
Zvyšování biologické dostupnosti léčiv pomocí formulace liquisolid systémů
Enhancing of drug bioavailability using liquisolid system formulation
One of the modern technologies of how to ensure sufficient bioavailability of drugs with limited water solubility is represented by the preparation of liquisolid systems. The functional principle of these formulations is the sorption of a drug in a liquid phase to a porous carrier (aluminometasilicates, microcrystalline cellulose, etc.). After addition of further excipients, in particular a coating material (colloidal silica), a powder is formed with the properties suitable for conversion to conventional solid unit dosage forms for oral administration (tablets, capsules). The drug is subsequently administered to the GIT already in a dissolved state, and moreover, the high surface area of the excipients and their surface hydrophilization by the solvent used, facilitates its contact with and release to the dissolution medium and GI fluids. This technology, due to its ease of preparation, represents an interesting alternative to the currently used methods of bioavailability improvement. The article follows up, by describing the specific aspects influencing the preparation of liquid systems, on the already published papers about the bioavailability of drugs and the possibilities of its technological improvement.
Key words:
liquisolid systems • bioavailability • porous carrier • coating material • preformulation studies
:
Jan Gajdziok; Barbora Vraníková
:
Ústav technologie lékÛ
; Veterinární a farmaceutická univerzita Brno, Farmaceutická fakulta
:
Čes. slov. Farm., 2015; 64, 55-66
:
Review Articles
Jednou z moderních technologií jak zajistit dostatečnou biologickou dostupnost léčiv s omezenou rozpustností ve vodném prostředí představuje příprava systémů kapalina v pevné fázi, neboli liquisolid systémů. Funkčním principem těchto formulací je nasorbování léčiva v kapalné formě na porézní nosič (aluminometasilikáty, mikrokrystalická celulosa atd.). Po přidání dalších pomocných látek, především obalovacího materiálu (koloidní oxid křemičitý), dochází ke vzniku prášku s vlastnostmi vhodnými pro převod na běžné jednotkové pevné lékové formy pro perorální podání (tablety, tobolky). Léčivo je v pak do organismu podáváno již v rozpuštěném stavu a navíc vysoký měrný povrch použitých excipientů a hydrofilizace jejich povrchů použitým rozpouštědlem usnadňuje jeho kontakt s a přechod do disolučního média i trávicích tekutin. Tato technologie představuje pro svou jednoduchost přípravy zajímavou alternativu k v současnosti používaným postupům. Článek navazuje popisem konkrétních aspektů ovlivňujících přípravu systémů kapalina v pevné fázi na již publikované práce o biologické dostupnosti léčiv a možnostech jejího technologického ovlivňování.
Klíčová slova:
liquisolid systémy • systémy kapalina v pevné fázi • biologická dostupnost • porézní nosič • obalovací materiál • preformulační studie
Úvod
Možnosti zlepšování nedostatečné biologické dostupnosti moderních léčiv, které v drtivé většině případů náleží dle biofarmaceutického klasifikačního systému do II. nebo IV. skupiny (látky s nízkou rozpustností a často i omezenou permeabilitou)1), patří mezi jedno z nejdiskutovanějších témat současné farmaceutické technologie. Jak popisuje předcházející článek2), mezi nejběžněji používané přístupy patří využívání zprostředkovaného rozpouštění a zvyšování smáčivosti3–5), micelární solubilizace léčiv5–7), použití kosolventů8–11), využití efektu hydrotropismu12, 13), tvorba cyklodextrinových komplexů5, 14–16), změna pH10, 17, 18), příprava pevných disperzí19, 20), užití interaktivní práškové směsi21, 22), mikrogranulační technologie18, 23), impregnace24), příprava samoemulgujících systémů25) nebo formulace mikro - a nanosuspenzí26), nanočásticové systémy27), lipozomální formulace28) a další. Jednou z průmyslově nejzajímavějších metod použitelných pro zvýšení biologické dostupnosti léčiva představuje formulace tzv. systémů kapalina v pevné fázi neboli liquisolid systémů.
Liquisolid systémy (LSS) jsou moderní lékové formulace schopné zajistit zvýšení biologické dostupnosti špatně rozpustných léčiv. Z historického hlediska se tyto systémy vyvinuly z tzv. práškových roztoků, které se získávají inkorporací léčiva rozpuštěného v netěkavém rozpouštědle do struktury inertního materiálu s velkým povrchem částic, jako je např. oxid křemičitý. Tyto přípravky však nebylo možné transformovat do jiné než práškové podoby, jelikož jejich vlastnosti neumožňovaly lisování do tablet ani jiné vhodné zpracování do finální jednotkové lékové formy. I přes snahu upravit lisovatelnost a tokové vlastnosti těchto systémů, nebyly práškové roztoky nikdy přizpůsobeny požadavkům farmaceutického průmyslu29, 30).
Systémy kapalina v pevné fázi jsou tedy technologickým vylepšením práškových roztoků. Základním principem jejich přípravy je nasorbování léčiva v kapalné fázi (roztok, suspenze, emulze nebo samoemulgující systém) na vysoce porézní nosič, který je následně obalen jemným materiálem s velkým povrchem částic (obalovací materiál), za vzniku suchého, volně tekoucího prášku s vlastnostmi vhodnými pro další zpracování (plnění do tobolek, lisování do tablet apod.)30).
Na zvyšování biologické dostupnosti léčiv formulovaných do podoby liquisolid systémů se podílí několik mechanismů. Prvním z nich je rozptýlení disperze léčiva na celém povrchu nosiče, díky čemu se léčivo snadněji uvolní z lékové formy. Navíc je vzhledem k přítomnosti obvykle hydrofilního rozpouštědla, nezbytného pro převedení účinné látky do kapalné podoby, zvýšena smáčivost přípravku disolučním médiem nebo gastrointestinální tekutinou. Hlavní princip zvyšování biologické dostupnosti pomocí těchto systémů však spočívá v přítomnosti léčiva v kapalné podobě, které se po podání do GIT již nemusí rozpouštět, čímž je obcházen často limitující krok absorpce léčiva, respektive jeho biodostupnosti31).
