
Biologická dostupnost léčiva a možnosti jejího ovlivňování
Bioavailability and factors influencing its rate
Bioavailability can be defined as the rate and range of active ingredient absorption, when it becomes available in the systemic circulation or at the desired site of drug action, respectively. Drug bioavailability after oral administration is affected by anumber of different factors, including physicochemical properties of the drug, physiological aspects, the type of dosage form, food intake, biorhythms, and intra - and interindividual variability of the human population. This article is the first from the series dealing with the bioavailability and methods leading to its improvement. The aim of the present paper is to provide an overview of aspects influencing the rate of bioavailability after oral administration of the active ingredient. Subsequentarticles will provide detailed descriptions of methods used for dug bioavailability improvement, which are here only summarized.
Keywords:
bioavailability • drug solubility • poorly soluble drugs • factors influencing bioavailability
Authors:
Barbora Vraníková; Jan Gajdziok
Authors‘ workplace:
Veterinární a farmaceutická univerzita Brno, Farmaceutická fakulta, Ústav technologie léků
Published in:
Čes. slov. Farm., 2015; 64, 7-13
Category:
Review Articles
Overview
Biologická dostupnost je definována jako rychlost arozsah, v nichž je léčivá látka absorbována adosáhne místa svého účinku, respektive systémové cirkulace. Míra biologické dostupnosti léčiva po perorálním podání je závislá na celé řadě skutečností, mezi něž je možné zařadit fyzikálně-chemické vlastnosti léčiva, fyziologické aspekty organismu, typ lékové formy, současný příjem potravy, biorytmy aintra - ainterindividuální rozdíly v lidské populaci. Tento článek je prvním ze série zabývající se biologickou dostupností ametodami jejího zlepšování, především uléčiv s nízkou rozpustností ve vodě. Cílem práce je přinést přehled faktorů, které mohou mít vliv na míru biologické dostupnosti léčiva po jeho perorálním podání. Následující články pak nabídnou popis, zde jen souhrnně vyjmenovaných, metod používaných při snaze zlepšit biologickou dostupnost špatně rozpustných účinných látek.
Klíčová slova:
biologická dostupnost • rozpustnost léčiv • špatně rozpustná léčiva • faktory ovlivňující biologickou dostupnost
Úvod
V současné farmakoterapii narůstá množství účinných látek, které mají po perorálním podání nedostačující biologickou dostupnost. Odhaduje se, že do této kategorie spadá až 40 % dnes běžně používaných léčiv a až 70 % nově syntetizovaných1). Doposud se nízká biologická dostupnost těchto léčiv řešila především zvyšováním jejich rozpustnosti fyzikální či chemickou úpravou dané účinné látky. Tyto zásahy jsou obecně značně problematické a nákladné, a často vedou k problémům se stabilitou, registrací, případně aplikačním komfortem podávaného přípravku. Z tohoto důvodu se jedním z trendů moderní farmaceutické technologie stala příprava lékových forem zajišťujících zvýšení biologické dostupnosti problematických léčivých látek, které zároveň poskytují vhodnou stabilitu a aplikační komfort přípravku. V odborné literatuře je popsáno několik postupů vedoucích ke zvýšení rozpustnosti, respektive biologické dostupnosti, mezi které je možné mimo jiné zařadit např. mikronizaci2), přípravu nanokrystalů3), formulaci pevných disperzí4), nebo zapracování těžce rozpustného léčiva do pevné lékové formy v kapalné formě za pomoci samoemulgujících systémů5), či systémů kapalina v pevné fázi6).
Původně se jako biologická dostupnost (angl. bioavailability) označovala vlastnost tablet, dražé a tobolek dostatečně rychle uvolňovat obsažené léčivé látky tak, aby byly k dispozici pro absorpci v trávicím ústrojí. Tato definice dnes odpovídá spíše termínu farmaceutická dostupnost, zatímco v současnosti je význam pojmu biologická dostupnost dle FDA (The U. S. Food and Drug Administration) mnohem širší a udává rychlost a rozsah, v nichž je léčivá látka absorbována a dosáhne místa svého účinku. Protože změření koncentrace léčiva v místě jeho účinku (např. na receptorech) je značně obtížné, je biologická dostupnost častěji definována jako rychlost a rozsah, ve kterých dosáhne účinná látka systémové cirkulace7–9).
Faktory ovlivňující biologickou dostupnost
Po perorálním podání, které je v současnosti nejrozšířenější a nejoblíbenější aplikační cestou většiny systémově působících léčiv, se na rozsahu, v jakém se látka dostane do systémového oběhu, podílí řada faktorů a procesů (obr. 1). Mezi faktory ovlivňující absorpci léčiv lze zařadit: fyzikálně-chemické faktory, které ovlivňující transport přes membrány, lokální podmínky měnící v místě absorpce rozpustnost léčiva, typ lékové formy, příjem a druh potravy, biorytmy a intra - a interindividuální rozdíly v lidské populaci. V rámci farmaceutické technologie je pak možné biologickou aktivitu léčivé látky ovlivnit na úrovni farmaceutické dostupnosti, tedy rychlosti rozpadu lékové formy, nebo rychlosti uvolňování a rozpouštění léčiva v gastrointestinálním traktu (GIT), a do jisté míry také na úrovni absorpce.
