. W. Pro: Warfarin should be the drug of choice for thromboprophylaxis in elderly patients with atrial fibrillation. Why warfarin should really be the drug of choice for stroke prevention in elderly patients with atrial fibrillation. Thromb. Haemost. 2008; 100, 14–15.
2. Lawren M. V., Zhanel G. Z. Clinical significance of bioequivalence and interchangeability of narrow therapeutic range drugs: focus on warfarins. J. Pharm. Pharmaceut. Sci. 1998; 3, 92–94.
3. Jaffer A., Bragg L. Practical tips for warfarin dosing and monitoring. Clev. Clin. J. Med. 2003; 70, 361–371.
4. Wittkowsky A. K. Generic warfarin: implications for patient care. Pharmacotherapy. 1997; 17, 640–643.
5. Cundiff D. K. Insufficient evidence supporting low-intensity warfarin for venous thromboembolism prophylaxis. 2003; Med. Gen. Med. 2003; 5(3), 2.
6. Warfarin sodium antitrust litigation. United states court of appeals for the third circuit. http://www2.ca3.uscourts.gov/opinarch/ 023603p.pdf (31. 7. 2014).
7. Hope K. A. Subtherapeutic INR values associated with a switch to generic warfarin. Clin. Pharmacol. Ther. 2003; 4, 215–221.
8. Swenson C., Fundak G. Observational cohort study of switching warfarin sodium products in a managed care organization. Am. J. Health. Syst. Pharm. 2000; 57, 452–455.
9. Sawoniak A. E., Shalansky A. F., Zed P. J., Sundreji R. Formulary considerations related to warfarin interchangeability. Can. J. Hosp. Pharm. 2002; 55, 215–218.
10. Franc A., Žaludek B., Goněc R. Method of producing dosage units a solid drug form containing warfarin sodium salts as active component. PLIVA-Lachema. Patent WO2005034919 (17. 10. 2003).
11. Franc A., Rabišková M., Goněc R. Impregnation: A progressive method in the production of solid dosage forms with low content of poorly soluble drugs. Eur. J. Parent. Pharm. Sci. 2011; 16, 85–93.
12. Benet L. Z., Goyan J. E. Bioequivalence and narrow therapeutic index drugs. Pharmacotherapy. 1995; 15, 433–440.
13. Franc A., Muselík J. Způsob přípravy pevné lékové formy se sodnou solí warfarinu ve formě klathrátu izopropanolu. VFU Brno. Patent CZ304136. (2. 10. 2013).
14. Muselík J., Franc A. Hodnocení obsahové stejnoměrnosti tablet s nízkým obsahem léčivé látky s úzkým terapeutickým indexem. Čes. Slov. Farm. 2012; 61, 271–275.
15. Franc A., Muselík J., Máslová R., Hadrabová J. Obsahová stejnoměrnost směsí a tablet obsahujících warfarin. Čes. slov. Farm. 2013; 62, 177–181
16. Muselík J., Franc A., Doležel P., Goněc R., Krondlová A., Lukášová I. Influence of process parameters on content uniformity of a low dose active pharmaceutical ingredient in a tablet formulation according to GMP. Acta Pharm. 2014; 64, 355–367.
Labels
Pharmacy Clinical pharmacologyArticle was published in
Czech and Slovak Pharmacy

2014 Issue 5
Most read in this issue
- Historie léčby diabetu v Československu do roku 1989
- Lyofilizácia liečiv na báze proteínov
- Kontinuální/celoživotní vzdělávání lékárníků v České republice 4. cyklus 2008–2011
- Příprava peletových jader pro řízené uvolňování glukosy k prevenci hypoglykémií u diabetiků