Oproti běžně používanému zapracování kapalných léčiv do pevné lékové formy (příprava želatinových perel, zatěsněných tvrdých tobolek) a ostatním metodám používaným ke zvyšování biologické dostupnosti léčiv se špatnou rozpustností poskytují LSS řadu výhod, mezi které lze zařadit: možnost zvýšení rozpustnosti léčiv omezeně rozpustných či prakticky nerozpustných ve vodě, jednoduchost zpracování, nízké výrobní náklady, možnost použití ve farmaceutickém průmyslu, minimalizaci vlivu pH na rychlost uvolňování léčiva, vyšší stabilitu přípravku, optimalizaci disolučního profilu a možnost přípravy lékové formy se zrychleným, konvenčním i řízeným uvolňováním léčivé látky32–37).
Hlavním omezením této technologie je požadavek na vysokou rozpustnost léčiva v netěkavém rozpouštědle v případě, že je použit roztok léčiva. Při použití většího množství léčiva v kapalné fázi může během lisování liquisolid prášků docházet k vytlačování kapalné fáze z tablet (tzv. liquid squeezing-out fenomén). Připravené tablety pak mají nevyhovující vzhled a mechanickou odolnost (pevnost, oděr). Mezi nevýhody této techniky je možné rovněž zařadit problematické zapracování vysokých dávek špatně rozpustných léčiv (např. karbamazepin, flutamid). K převedení těchto látek do kapalné fáze je zapotřebí větší množství netěkavého rozpouštědla, což má za následek navýšení množství nosiče a obalovacího materiálu potřebných pro přípravu prášku s vhodnými tokovými vlastnostmi a dobrou stlačitelností. To následně způsobí nárůst velikosti tablety nad hranici pohodlné aplikace (polykání). Jedním z možných řešení tohoto problému je použití nosičů s velkým povrchem a vysokou sorpční schopností (např. Neusilin®)36, 38–40).
V odborné literatuře již byla popsána řada těchto formulací se zapracovaným kapalným léčivem – klofibrát36), roztokem nebo suspenzí špatně rozpustného léčiva ve vybraném hydrofilním rozpouštědle – atorvastatin41), griseofulvin42), karbamazepin43), indomethacin39), apod. či kapalným samoemulgujícím systémem – cyklosporin A44). Navíc se touto technikou připravily i orálně dispergovatelné tablety s obsahem aceklofenaku45) a tablety s řízeným uvolňováním léčiv dobře rozpustných ve vodě – propranolol33), tramadol46), theofylin47).
Formulace liquisolid systémů
Příprava liquisolid systémů probíhá obvykle v několika krocích. Léčivo se nejdříve rozpustí nebo disperguje v požadovaném množství netěkavého rozpouštědla a vzniklá disperze se postupně rovnoměrně sorbuje na přesně vypočítané množství nosiče. Pro zajištění homogenního rozložení disperze léčiva je ji možné na částice nosiče nanést pomocí nástřiku ve fluidním zařízení nebo mísením ve vysokoobrátkových mixérech. Homogenizace probíhá ve třech základních fázích. Během první dochází k promíchávání směsi, aby se zajistilo homogenní rozložení léčiva v kapalné fázi v celém množství směsi práškového nosiče. Ve druhém kroku se vzniklá směs nechá ustát, aby došlo k dokonalé sorpci a rozložení léčiva v porézní struktuře nosného materiálu. Během třetí fáze homogenizace jsou částice nosiče s léčivem obaleny vhodným obalovacím materiálem a následně přidány pomocné látky nezbytné pro přípravu finální lékové formy (nejčastěji tablety)48).
Preformulační studie
Provedení preformulačních studií před samotnou přípravou liquisolid systémů je nezbytné pro získání práškové směsi s vhodnými tokovými vlastnostmi a následně finální formulace, která splňuje všechny požadavky kladené obecně na tablety, tobolky nebo granuláty. Tyto studie úzce souvisejí s výběrem vhodného netěkavého rozpouštědla k dispergaci léčiva a výpočtem správného množství práškových pomocných látek – nosiče a obalovacího materiálu49).
Důležitým parametrem, který je tedy nutné před samotnou přípravou LSS určit, je rozpustnost léčiva. Zkouška rozpustnosti léčiva se obvykle provádí připravením nasyceného roztoku v různých netěkavých rozpouštědlech pomocí přídavku nadbytku testované látky. Následně po ustavení rovnováhy a nasycení roztoku je množství rozpuštěné účinné látky kvantifikováno vhodnou analytickou metodou29, 35). Fyzikálně-chemické vlastnosti (polarita, viskozita, molekulová hmotnost, chemická struktura a lipofilita) zvoleného rozpouštědla mohou mít vliv na profil uvolňování léčiva z liquisolid tablet50). Obecně lze očekávat, že rozpouštědla s vyšší rozpouštěcí schopností pro zvolené léčivo jsou vhodnější pro formulaci LSS se zrychleným uvolňováním35).
Určení úhlu skluzu (angle of slide) – θ patří mezi specifické zkoušky používané k hodnocení tokových vlastností práškových materiálů51). Spireas et al.52) ve své práci považují tento test za preferovanou metodu k hodnocení toku prášků s částicemi menšími než 150 ∝m. Prakticky se úhel skluzu určuje pomocí umístění definovaného množství práškového materiálu na jeden konec vyleštěné nerezové destičky. Tento konec se následně postupně zvedá, dokud nedojde ke sklouznutí materiálu po destičce. Úhel, který svírá destička s vodorovným podstavcem, je pak označován jako úhel skluzu. Experimentálně bylo zjištěno, že jeho optimální hodnota pro další zpracování (lisování do tablet, plnění do tobolek) liquisolid směsi se pohybuje okolo 33° 35, 53).