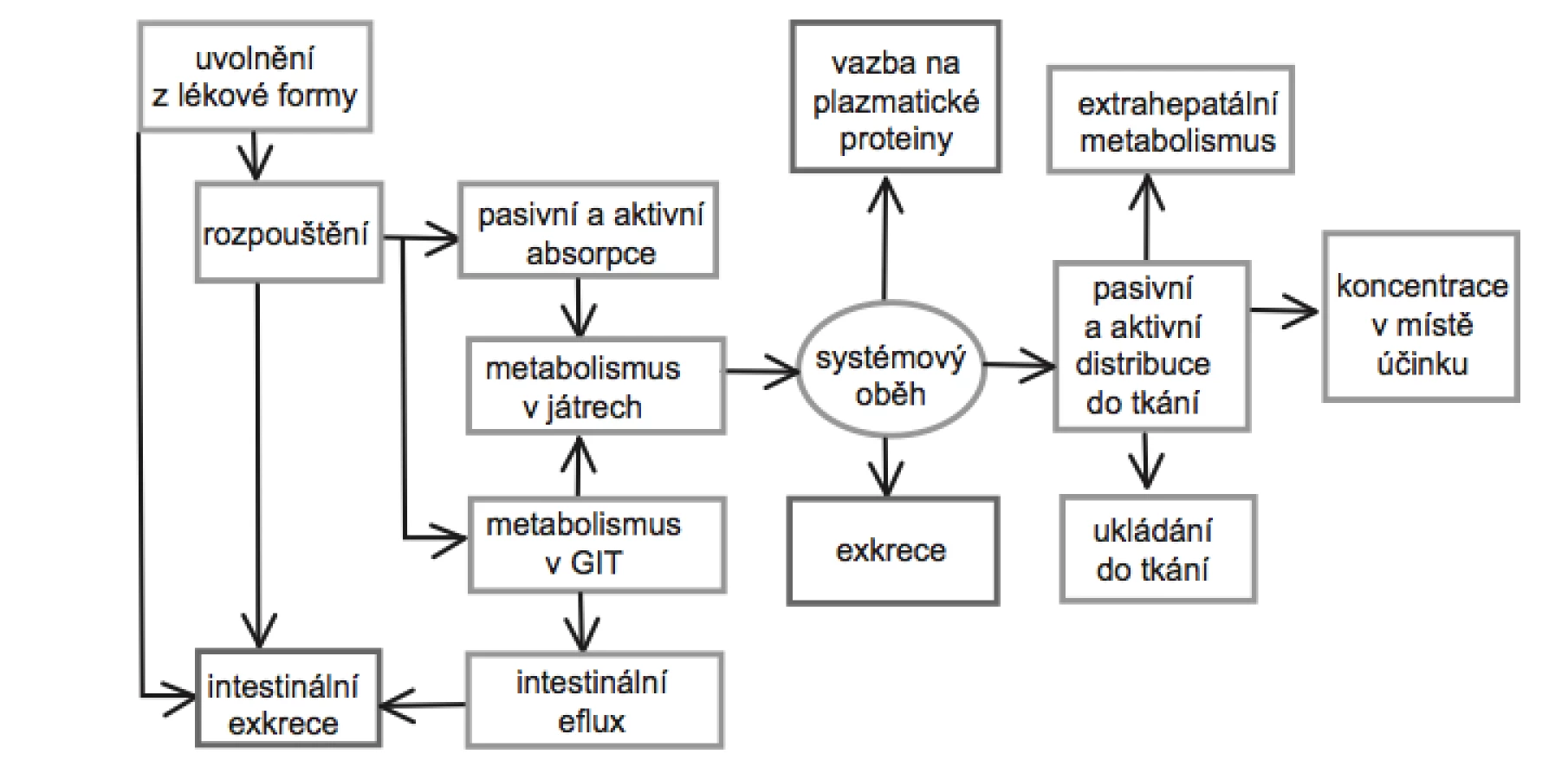
Fyzikálně-chemické vlastnosti léčiva
Mezi fyzikálně-chemické vlastnosti léčiv, které ovlivňují jejich biologickou dostupnost, je možné zařadit rozpustnost ve vodě a v tucích, acidobazické vlastnosti (hodnoty pH a pKa), rozdělovací koeficient, molekulovou hmotnost, tvar molekuly a míru schopnosti vázat se na plazmatické proteiny9, 11). Na důležitost fyzikálně-chemických vlastností léčivé látky poukazuje Lipinského „pravidlo tří pětek“ 12). Podle tohoto pravidla je léčivo obvykle těžce vstřebatelné, pokud je jeho molekulová hmotnost větší než 500, dekadický logaritmus rozdělovacího koeficientu oktanol/voda je větší než 5 a molekula má 5 a více vodíkových donorů12, 13).
Rozpustnost
Rozpustnost léčiva ve vodě patří mezi nejdůležitější parametry ovlivňující dosažení jeho požadované koncentrace v systémové cirkulaci, která je schopná v organismu vyvolat zamýšlenou farmakologickou odpověď. Z fyzikálně-chemického hlediska je možné léčiva rozdělit dle platného Lékopisu do sedmi skupin na základě jejich rozpustnosti14). Toto členění je však z farmakologického hlediska obvykle nedostačující, neboť zde není zohledněna terapeutická dávka léčiva. V případě některých látek je možné získat farmakologickou odpověď již po podání velmi malého množství léčiva, zatímco u jiných je podání vyšší dávky nezbytné. Z toho vyplývá, že i nízká dávka špatně rozpustného léčiva se může v GIT zcela rozpustit, zatímco vyšší dávka relativně dobře rozpustné léčivé látky se zde naopak rozpustit nemusí13).
Z tohoto důvodu byl vytvořen biofarmaceutický klasifikační systém (BCS), který rozděluje léčivé látky do čtyř tříd na základě jejich rozpustnosti ve vodném prostředí a gastrointestinální prostupnosti (permeabilitě) (obr. 2). Za vysoce rozpustná léčiva se v rámci BCS považují ta, jejichž nejvyšší dávka se rozpustí v 250 ml vodného pufru obvykle v rozmezí pH 1–8, zatímco jako vysoce prostupné se označují látky, které mají u člověka míru absorpce z GIT nejméně 90 % z podané dávky15).