Mezi důležité preformulační parametry při přípravě LSS patří tzv. retenční potenciál (tokový retenční potenciál Φ a lisovací retenční potenciál Ψ). Tokový retenční potenciál (flowable liquid retention potential) popisuje schopnost prášku zadržovat ve své struktuře specifické množství kapalné fáze při zachování akceptovatelných tokových vlastností. Hodnota tokového retenčního potenciálu se před formulací LSS určuje jak pro nosič, tak i obalovací materiál (ΦN – retenční potenciál nosiče, ФO – retenční potenciál obalovacího materiálu), a je definována jako maximální hmotnost kapaliny (max m), která může být zadržována jednotkovou hmotností práškového materiálu (N – nosič, O – obalovací materiál) při zachování dobrých tokových vlastností prášku s nasorbovanou kapalinou [1]40). Tokový retenční potenciál je závislý především na procesu absorpce (viskozitě, polaritě kapalné fáze, atd.) a struktuře použitých práškových materiálů54).

[1]
V průběhu tohoto testování se práškové pomocné látky (nosič, obalovací materiál) smíchávají s měnícím se množstvím vybraného netěkavého rozpouštědla a stanovují se úhly skluzu jednotlivých směsí. Poměr hmotnosti kapalina/pevná látka směsi s úhlem skluzu blízko 33° se považuje za tokový retenční potenciál daného práškového materiálu29, 32, 49, 55).
Dalším parametrem určovaným před zpracováním LSS je tzv. absorpční faktor (liquid load factor) – Lf, který je definován jako poměr mezi hmotností kapalné fáze (m) a hmotností nosiče (N) [2]. Na rozdíl od tokového retenčního potenciálu (Φ) zde m nepředstavuje maximální možné množství kapalné fáze, ale množství léčiva v kapalné fázi, které má být ve vztahu k dávce a koncentraci roztoku/disperze skutečně zapracováno do finální podoby jednotkové lékové formy. Tato hodnota může, ale nemusí odpovídat maximálnímu množství kapaliny nasorbovatelné daným nosičem40, 56).

[2]
Pro další výpočty teoretických aspektů souvisejících s formulací LSS je nutné najít správný poměr mezi nosičem a obalovacím materiálem (R). Tento poměr se určuje experimentálně pomocí stanovení úhlu skluzu s hodnotou 33° (nebo jiné tokové vlastnosti) pro směs nosič (N)/obalovací materiál (O) [3]. Experimentálně bylo ověřeno, že se tato hodnota pro nejčastěji používané nosiče a obalovací materiály pohybuje mezi 10–10048).

[3]
Tento poměr R, absorpční faktor při zachování dobrých tokových vlastností LfΦ a retenční potenciály ΦN a ФO dané formulace jsou ve vzájemném vztahu dle [4]29, 30, 35, 36):

[4]
Dle Spirease et al. může být hodnota Φ určena procesem, zvaným liquisolid flowability test (LSF)32). LSF test prakticky probíhá zpravidla tak, že se vytvoří několik práškových směsí skládajících se z 25–30 g nosiče a obalovacího materiálu zastoupených v různých poměrech R. Postupně se standardním mísením k systémům přidává rozpouštědlo, ve kterém se bude léčivo při přípravě liquisolid produktu rozpouštět. Po každém přídavku rozpouštědla se zaznamenávají tokové vlastnosti (nejčastěji úhel skluzu a hmotnostní rychlost sypání) a konzistence prášku pomocí vhodného zařízení. Nejvyšší hodnota poměru hmotností kapalné a pevné fáze (w/w) v každém ze systémů, která splňuje podmínku požadovaných tokových vlastností prášků, tzn. podle Spirease hmotnostní rychlost sypání ne horší než 4 g/s bez jakéhokoliv zadržování prášku v násypce a úhel skluzu okolo 33°, je brána jako absorpční faktor prášku při zachování jeho dobrých tokových vlastností (LfΦ). Matematickou úpravou se získají hodnoty tokových retenčních potenciálů (ΦN a ΦO) pro nosič a obalovací materiál [4]32).
Jako příklad využití hodnocení těchto parametrů lze uvést formulaci liquisolid tablet s obsahem rofekoxibu, u nichž se prokázalo, že hodnota absorpčního faktoru je nepřímo úměrná pevnosti vyrobených tablet. Tento jev lze vysvětlit tím, že zvyšující se absorpční faktor některých vzorků spojený s nutností použít vyšší množství špatně stlačitelného obalovacího materiálu vede ke snížené pevnosti tablet49). Stejný trend byl potvrzen ve studii týkající se hodnocení LSS tablet s obsahem griseofulvinu jako modelového léčiva. Vzorky s nižší hodnotou absorpčního faktoru vykazovaly vyšší pevnost tablet z důvodu vyššího zastoupení nosiče Avicelu® PH 102 vedoucího k lepší plastické deformovatelnosti tabletovací směsi a vzniku vodíkových můstků mezi částicemi57).
Test lisovatelnosti liquisolid systémů (liquisolid compressibility test – LSC) se používá k určení tzv. lisovacího retenčního potenciálu (compressible liquid retention potential – ψ) a k charakterizaci zhušťovacích a lisovacích vlastností připravených směsí58). Lisovací retenční potenciál lze popsat jako maximální hmotnost kapalné fáze, která může být zadržována práškovým materiálem (nosič, obalovací materiál) při zachování jeho dobrých lisovacích vlastností [5]. Je tedy jakousi obdobou tokového retenčního potenciálu (Φ), který ovšem souvisí s tokovými vlastnostmi materiálu. Celkově lze říci, že se jedná o množství tekutiny, která může být přítomna v tabletě s dostatečnou mechanickou odolností bez toho, aby v průběhu lisování docházelo k projevům vytlačování kapaliny ze struktury výlisku (liquid squeezing out fenomén)53).

[5]
Stejně jako závislost poměru excipientů práškové směsi R a absorpčního faktoru směsi LfΦ, při zachování dobrých tokových vlastností, jsou vyjádřeny vztahem rovnice [4], tak lze vypočítat obdobný absorpční faktor Lfψ pro LSS systémy s akceptovatelnou lisovatelností [6].