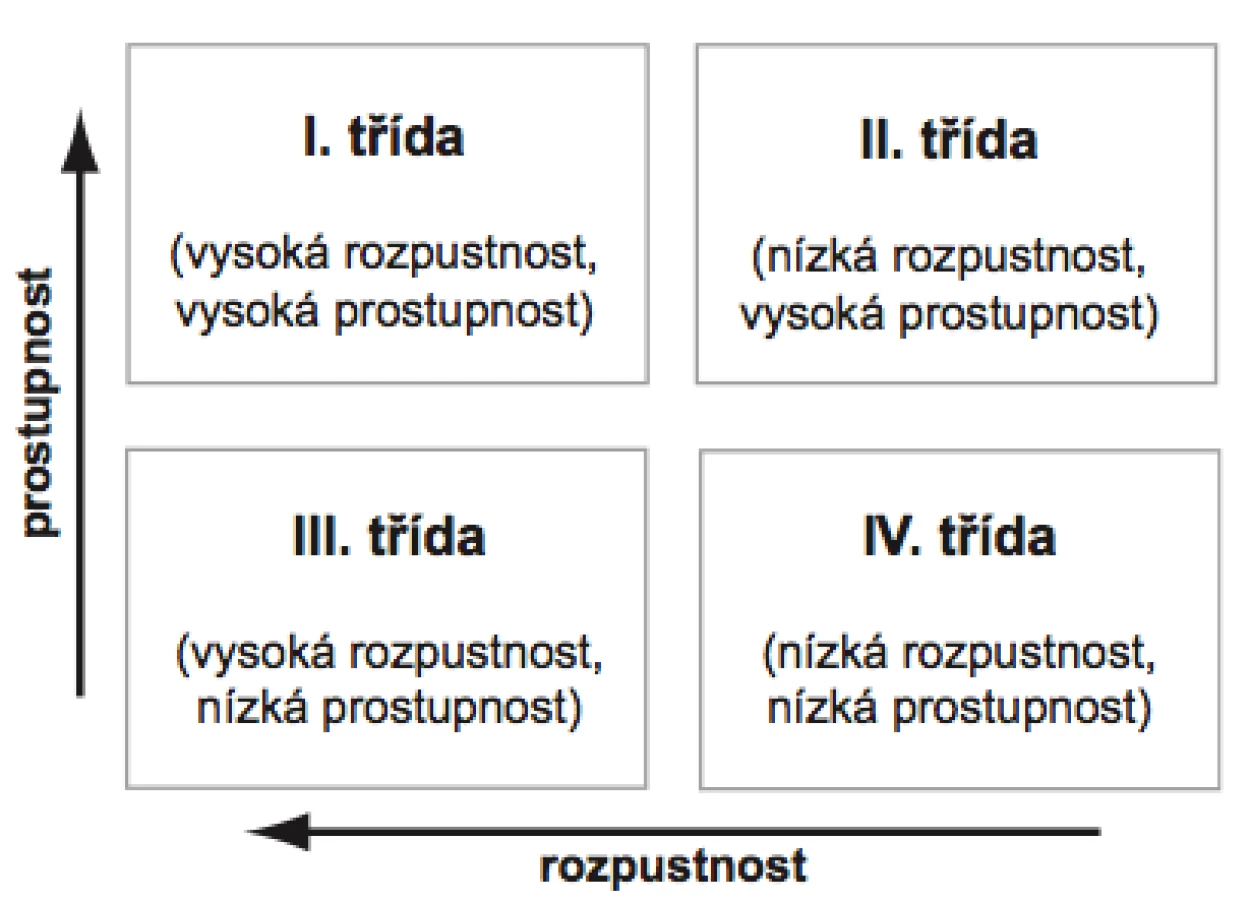
BCS byl navržen již v devadesátých letech 20. století Amidonem et al.16) se záměrem usnadnit registraci nových přípravků díky možnosti záměny in vivo bioekvivalenčních studií za specifické in vitro zkoušky, ze kterých je možné předvídat chování přípravku in vivo za předpokladu, že se jedná o lékovou formu s běžným uvolňováním léčivé látky (více než 85 % uvolněné látky během prvních 30 minut) určenou pro systémovou absorpci17).
Rozpustnost léčiva v tucích naopak určuje jeho schopnost difundovat přes lipofilní membrány. Míra absorpce pasivním transportem, respektive prostou difuzí, klesá se snižující se rozpustnosti účinné látky v lipidech8, 18).
Acidobazické vlastnosti
Většina léčivých látek podávaných do organismu jsou slabé kyseliny nebo slabé báze, které se mohou vyskytovat v disociované či nedisociované formě, přičemž platí, že se lépe absorbují látky v nedisociované podobě. Struktura molekuly určuje, zda a v jakém prostředí zůstane léčivo natolik apolární (rozpustné v tucích), aby mohlo procházet lipofilními buněčnými membránami. Lipofilitu a hydrofilitu léčiva je možné charakterizovat pomocí disociační konstanty (pKa), tj. hodnoty pH, při němž je 50 % léčiva v ionizované (hydrofilní) podobě11, 19).
Rozdělovací koeficient
Rozdělovací koeficient (log P) mezi polární (vodnou) a nepolární fází (nejčastěji oktanol) udává poměr, v jaké se léčivo rozdělí mezi lipidovou dvojvrstvu biomembrány a vodnou fázi. Poměr koncentrace léčiva v lipofilní fázi ke koncentraci v hydrofilní fázi má z hlediska absorpce mít hodnotu okolo jedné, protože membrány, kterými léčivá látka prochází, mají hydrofilně-lipofilní charakter11, 19). V případě, že je léčivo příliš lipofilní, tzn., pokud je jeho rozdělovací koeficient vyšší než 5, může být zadrženo v buněčné membráně20.
Velikost a tvar molekuly
Rychlost difuze látky je ovlivněna velikostí její molekuly. Velké molekuly difundují výrazně pomaleji než molekuly malé. Většina terapeuticky používaných látek má molekulovou hmotnost v rozmezí 100–1000. Větší molekulovou hmotnost mají např. peptidy (až 10 000), trombolytické enzymy (50 000) či moderní biologická léčiva, jako jsou např. monoklonální protilátky cetuximab (170 000) a rituximab (144 187)8, 19, 21).
Vazba na plazmatické proteiny
Rozsah vazby na plazmatické bílkoviny je závislý na koncentraci reagujících látek a afinitě léčiva k proteinům. Uvádí se, že lipofilní látky mají obvykle vyšší tendenci vázat se na plazmatické bílkoviny (především albumin) než látky hydrofilní22). Tato vazba vzniká rychle a je reverzibilní, tzn., že každá změna koncentrace volného, nenavázaného léčiva bezprostředně vyvolá odpovídající změnu koncentrace navázané účinné látky. Vazba na plazmatické bílkoviny představuje depotní systém, který na jedné straně snižuje intenzitu účinku, avšak na straně druhé prodlužuje jeho trvání8, 23).
Faktory organismu
Vyprazdňování žaludku a motilita střev
Rychlost jakou je žaludek vyprazdňován a motilita střev výrazně ovlivňují rychlost a rozsah absorpce léčiva24). Odborná literatura udává rozdílné informace o tom, zda zpomalení vyprazdňování žaludku zvyšuje, či naopak snižuje míru absorpce léčiva, respektive jeho biologickou dostupnost. Například Maerrick et al.25) uvádí, že zpomalené vyprazdňování žaludku snižuje u pacientů terapeutickou odezvu levodopy. Naopak Alhamami24) ve své studii uvádí, že zpomalení vyprazdňování žaludku zvyšuje u králíků biologickou dostupnost salicylátu sodného. Z těchto údajů je možné vyvodit, že vliv rychlosti vyprazdňování žaludečního obsahu na biologickou dostupnost je závislý na místě absorpce léčiva v GIT. U látek vstřebávajících se v žaludku dochází ke zlepšení jejich biologické dostupnosti se sníženou rychlostí vyprazdňování, zatímco léčiva absorbující se ve střevech vykazují naopak snížení biologické dostupnosti24, 25).
Vyprazdňování žaludku je obvykle úzce spjato s příjmem potravy a je závislé především na11, 24, 26):
- objemu jídla – zvětšený objem zpočátku zvyšuje, později snižuje rychlost vyprazdňování žaludku
- složení stravy – tuky a bílkoviny zpomalují vyprazdňování žaludku
- viskozitě potravy – zvyšující se viskozita snižuje rychlost vyprazdňování
- teplotě stravy – teplá jídla urychlují vyprazdňování
- léčivých látkách – např. analgetika snižují, naopak NaHCO3 nebo ranitidin zvyšují rychlost vyprazdňování žaludku
- alkoholu – konzumace alkoholu snižuje rychlost vyprazdňování žaludku
Motilita obsahu střev pozitivně působí na rozpad kusových léků, rozpouštění i difuzi rozpuštěné léčivé látky směrem ke střevní membráně a ovlivňuje tak jeho absorpci. Na motilitu střev může mít vliv viskozita jídla (s vyšší viskozitou se snižuje motilita) nebo aplikace léčivých látek ze skupiny cholinolytik a spazmolytik (snižují motilitu) či prokinetik (zvyšují motilitu)11, 25).
pH prostředí žaludku a střev
Jak již bylo zmíněno dříve, rozpustnost a míra absorpce celé řady léčivých látek jsou závislé na pH prostředí. Jeho hodnota se však výrazně mění napříč celým GIT. pH žaludku se obvykle pohybuje v rozmezí 1,5–2,9 (na lačno), ale může stoupat až k hodnotám okolo 6,7 (po jídle)27, 28). V duodenu se pH pohybuje v rozmezí 6,0–6,527, 29), v jejunu okolo 6,829) a v ileu pak dosahuje až hodnoty 7,430). V tlustém střevě nabývá hodnot 5,5–8,0, přičemž nižší pH se objevuje na začátku kolonu a je způsobeno vznikem kyselých fermentačních produktů bakteriální flóry31).
Přítomnost enzymů
V odlišných částech GIT se vyskytuje rovněž rozdílné množství tekutin a trávicích enzymů. Žaludek produkuje denně 1,5–2,0 litry tekutiny obsahující kyselinu chlorovodíkovou, pepsin, mucin a žaludeční lipázu. Do tenkého střeva se pak denně dostane okolo 1,0–1,5 litru pankreatických šťáv s obsahem trávicích enzymů (jako je např. amyláza, trypsin, chymotrypsin, karboxypeptidáza, lipáza, esteráza a ribonukleáza) a 0,5–1 litru žluči obsahující žlučové soli, které napomáhají rozpouštění a absorpci lipofilních látek29).
Hlavním úkolem enzymů je natrávení základních složek potravy, jako jsou bílkoviny, sacharidy, tuky a nukleotidy, což je důvodem proč léčiva na bázi proteinu a genetických materiálů (pro genovou terapii) nemohou být podávána perorálně, pokud není použitá speciální technologie přípravy lékové formy, která jejich rozkladu zabraňuje20).
Metabolizace léčiva a first pass efekt
Jak ukazuje obrázek 1, prochází léčivo před dosažením systémové cirkulace, popřípadě místa svého účinku celou řadou procesů. Část léčiva podaného perorálně však vůbec není absorbována z důvodu chemické degradace, inaktivace díky vzniku vazeb nebo komplexů, mikrobiální biotransformace apod. Další část léčiva může být metabolizována v průběhu transportu přes stěnu GIT. Nezměněné léčivo, které se dostane do portální žíly, může být biotransformováno v játrech nebo navráceno zpět do střev biliární exkrecí. Z toho vyplývá, že množství léčiva, které dosáhne krevního oběhu, je mimo jiné také závislé na míře metabolizace v GIT a first pass efektu při průchodu játry32).
Léková forma
V minulosti byla biologická dostupnost úzce, pokud ne výlučně, spojována s lékovou formou. Nyní je známo, že biologická dostupnost je více komplexní a je možné jí ovlivnit na více úrovních, avšak výběr vhodné lékové formy zůstává nezbytnou součástí zajištění terapeutického účinku léčiva32). Účinná látka podána ve formě vodného roztoku je absorbována rychleji než látka podána v olejovém roztoku nebo pevné lékové formě (např. tablet, tobolek aj.)33). V současné době je možné připravit lékovou formu, která obchází agresivní prostředí žaludku, řídí uvolňování léčiva nebo naopak urychluje jeho uvolňování a rozpouštění v gastrointestinálních tekutinách20).
Potrava
Biologická dostupnost je často ovlivněna konzumací potravy, a to především u léčiv s nízkou mírou absorpce. Jídlo může zasahovat nejen do rychlosti rozpadu lékové formy, uvolnění, rozpouštění (ovlivněním pH žaludku či střev, množstvím uvolněné žluči), vstřebávání léčiva (zvýšením prokrvení v místě absorpce) a průchodu léčiva trávicím traktem (především vyprazdňování žaludku), ale může mít také vliv na metabolickou přeměnu léčiv v GIT a v játrech34). Některé složky potravy mohou navíc s účinnou látkou vytvářet neabsorbovatelné nebo nerozpustné komplexy (např. tetracykliny a Ca2+), a tím rovněž snižovat její biologickou dostupnost11, 35).