[6]
Pro stanovení hodnoty ψ pro nosič a obalovací materiál se připraví několik vzorků prášků obsahujících směs nosiče a obalovacího materiálu v různých poměrech R. K jednotlivým směsím se postupně přidává rozpouštědlo, ve kterém se bude léčivo při přípravě liquisolid produktu rozpouštět (obsah rozpouštědla = Cm). Po přídavku rozpouštědla se z jednotlivých směsí vylisují tablety určité hmotnosti s nejvyšší možnou pevností Sc a vypočítá se jejich kompaktnost (pactisity) Ω prezentovaná destrukční silou vyjádřenou na jednotku gramu tablety. Dále se určí vnitřní kompaktnost (intrinsic pactisity) Ω0 a definuje se index pórovitosti σi [7]. Z těchto veličin se vyjádří minimální hodnoty lisovacího retenčního potenciálu směsí ψmix [8], a z něho lze logaritmickým vyjádřením získat absorpční faktor směsi (Lfψ) při zachování její dobré stlačitelnosti [9]. Takto se postupuje u všech systémů s poměrem R. Matematickou úpravou hodnot Lfψ všech připravených systémů lze získat retenční potenciál Ψ pro nosič a obalovací materiál, kdy hodnota Ψ představuje obsah rozpouštědla Cm ve směsi, z níž se získají výlisky s kompaktností 20 kg/g32, 38, 59).

[7]
kde Ω je maximální destrukční síla přepočtena na 1 g tablety, Ω0 je vnitřní kompaktnost, σi je index pórovitosti, Cm je poměr hmotností kapalné/pevné fáze (obsah rozpouštědla):

[8]
kde Ψmix je lisovací retenční potenciál směsi, Ω0 je vnitřní kompaktnost, σi je index pórovitosti, 20 je hodnota kompaktnosti Ω = 20 kg/g.

[9]
kde Lψf je absorpční faktor směsi při její dobré stlačitelnosti, ψmix je lisovací retenční potenciál směsi, R je poměr hmotnosti nosiče a obalovacího materiálu.
Optimální absorpční faktor (LfO)
Výše uvedené postupy lze využít pro výpočet optimálního absorpčního faktoru LfO. Pro systém vykazující vhodné tokové vlastnosti a lisovatelnost vychází buď ze vztahu pro výpočet LfΦ, nebo Lψf podle toho, který udává nižší hodnotu.
Tímto lze dospět k výpočtu požadovaného množství nosiče (NO) a obalovacího materiálu (OO) nutného k přípravě směsi s vhodnými vlastnostmi nutnými k vylisování tablet, při přídavku daného množství kapalného léčiva (m) [10]50, 60).

kde NO je optimální množství nosiče (g), OO optimální množství obalovacího materiálu (g), m množství kapalného léčiva (g), R poměr hmotností nosiče a obalovacího materiálu (w/w).
Pomocné látky
Důležitou součástí formulace LSS je výběr vhodných pomocných látek. Kromě klasických pomocných látek běžně používaných pro přípravu pevných lékových forem (plniva, pojiva, kluzné a antiadhezivní látky atd.) se při přípravě liquisolid systémů uplatňují tři hlavní skupiny látek – netěkavá rozpouštědla, nosiče a obalovací materiály29).
Rozpouštědla
Během přípravy liquisolid systémů je možné použít řadu netěkavých, nízkoviskózních rozpouštědel, která jsou obvykle mísitelná s vodou. Bylo prokázáno, že výběr rozpouštědla má výrazný vliv na rychlost uvolňování léčiva z hotových LSS33, 38). Pro zrychlené uvolňování jsou volena rozpouštědla, ve kterých je léčivo co nejlépe rozpustné. V případě přípravy LSS s prodlouženým uvolňováním se používá rozpouštědlo s nízkou schopností rozpouštět vybrané léčivo48).
Podle jiné hypotézy však vyšší rozpustnost léčiva v daném rozpouštědle zvyšuje jeho schopnost vázat molekuly léčiva, a tím snížit množství uvolněné léčivé látky, resp. prodloužit dobu jejího uvolňování61). Adibkia et al.61) proto zkoumali vliv rozpouštědla na rychlost uvolňování léčiva z liquisolid tablet s obsahem diltiazemu. Ve své studii prokázali, že diltiazem je nejlépe rozpustný v polysorbátu 80 a nejméně pak v polysorbátu 20. Podle výsledků disolučního testu bylo uvolňování léčiva nejvíce prodlouženo právě u liquisolid tablet obsahujících jako kapalné vehikulum polysorbát 80 (48 % léčiva uvolněno po 7,5 hodinách), avšak druhé nejlepší výsledky vykazoval vzorek s obsahem polysorbátu 20 (67 % uvolněného léčiva po 7,5 hodinách). Z těchto výsledků vyplývá, že rychlost uvolňování léčiva je závislá na celé řadě faktorů, jako jsou např. polarita, viskozita, molekulová hmotnost, tvorba micel, dielektrická konstanta a hydrofilně-lipofilní rovnováha (HLB) rozpouštědla61, 62).
Mezi netěkavá rozpouštědla, která se často používají pro přípravu LSS, lze zařadit např. propylenglykol (PG), kapalné makrogoly (PEG 200–600), polysorbáty (Tweeny), glycerol, poloxamery (Synperonic® PE/L 61), glyceromakrogol-ricinoleát (Cremophor® EL), propylenglykol monokaprylát (Capryol® 90), makrogol-15-hydroxystearát (Solutol® HS 15) a další48, 63).
Propylenglykol je ve farmaceutickém průmyslu běžně používán jako stabilizátor vitaminů, zvlhčovadlo a kosolvent při přípravě mastí64). Hlavní úlohou propylenglykolu v přípravcích je rozpustit léčivo, a vytvořit tak homogenní disperzi aktivních látek ve formulaci65). Příkladem použití propylenglykolu pro přípravu systémů kapalina v pevné fázi mohou být liquisolid tablety s obsahem valsartanu. V této studii Chella et al. zvažovali několik rozpouštědel (PG, polysorbát 20 a 80, PEG 200, 400 a 600), přičemž nejlépe rozpustný byl valsartan v propylenglykolu, a proto bylo toto rozpouštědlo vybráno pro přípravu liquisolid tablet se zrychleným uvolňováním léčiva66).