Vliv potravy na míru biologické dostupnosti je velice komplexní, a proto jej není možné určit bez provedení studií na konkrétním léčivu34). Například Moses et al.36) prokázali, že jídlo výrazně snižuje biologickou dostupnost anticholinergika propantelinu. Naopak biologická dostupnost anthelmintika triclabendazolu se zvýšila až na dvojnásobek, pokud bylo léčivo podáno po jídle37).
FDA vydala seznam doporučení týkajících se testování vlivu příjmu potravy na biologickou dostupnost a bioekvivalenčních studií po podání jídla. Měření se doporučuje provádět jak nalačno (0,5 h před jídlem nebo 2 h po jídle), tak i po podání jídla. Strava pro provedení těchto studií by měla mít vysokou kalorickou hodnotu (800–1000 kcal) z toho 50 % tuků (500–600 kcal), přibližně 150 kcal proteinů a 250 kcal sacharidů35).
Biorytmy
Téměř všechny fyziologické funkce, včetně absorpce léčiva, jsou závislé na čase, přičemž nejvýznamnější jsou výkyvy v průběhu dne (cirkadiánní rytmy). Existence cirkadiánních rytmů byla prokázána u srdeční frekvence, tělesné teploty, krevního tlaku, prokrvení orgánů, hladiny hormonů v plazmě, koncentrace neurotransmiterů a druhých poslů (např. kortizolu, melatoninu, insulinu, prolaktinu apod.), množství kolujících červených a bílých krvinek a krevních destiček, funkce plic (minutový objem, usilovně vydechnutý objem) jater (metabolismus, průtok krve, first pass efekt) a ledvin (glomerulární filtrace, pH a objem moči, vylučování elektrolytů), gastrointestinální motility, kyselosti žaludku, času potřebného k vyprázdnění žaludku a řady dalších fyziologických funkcí. Rovněž nástup některých nemocí a jejích symptomů je závislý na denním rytmu, např. astmatické záchvaty jsou nejčastější kolem 4. hodiny ranní, nebo srdeční infarkty jsou nejpočetnější v ranních hodinách11, 38, 39). I s těmito faktory je tedy nutné počítat při předvídání biologické dostupnosti podaného léčiva.
Intra - a interindividuální variabilita organismů
Intra - a interindividuální rozdíly v účinnosti léčiv jsou významnou součástí variability jejich biologické dostupnosti. Z velké části se jedná o geneticky podmíněné změny v metabolizaci léčivých látek (např. aktivita N-acetyltransferázy, hydrolázy, genetický polymorfismus CYP2D6 apod.), na jejichž základě může být lidská populace rozdělena na tzv. rychlé, pomalé nebo intermediární metabolizátory. Na míře biologické dostupnosti se podílí rovněž věk pacienta. U dětských pacientů je biotransformace pomalejší než u pacientů dospělých. Naopak u starších osob dochází ke zpomalování metabolismu některých léčivých látek, snížení jaterního průchodu krve a snížení aktivity biotranformačních enzymů v játrech8, 40). K nejvýznamnějším interindividuálním rozdílům se řadí onemocnění, popřípadě souběžně probíhající další onemocnění (polymorbidita). Neméně se uplatňují i poruchy funkce orgánů, např. poruchy trávicího systémů, změny ve vazbě léčiv nebo snížená funkce eliminačních orgánů. Vedle těchto faktorů působí na míru biologické dostupnosti také vlivy vnějšího prostředí, např. stravovací návyky, kouření, lékové interakce a expozice toxickým látkám40).
Doposud byla provedena řada studií41–43) zabývající se vlivem intra - a interindividuální variability na biologickou dostupnost a účinek léčiva, ale jeho rozsah stále není možné plně předvídat. V případě vyšší pravděpodobnosti výskytu této variability je vhodné zvážit možnost individualizované terapie.
Metody zvyšování biologické dostupnosti
V současnosti se používá řada metod s cílem zajistit zvýšenou biologickou dostupnost podané účinné látky. Tyto lze rozdělit do tří základních skupin:
1. Chemické metody úpravy léčivé látky
- soli13, 44)
- hydráty13)
- glykosylované deriváty45)
- kokrystaly46, 47)
- proléčiva48)
- chelatace49)
2. Fyzikální metody
- krystalický polymorf nebo amorf50–53)
- řízená krystalizace (sonokrystalizace, krystalizace ze superkritických médií)54–57)
- lyofilizace58, 59)
- sprejové sušení60–63)
- mikronizace léčivé látky53, 64, 65)
- nanonizace66)
3. Technologické možnosti zvyšování biologické dostupnosti léčiv
- zprostředkované rozpouštění13)
- zvýšení smáčivosti67–69)
- micelární solubilizace69–71)
- použití kosolventů72–75)
- hydrotropní látky76, 77)
- tvorba cyklodextrinových komplexů44, 53, 69, 78)
- změna pH74, 79, 80)
- příprava pevných disperzí81, 82)
- užití interaktivní práškové směsi83, 84)
- mikrogranulace80, 85)
- impregnace86)
- samoemulgující systémy87)
- mikro - a nanosuspenze88)
- formulace liquisolid systémů89, 90)
- nanočásticové systémy91)
- lipozomální formulace92)
- použití enhancerů absorpce93, 94)
Střet zájmů: žádný.
Došlo 21. ledna 2015
Přijato 16. února 2015
PharmDr. Jan Gajdziok, Ph.D. • B. Vraníková
Veterinární a farmaceutická univerzita Brno, Farmaceutická fakulta, Ústav technologie léků
Palackého třída 1/3, 612 42 Brno
e-mail: gajdziokj@vfu.cz
Sources
1. Kawabata Y., Wada K., Nakatani M., Yamada S., Onoue S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. Int. J. Pharm. 2011; 420, 1–10.
2. Rasenack N., Müller B. W. Dissolution rate enhancement by in situ micronization of poorly water-soluble drugs. Pharm. Res. 2002; 19, 1894–1900.