Kapalné makrogoly (polyethylenglykoly) jsou široce používány jako rozpouštědla a látky zvyšující rozpustnost léčivých i pomocných látek v kapalných a polotuhých přípravcích. Pokud jsou použity spolu s emulgátory, mohou plnit rovněž funkci stabilizátorů emulgujících systémů. Nízká molekulová hmotnost polyethylenglykolů, především pak PEG 200, může mít za následek zvýšení rychlosti uvolňování léčiva, což je výhodné především během formulace systémů kapalina v pevné fázi se zrychleným uvolňováním léčiva. Naopak PEG s vyšší molekulovou hmotností mohou navýšit účinnost pojiv, a tím zpomalit rozpad tablet, resp. rychlost uvolňování léčiva59). Dle Jabbara et al. byla rozpustnost piroxikamu při snaze o jeho formulování do podoby LSS vyšší v PEG 400 ve srovnání s PG, což může být způsobeno přítomností delších nepolárních vazeb v PEG 400 a interakcí tohoto rozpouštědla s hydrofobní části léčiva67). Příkladem použití kapalných makrogolů pro přípravu liquisolid systémů jsou také tablety s obsahem griseofulvinu. V této studii se griseofulvin dispergoval v PEG 300 a následně smísil s nosičem a obalovacím materiálem. Zkouška disoluce tablet připravených z těchto směsí ukázala, že uvolňování léčiva ze všech připravených liquisolid systémů bylo rychlejší v porovnání s konvenčními tabletami. Z hodnocení dále vyplynulo, že rychlost uvolňování je vyšší s rostoucím množstvím léčiva rozpuštěného v PEG 300, přičemž nejrychlejší disoluční profil vykazovaly tablety obsahující již zcela rozpuštěný griseofulvin50).
Oxyethylenované sorbitanové estery vyšších mastných kyselin (polysorbáty, Tweeny) jsou široce používány ve farmaceutickém a kosmetickém průmyslu jako emulgátory, dispergující látky a stabilizátory, především díky jejich efektivitě v nízké koncentraci, inertnosti vůči většině aktivních substancí a relativně nízké toxicitě. Nejčastěji používaným sorbitanovým esterem pro přípravu LSS je polysorbát 8048). Dle Yadav et al. se při přípravě orálně dispergovatelných tablet obsahujících aceklofenak LSS technikou za použití polysorbátu 80 zlepšil disoluční profil tohoto léčiva a systém vykazoval také lepší tokové vlastnosti ve srovnání s ostatními rozpouštědly (PG nebo PEG 400)45). Polysorbát 80 se dále použil např. ve studii zabývající se přípravou liquisolid systémů s prodlouženým uvolňováním trimetazidinu. Pro jeho převedení do kapalné podoby se zvažovala tři rozpouštědla, polysorbát 80, glycerol a PEG 400, přičemž nejméně rozpustný byl trimetazidin právě v polysorbátu 80, díky čemuž byl zvolen jako nejvhodnější rozpouštědlo pro tuto formulaci. Výsledky disoluce ukázaly, že použití polysorbátu 80 v kombinaci s přídavkem Eudragitu® L-100 k nosiči (mikrokrystalická celulosa) je možné docílit prodlouženého uvolňování trimetazidinu z liquisolid tablet68).
Mahajan et al. testovali vliv netěkavého rozpouštědla na rychlost uvolňování glipizidu z LSS. Pro převedení léčiva do kapalné fáze se použily PG, PEG 200 a PEG 400. Ve srovnání s komerčně dostupnými tabletami došlo u všech tří rozpouštědel ke zrychlení uvolňování glipizidu z LSS tablet. Nejrychlejšího uvolnění bylo dosaženo u liquisolid tablet s obsahem PEG 400, ve kterém byl glipizid nejlépe rozpustný69).
Saeedi et al. použili glycerol a PEG 200 jako netěkavá rozpouštědla pro přípravu liquisolid výlisků obsahujících jako modelové léčivo indometacin. Uvolňování léčiva bylo v případě obou rozpouštědel rychlejší než u klasických tablet, avšak mezi jednotlivými liquisolid systémy nebyly pozorovány významnější rozdíly v uvolňování léčiva70).
Celkově lze shrnout, že výběr vhodného rozpouštědla má zásadní vliv na vlastnosti LSS (rychlost disoluce, mechanická odolnost atd.) a také na ovlivňování dostupnosti léčiva podaného v podobě tohoto systému. Z principu přípravy LSS je totiž, kromě látek mísitelných s vodou a umožňujících podání již rozpuštěného léčiva, možné jako rozpouštědlo použít i tenzid, nebo lipofilní látku, a na porézní nosič sorbovat samoemulgující formulace zajišťující zvýšenou biodostupnost některých léčiv.
Nosiče
Při přípravě systémů kapalina v pevné fázi hrají nosiče hlavní úlohu během převádění léčiva v kapalné fázi do podoby suchého, nepřilnavého, volně tekoucího a dobře stlačitelného prášku vhodného pro další zpracování. Z chemického hlediska se jedná o různorodou skupinu látek, ale všechny je možné charakterizovat jako porézní materiály s vysokou hodnotou specifického měrného povrchu (SSA) a schopností absorbovat kapaliny40). Bylo prokázáno, že vlastnosti nosiče, především pak jeho SSA a adsorpční kapacita, ovlivňují množství léčiva v kapalné podobě, které je možné zapracovat do liquisolid systémů53). Výběr nejvhodnějšího nosiče pro přípravu systémů kapalina v pevné fázi tedy závisí především na jeho adsorpční kapacitě, SSA, tokových vlastnostech a stlačitelnosti30).