3. Keck C. M., Müller R. H. Drug nanocrystals of poorly soluble drugs produced by high pressure homogenization. Eur. J. Pharm. Biopharm. 2006; 62, 3–16.
4. Vasconcelos T., Sarmento B., Costa P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007; 12, 1068–1075.
5. Gursoy R. N., Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomedicine and Pharmacotherapy. 2004; 58, 173–182.
6. Vraníková B., Gajdziok J., Vetchý D., Kratochvíl B., Seilerová L. Systémy kapalina v pevné fázi jako moderní trend zvyšování biologické dostupnosti léčiva. Chem. Listy 2013; 107, 681–687.
7. Lüllmann H., Mohr K., Wehling M. Farmakologie a toxikologie. 2. vydání Praha: Grada Publishing 2004.
8. Lincová, D., Farghali, H. Základní a aplikovaná farmakologie. 2. vydání. Praha: Galén 2007.
9. Allam A. N., El Gamal, S. S., Naggar V. F. Bioavailability: a pharmaceutical review. Int. J. Nov. Drug Deliv. Tech. 2001; 1, 80–96.
10. van de Waterbeemd H., Testa B., Mannhold R., Kubinyi H., Folkers G. Drug Bioavailability: Estimation of solubility, Permeability, Absorption and Bioavailability. 2nd Edition. Weinheim: John Wiley & Sons 2009.
11. Komárek P., Rabišková M. Technologie léků. 3. přepracované a doplněné vydání. Praha: Galén 2006.
12. Lipinski C. A., Lombardo F., Dominy B. W., Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001; 46, 3–26.
13. Okáčová L., Vetchý D., Franc A., Rabišková M., Kratochvíl B. Zvýšení biodostupnosti těžce rozpustných léčivých látek jejich modifikací. Chem. Listy 2010; 104, 21–26.
14. Český lékopis 2009. 1. vydání. Praha: Grada Publishing 2009.
15. Yazdanian M., Briggs K., Jankovsky C., Hawi A. The „high solubility“ definition of the current FDA guidance on biopharmaceutical classification system may be too strict for acidic drugs. Pharm. Res. 2004; 21, 293–299.
16. Amidon G. L., Lennernäs H., Shah V. P., Crison J. R. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995; 12, 413–420.
17. Chavda H. V., Patel C. N., Anand I. S. Biopharmaceutics classification system. Syst. Rev. Pharm. 2010; 1 : 62–69.
18. van De Waterbeemd H., Smith D. A., Beaumont K., Walker D. K. Property-based design: optimization of drug absorption and pharmacokinetics. J. Med. Chem. 2001; 44, 1313–1333.
19. Dostálek M. Farmakokinetika. Praha: Grada Publishing 2006.
2 0. Li X., Hu M. Oral bioavailability: basic principles, advanced concepts, and applivations. 1st edition. Weinheim: John Wiley & Sons 2011.
21. Farsa O. Terapeutické monoklonální protilátky v léčbě a ve vývoji. Chem. Listy 2013; 107, 464–470.
22. Kratochwil N. A., Huber W., Müller F., Kansy M., Gerber P. R. Predicting plasma protein binding of drugs: a new approach. Biochem. Pharmacol. 2002; 64, 1355–1374.
23. Lüllmann, H., Mohr, K., Hein, Lutz. Barevný atlas farmakologie. 3. vyd. Praha: Grada Publishing 2007.
24. Alhamami O. M. Delay in gastric emptying rate enhances bioavailability of sodium salicylates in rabbit. Arch. Pharm. Res. 2007; 30, 1144–1148.
25. Mearrick P. T., Wade D. N., Birkett D. J., Morris J. Metoclopramide, gastric emptying and L-dopa absorption. Aust. N. Z. J. Med. 1974; 4, 144–148.
26. Amir I., Anwar N., Baraona E., Lieber C. S. Ranitidine increases the bioavailability of imbibed alcohol by accelerating gastric emtying. Life Sci. 1996; 58, 511–518.
27. Dressman J. B., Berardi R. R., Dermentzoglou L. C., Russell T. L., Schmaltz S. P., Barnett J. L., Jarvenpaa K. M. Upper gastrointestinal (GI) pH young healthy men and women. Pharm. Res. 1990; 7, 756–761.
28. Augustijns P., Brewster M. Solvent systems and their selection in pharmaceutics and biopharmaceutics. Berlin: Springer Science & Business Media 2007.
29. Zhou D., Qiu Y. Oral Absorption and the Biopharmaceutics Classification System. Journal of validation technology 2009; 15, 62–72.
30. Fallingborg J. Intraluminal pH of the human gastrointestinal tract. Dan. Med. Bull. 1999; 46, 183–196.
31. Dvořáčková K., Franc A., Kejdušová M. Směrování léčiv do tlustého střeva. Chem. Listy 2013; 107, 522–529.
32. Kwan K. C. Oral bioavailability and first-pass effects. Drug Metab. Dispos. 1997; 25, 1329–1336.
33. Levy G., Gumtow R. H., Rutowski J. M. The Effect of Dosage form upon the Gastrointestinal Absorption Rate of Salicylates. Can. Med. Assoc. J. 1961; 85, 414–419.
34. Melander A. Influence of food on the bioavailability of drugs. Clin. Pharmacokinet. 1978; 3, 337–351.
35. Guidance for Industry: Food-Effect Bioavailability and Fed Bioequivalence Studies. http://www.fda.gov/downloads/Regulatory Information/Guidances/UCM126831.pdf. (12. 1. 2015).
36. Moses D. K., Charles B. G., Ravenscroft P. J., Whyte I. M. Food reduces the oral bioavailability of propantheline bromide in healthy subjects. Br. J. Clin. Pharmacol. 1983; 16, 758–759.
37. Lecaillon J. B., Godbillon J., Campestrini J., Naquira C., Miranda L., Pacheco R., Mull R., Poltera A. A. Effect of food on the bioavailability of triclabendazole in patients with fascioliasis. Br. J. Clin. Pharmacol. 1998; 45, 601–604.