Ideální nosič vhodný pro formulaci LSS by měl vykazovat tyto vlastnosti71):
- vysoká sorpční kapacita
- umožnit snadné zpracování do finální lékové formy běžnými postupy
- vysoká stabilita při skladování
- žádný negativní vliv na stabilitu léčiva
- umožnit kompletní uvolnění léčiva v organismu
- nulová toxicita
Mikrokrystalická celulosa (MCC) (obr. 1) je stále nejčastěji používaným nosičem pro přípravu liquisolid systémů vzhledem k jejímu dlouhodobému využití ve farmaceutickém průmyslu, vysoké stabilitě a snadné dostupnosti. Dřívější studie ukázaly, že jiné nosiče, jako je např. laktosa (SSA ≈ 0,35 m2/g), sorbitol (SSA ≈ 0,37 m2/g) nebo škrob (SSA ≈ 0,60 m2/g)31), jsou pro převedení léčiva do pevné podoby zapotřebí ve větším množství než MCC, což je způsobeno jejím vyšším měrným povrchem (SSA ≈ 1,18 m2/g)35). Mikrokrystalická celulosa se použila během přípravy LSS s obsahem aceklofenaku72), diazepamu73), furosemidu74), piroxikamu67), rosuvastatinu75), valsartanu76), prednisolonu32) a řady dalších léčiv. Nicméně v současné době je dostupná celá řada látek (např. magnezium aluminometasilikáty, bezvodý hydrogenfosforečnan vápenatý, mezoporézní silikáty aj.), které mají několikanásobně vyšší hodnoty SSA a značně vyšší absorpční kapacitu.

Bezvodý hydrogenfosforečnan vápenatý se ve farmaceutické technologii používá nejčastěji jako plnivo pro přípravu tablet a tobolek. Fujicalin® (obr. 2) je jeho volně tekoucí sféricky aglomerovaná forma vhodná pro přímé lisování78). Díky unikátnímu procesu přípravy má Fujicalin® v porovnání s běžně používaným hydrogenfosforečnanem vápenatým (SSA ≈ 0,30 m2/g) výrazně větší povrch (SSA ≈ 40 m2/g)79, 80). Velký měrný povrch, vysoká pórovitost a schopnost absorbovat až 1,2 ml kapaliny na 1 g Fujicalinu® při zachování práškové podoby z něj činí nosič vhodný pro přípravu systémů kapalina v pevné fázi50) i pevných samoemulgujících systémů81). Příkladem jeho využití pro přípravu systémů kapalina v pevné fázi mohou být liquisolid tablety s obsahem olmesartan medoxomilu82) a tokoferol acetátu50).

Mezoporézní silikáty jsou látky dostupné v práškové podobě, u nichž rozpustnost závisí na koncentraci silanolových skupin na povrchu materiálu (vyšší koncentrace značí vyšší hydrofilitu)83). Jedná se o materiály objevené v devadesátých letech 20. století. Široké uplatnění mezoporézních silikátů je dáno jejich výhodnými vlastnostmi, jako je dobrá mechanická odolnost, vysoká tepelná stabilita, velký specifický povrch (až 1500 m2/g), značná porózita (objem póru ≈ 0,5–1,5 cm3/g), vysoká adsorpční kapacita (až 7 g/g), zdravotní nezávadnost a snadná modifikovatelnost83–85). Vlastnosti materiálu je navíc možné dle potřeby upravit modifikací volných silanolových skupin na povrchu různými funkčními skupinami. Mezoporézní silikáty (obr. 3) byly původně objeveny jako molekulová síta86). Ve farmaceutické technologii se nejdříve využívaly při přípravě lékových forem s řízeným uvolňováním léčiva87). Jejich schopnost zvyšovat biologickou dostupnost špatně rozpustných léčiv byla odhalena až o několik let později88, 89). S ohledem na jejich extrémně velký povrch, velikost a objem pórů vykazují mezoporézní silikáty vysoký potenciál pro formulaci liquisolid systémů.

Chen et al.84) použili mezoporézní silikáty pro přípravu LSS s obsahem karbamazepinu. Výsledky ukázaly, že uvolňování léčiva z těchto liquisolid systémů je rychlejší v porovnání s komerčně dostupnými tabletami. V jiné studii91) byly použity duté mezoporézní silikáty (hollow mesoporous silicas – HMS) jako nosiče pro roztok karbamazepinu v PEG 400. Výrazné navýšení množství nasorbovaného léčiva v kapalné fázi a zrychlený disoluční profil karbamazepinu dokázali, že HMS představují vhodné nosiče pro roztoky léčiv.
Nejznámější magnesium aluminometasilikát Neusilin® je komerčně dostupný v jedenácti variantách, které se vzájemně liší formou (prášek, granulát), sypnou hustotou, obsahem vody, velikostí částic, hodnotou specifického měrného povrchu a pH (alkalické, neutrální)92). Tato vysoká variabilita ve vlastnostech z něj dělá pomocnou látku vhodnou pro celou řadu aplikačních forem (tab. 1 a 2).
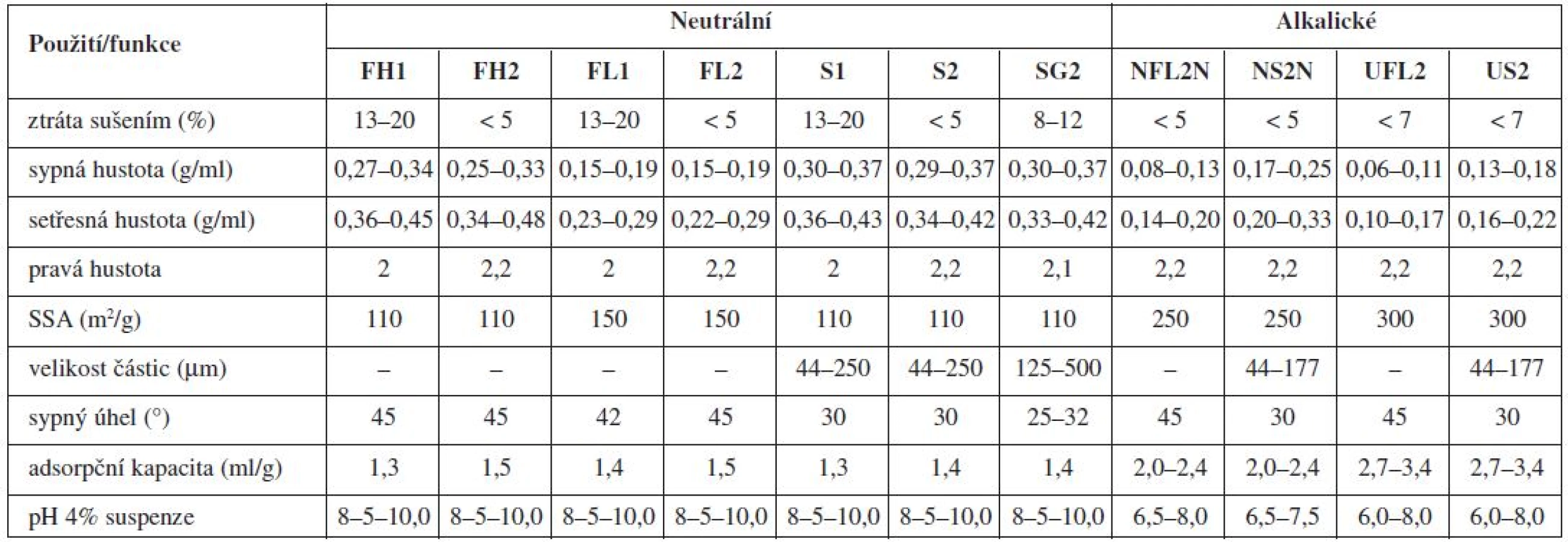
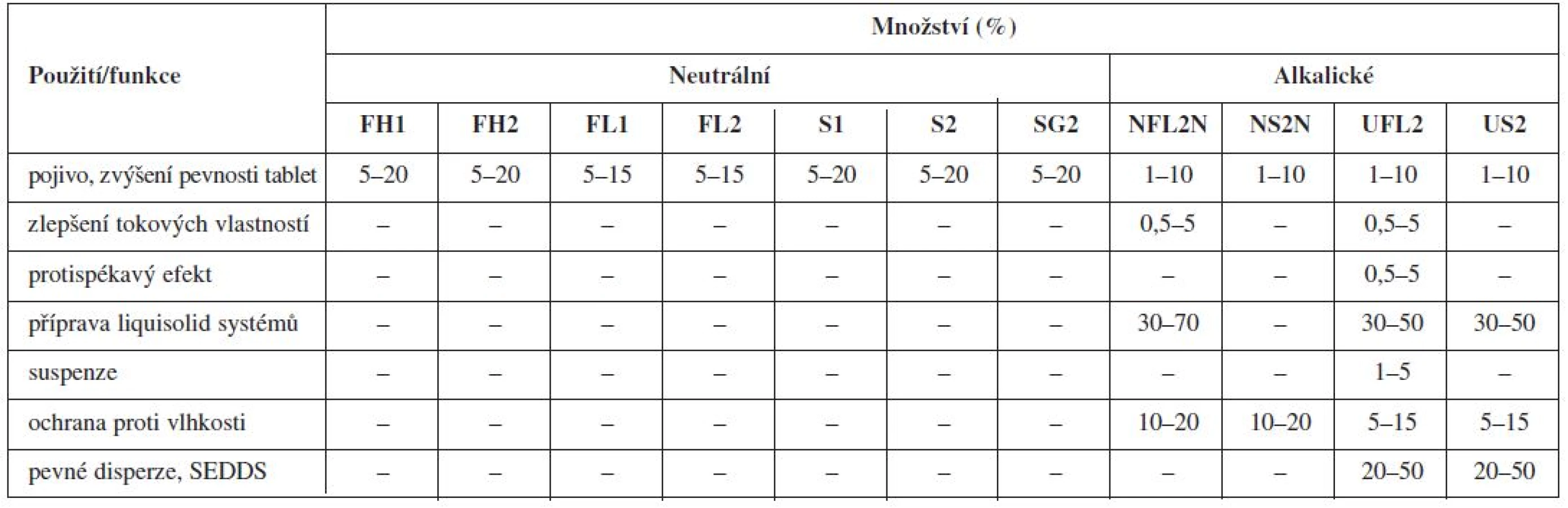
Použití magnezium aluminometasilikátů se stalo předmětem řady studií. Jejich adsorpčních vlastností se využilo např. v tabákovém průmyslu, kde byly aluminometasilikáty přidávány do cigaretových filtrů za účelem absorpce aldehydů (především formaldehydu)93). Ve farmaceutické technologii byly použity jako nosiče při přípravě pevných disperzí s cílem zvýšit rozpustnost léčiva, ke granulaci olejových formulací a ke zlepšení stability přípravků94).
Na základě zkušeností s Neusilinem jako s pomocnou látkou v různých perorálních formulacích bylo zjištěno, že je možné denně aplikovat až 1,05 g Neusilinu®. Navíc alkalické typy Neusilinu® jsou používány jako antacida s maximální dávkou 4 g/den95).
V systémech kapalina v pevné fázi mohou Neusiliny® plnit nejen úlohu nosiče (granulovaná forma), ale také obalovacího materiálu (prášková forma). V porovnání s běžně používanou mikrokrystalickou celulosou jsou Neusiliny® schopné adsorbovat až 3krát větší množství kapaliny, avšak rozpad vzniklých tablet je pomalejší kvůli jejich špatným desintegračním vlastnostem50).
Neusiliny® US2 a UFL2 jsou pro svou vysokou schopnost sorpce velkého množství kapalných látek nadějnými pomocnými látkami pro formulace pevných lékových forem z léčiv povahy olejů a léčivých látek převedených do kapalné fáze za účelem zvýšení jejich rozpustnosti. Díky jejich velkému měrnému specifickému povrchu a pórovitosti se jejich adsorpční kapacita pohybuje v rozmezí 2,7–3,4 ml/g pro olejovité kapaliny a 2,4 až 3,1 ml/g pro vodné roztoky. Navíc pro jejich neutrální charakter, lze na rozdíl od tradičních magnesium aluminosilikátů, jejichž pH je alkalické, předpokládat kompatibilitu s širší paletou léčiv92).