38. Lemmer B. Relevance for chronopharmacology in practical medicine. Semin. Perinatol. 2000; 24, 280–290.
39. Griffett K., Burris T. P. The mammalian clock and chronopharmacology. Bioorg. Med. Chem. Lett. 2013; 23, 1929–1934.
40. Ptáček R., Bartůněk P. a kol. Etické problémy medicíny na prahu 21. století. 1. vydání. Praha: Grada Publishing 2014.
41. Kovarik J. M., Mueller E. A., van Bree J. B., Tetzloff W., Kutz K. Reduced inter - and intraindividual variability in cyclosporine pharmacokinetics from a microemulsion formulation. J. Pharm. Sci. 1994; 83, 444–446.
42. Lebbe C., Beyeler C., Gerber N. J., Reichen J. Intraindividual variability of the bioavailability of low dose methotrexate after oral administration in rheumatoid arthritis. Ann. Rheum. Dis. 1994; 53, 475–477.
43. Grahnén A., Hammarlund M., Lundqvist T. Implications of intraindividual variability in bioavailability studies of furosemide. Eur. J. Clin. Pharmacol. 1984; 27, 595–602.
44. Kataria M. K., Bhandari A. Solubility and dissolution enhancement: technologies and research emerged. Journal of Biological and Scientific Opinion 2013; 1, 105–116.
45. Kusumi S., Tomono S., Okuzawa S., Kaneko E., Ueda T., Sasaki K., Takahashi D., Toshima K. Total synthesis of vineomycin B2. J. Am. Chem. Soc. 2013; 135, 15909–15912.
46. Vishweshwar P., McMahon J. A., Bis J. A., Zaworotko M. J. Pharmaceutical Co-Crystals. J. Pharm. Sci. 2006; 95, 499–516.
47. Hetal T., Bindesh P., Sneha T. A review on techniques for oral bioavailability enhancement of drugs. International Journal of Pharmaceutical Sciences Review and Research 2010; 4, 203–223.
48. Offermanns S., Rosenthal W. Encyclopedia of molecular pharmacology. 2nd edition. Berlin: Springer Science & Business Media 2008.
49. Djokic S., Vajtner Z., Krnjevic H., Lopotar N., Kolacny-Babic L. Complexes and chelates of azithromycin with bivalent and/or trivalent metals and their use as antiulcer. 1990; US5498699A.
50. Hancock B. C., Parks M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm. Res. 2000; 17, 397–404.
51. Pudipeddi M., Serajuddin A. T. M. Trends in solubility of polymorphs. J. Pharm. Sci. 2005; 94, 929–939.
52. Kaur J., Aggarwal G., Gurpreet S., Rana A. C. Improvement of drug solubility using solid dispersion. International Journal of Pharmacy and Pharmaceutical Sciences. 2012; 4, 47–53.
53. Gowardhane A. P., Kadam N. V., Dutta S. Review on enhancement of solubilisation precess. Journal of Pharmacy and Phytotherapeutics 2013; 2, 28–38.
54. Maheshwari M., Jahagirdar H., Paradkar A. Melt sonocrystallization of ibuprofen: Effect on crystal properties. European Journal of Pharmaceutical Sciences. 2005; 25, 41–48.
55. Chuchvalec P., Novák J. P. Kritické veličiny látek a jejich predikce. Chem. Listy 2007; 101, 989–993.
56. Kim J., Kim H., Ju C. Micronization and characterization of drug substances by RESS with supercritical CO2. Korean J. Chem Eng. 2010; 27, 1139–1144.
57. Sander J. R. G., Zeiger B. W., Suslick K. S. Sonocrystallization and sonofragmentation. Ultrason. Sonochem. 2014; 21, 1908–1915.
58. Nireesha G., Divya L., Sowmya C., Venkateshan N., Babu M. N., Lavakumar V. Lyophilization/freeze drying – an review. International Journal of Novel Trends in Pharmaceutical Sciences 2013; 3, 87–98.
59. Yasmin R., Tan A., Bremmell K. E., Prestidge C. A. Lyophilized silica lipid hybrid (SLH) carriers for poorly water-soluble drugs: physicochemical and in vitro pharmaceutical investigations. J. Pharm. Sci. 2014; 103, 2950–2959.
60. Sahoo N. G., Abbas A., Judeh Z., Li C. M., Yuen K. H. Solubility enhancement of a poorly water-soluble anti-malarial drug: Experimental design and use of a modified multifluid nozzle pilot spray drier. J. Pharm. Sci. 2009; 98, 281–296.
61. Sollohub K., Cal K. Spray drying technique: II. current applications in pharmaceutical technology. J. Pharm. Sci. 2010; 99, 587–597.
62. Walters R. H., Bhatnagar B., Tchessalov S., Izutsu K., Tsumoto K., Ohtake S. Next generation drying technologies for pharmaceutical applications. J. Pharm. Sci. 2014; 103, 2673–2695.
63. Vandana K. R., Raju Y. P., Chowdary V. H., Sushma M., Kumar V. N. An overview on in situ micronization technique – an emerging novel concept in advanced drug delivery. Saudi Pharm. J. 2014; 22, 283–289.
64. Joshi J. T. A review on micronization techniques. Journal of Pharmaceutical Science and Technology 2011; 3, 651–681.
65. Bansal K., Pant P., Rao P. R. T., Padhee K, Sathapathy A., Kochhar P. S. Micronization and dissolution enhancement of norethindrone. International Journal of Research in Pharmacy and Chemistry 2011; 1, 315–319.
66. Chen H., Khemtong C., Yang X., Chang X., Gao J. Nanonization strategies for poorly water-soluble drugs. Drug Discov. Today 2011; 16, 354–360.
67. Balakrishnan A., Rege B. D., Amidon G. L., Polli E. Surfactant-mediated dissolution: Contributions of solubility enhancement and relatively low micelle diffusivity. J. Pharm. Sci. 2004; 93, 2064–2075.
68. Bajaj H., Bisht S., Yadav M., Singh V. Bioavaolability enhancement: a review. International Journal of Pharma and Bio Sciences 2011; 2, 202–216.
69. Sikarra D., Shukla V., Kharia A. A., Chatterjee D. P. Techniques for solubility enhancement of poorly soluble drugs: an overview. Journal of Medical Pharmaceutical and Allied Sciences 2012; 1, 1–22.
70. Li P., Yhao L. Solubilization of flurbiprofen in pH-surfactant solutions. J. Pharm. Sci. 2003; 92, 951–956.
71. Rangel-Yagui C. O., Pessoa Jr. A., Taveres L. C. Micellar solubilization of drugs. J. Pharm. Pharm. Sci. 2005; 8, 147–163.
72. Zhu J., Bierwagen G. P. The surface chemistry of water-reducible polymer solutions/dispersions – 1. Surface tension behavior. Prog. Org. Coat. 1995; 26, 87–100.
73. Kawakami K., Oda N., Miyoshi K., Funaki T., Ida Y. Solubilization behavior of a poorly soluble drug under combined use of surfactants and cosolvents. Eur. J. Pharm. Sci. 2006; 28, 7–14.
74. Vemula V. R., Lagishetty V., Lingala S. Solubility enhancement techniques. International Journal of Pharmaceutical Sciences Review and Research 2010; 5, 41–51.
75. Jangher A., Griffiths P. C., Paul A., King S. M., Heenan R. K., Schweins R. Polymeric micelle disruption by cosolvents and anionic surfactants. Colloid and Surface A. 2011; 391, 88–94.
76. Patil A. E., Devtalu S. V., Bari M. M., Barthel S. D. A review on: novel solubility enhancement technique hydrotropy. Indo American Journal of Pharmaceutical Research 2013; 3, 4670–4679.
77. Kumar V. S., Raja C., Jayakumar C. A review on solubility enhancement using hydrotropic phenomena. International Journal of Pharmacy and Pharmaceutical Sciences 2014; 6, 1–7.
78. Terao K., Nakata D., Fukumi H., Schmid G., Arima H., Hirayama F., Uekama K. Enhancement of oral bioavailability of coenzyme Q10 by complexation with γγ-cyclodextrin in healthy adults. Nutr. Res. 2006; 26, 503–508.
79. Dvořáčková K. Principy uvolňování léčiv z perorálních matricových tablet obsahujících hypromelosu. Chem. Listy 2009; 103, 66–72.
80. Okáčová L., Vetchý D., Franc A, Rabišková M. Zvýšení biodostupnosti těžce rozpustných léčivých látek technologickými postupy usnadňujícími jejich rozpouštění. Chem. Listy 2011; 105, 34–40.
81. Sinha S., Ali, M., Baboota S., Ahuja A., Kumar A., Ali J. Solid dispersion as an approach for bioavailability enhancement of poorly water-soluble drug ritonavir. AAPS PharmSciTech. 2010; 11, 518–527.
82. Baek H. H., Kim D. H., Kwon S. Y., Rho S. J., Kim D. W., Choi H. G., Kim Y. R., Yong C. S. Development of novel ibuprofen-loaded solid dispersion with enhanced bioavailability using cycloamylose. Arch. Pharm. Res. 2012; 35, 683–689.
83. Zatloukal Z. Interaktivní práškové směsi. Čes. slov. Farm. 2004; 53, 165–171.
84. Allahham A., Stewart P. J. Enhancement of the dissolution of indomethacin in interactive mixtures using added fine lactose. Eur. J. Pharm. Biopharm. 2007; 67, 732–742.
85. Watano S., Imada Y., Hamada K., Wakamatsu Y., Tanabe Y., Dave R. N., Pfeffer R. Microgranulation of fine powders by a novel rotating fluidized bed granulation. Powder Technol. 2003; 131, 250–255.
86. Planinšek O., Kovačič B., Vrečer F. Carvediol dissolution improvement by preparation of solid dispersions with porous silica. Int. J. Pharm. 2011; 406, 41–48.
87. Krupa A., Jachowicz R., Kurek M., Figiel W., Kwiecień M. Preparation of solid self-emulsifying drug delivery systems using magnesium aluminometasilicates and fluid-bed coating proces. Powder Technol. 2014; 266, 329–339.
88. Yang H., Teng F., Wang P., Tian B., Lin X., Hu X., Zhang L., Zhang K., Zhang Y., Tang Y. Investigation of a nanosuspension stabilized by Soluplus® to improve bioavailability. Int. J. Pharm. 2014; 477, 88–95.
89. Kulkarni S. A., Aloorkar N. H., Mane M. S. Liquisolid systems: a review. International Journal of Pharmaceutical Sciences and Nanotechnology 2010; 3, 795 – 802.
90. Kavitha K., Lova Raju K. N. S., Ganesh N. S., Ramesh B. Effect of dissolution rate by liquisolid compacts approach: an overview. Der Pharmacia Lettre 2011; 3, 71–83.
91. Shaikh J., Ankola D. D., Beniwal V., Singh D., Kumer M. N. Nanoparticle encapsulation improves oral bioavailability of curcumin by at least 9-fold when compared to curcumin administered with piperine as absorption enhancer. Eur. J Pharm. Sci. 2009; 37, 223–230.
92. Chen Y., Lu Y., Chen J., Lai J., Sun J., Hu F., Wu W. Enhanced bioavailability of the poorly water-soluble drug fenofibrate by using liposomes containing a bile salt. Int. J. Pharm. 2009; 376, 153–160.
93. Petersen S. B., Nolan G., Maher S., Rahbek U. L., Gultdbrandt M., Brayden D. J. Evaluation of alkylmaltosides as intestinal permeation enhancers: comparison between rat intestinal mucosal sheets and Caco-2 monolayers. Eur. J. Pharm. 2012; 47, 801–712.
94. Thanou M., Verhoef J. C., Junginger H. E. Chitosan and its derivatives as intestinal absorption enhancers. Adv. Drug Deliv. Rev. 2001; 50, S91–101.
Labels
Pharmacy Clinical pharmacologyArticle was published in
Czech and Slovak Pharmacy

2015 Issue 1-2
Most read in this issue
- Biologická dostupnost léčiva a možnosti jejího ovlivňování
- Účinnost fytoterapie v podpůrné léčbě diabetes mellitus typu 2 Borůvka černá (Vaccinium myrtillus)
- Užívání vybraných OTC přípravků: srovnání Řecko a Česká republika
- Vliv velikosti otvoru kónické testovací násypky na parametry rovnice sypání sorbitolu a jeho velikostních frakcí