Pro přípravu liquisolid systémů je nejčastěji používaným aluminometasilikátem Neusilin® US250, 96), který je připravován sprejovým sušením, díky čemuž má extrémně velký specifický měrný povrch (≈ 300 m2/g), vysokou absorpční kapacitu (až 3,4 ml/g), vysokou pórovitost i dobré tokové a lisovací vlastnosti91). Hentzschel et al. zjistili, že užití Neusilinu® umožňuje získat tablety s obsahem griseofulvinu s výrazně nižší hmotností, než když se použije jako nosič a obalovací materiál MCC a koloidní oxid křemičitý96).
Ve studii publikované Hentzschel et al. byl sledovanou látkou formulovanou do tablet tokoferol acetát (TA), který byl jako kapalné léčivo v acetonovém roztoku přidáván ke směsi nosiče (Avicel®, Fujicalin®, Neusilin® US2) a obalovacího materiálu (Aerosil®), který zde byl zastoupen v poměru 1 : 20 (Aerosil®: nosič). Všechny formulace včetně Neusilinu® neobsahující TA vykazovaly nevhodné tokové vlastnosti projevující se vysokou hodnotou Hausnerova poměru a špatnou hmotnostní rychlosti sypání, což může být zapříčiněno velkým množstvím Aerosilu®, který obalením jemných suchých částic nosičů zvýšil působící kohezivní a adhezivní síly. Přídavkem TA se tokové vlastnosti zlepšovaly, ale pouze do určitého množství. U Avicelu® to bylo pouze 8 % přidaného léčiva, neboť při vyšším množství se začaly tvořit lepivé aglomeráty a tokové vlastnosti se zhoršovaly. Fujicalin® a Neusilin® adsorbovali až 20 %, resp. 55 % (TA). Z hlediska vlastností tablet jejich pevnost klesala se zvyšujícím se množstvím adsorbovaného TA, takže tablety s vhodnými mechanickými vlastnostmi se získaly s 50% obsahem TA u Neusilinu®, 8% u Avicelu® a 12% u Fujicalinu®. Na základě těchto výsledků byl pro další zkoumání jako nosič vybrán Neusilin® US2 a obalovací materiál buďto Aerosil®, Florite® nebo stejný typ Neusilinu®. Směs s Aerosilem® jako obalovacím materiálem absorbovala maximálně 50 % TA pro získání tablet vhodné pevnosti, na druhou stranu tablety s Florite® nebo Neusilinem® US2, jako obalovacím materiálem, mohly pojmout až 55 % TA. Z uvedeného vyplývá, že použití Neusilinu jako nosiče i obalovacího materiálu místo Avicelu® a Aerosilu® umožnilo navýšení obsahu TA v tabletách z 8 % na 55 %50).
V další studii Hentzschel et al. zjistili, že absorpční faktor směsi Avicelu® a Aerosilu® (20 : 1) v LSS systému dosahuje hodnoty Lf = 0,22, zatímco u Neusilinu® ve funkci nosiče i obalovacího materiálu je Lf = 1,58 při zachování vhodných tokových vlastností směsi. Následně se připravily směsi těchto látek s griseofulvinem (roztok nebo suspenze v PEG 300). Z formulací s Neusilinem®, který nahradil jak Avicel®, tak Aerosil®, se podařilo díky vyššímu adsorpčnímu faktoru získat tablety nižší hmotnosti, (z 2026 mg klesla hmotnost na 600 mg) při stejném obsahu 3 mg grisoefulvinu. Použitím Neusilinu® je tedy možné výrazně snížit hmotnost LSS tablet se stejným množstvím léčiva, čímž se dosahuje i lepší compliance pacienta a vyšší ekonomické efektivity procesu výroby42).
S ohledem na vysoké hodnoty SSA může být několik dalších pomocných látek použito jako potenciální nosič v LSS. Mezi tyto látky patří např. kaolin, diosmektit a uhličitan hořečnatý48).
Obalovací materiál
Obalovací materiál vhodný pro přípravu liquisolid systémů by měl mít velmi jemné částice (0,01–5,0 ∝m) s vysokou absorpční schopností, které se obalením vlhkých částic nosiče a absorpcí přebývající kapaliny podílejí na vzniku suchého prášku s dobrými tokovými vlastnostmi29, 40). Do skupiny obalovacích materiálů se obvykle řadí látky s vysokým měrným povrchem a absorpční kapacitou, které nelze použít jako nosiče kvůli nevyhovujícím tokovým vlastnostem a špatné stlačitelnosti.
Koloidní oxid křemičitý se běžně používá při přípravě pevných perorálních lékových forem jako kluzná a antiadhezivní látka zlepšující tokové vlastnosti práškových směsí nebo granulátů95). Komerčně je dostupný ve dvou základních typech: hydrofilním a hydrofobním. Hydrofobní typy jsou charakterizovány nízkou absorpční kapacitou, výbornou dispergovatelností a schopností upravovat reologické vlastnosti97). Pro přípravu systémů kapalina v pevné fázi se používají hydrofilní typy koloidního oxidu křemičitého vzhledem k jejich vyšší absorpční schopnosti.
V minulosti byl oxid křemičitý používán jako nosič při přípravě předchůdců liquisolid systémů tzv. práškových roztoků. V současné době je při přípravě LSS nejčastěji používaným obalovacím materiálem. Příkladem jeho využití můžou být liquisolid systémy s obsahem atorvastatinu41), naproxenu51), famotidinu98), rofecoxibu49) a řadě dalších.
Křemičitan vápenatý se používá k absorpci olejů, kapalných samoemulgujících systémů a kapalných léčiv a jako antiadhezivní látka50, 99). V rámci liquisolid systémů byl použit pro přípravu tablet s obsahem repaglinidu100).
Dále mohou být jako obalovací materiály použity také práškové formy magnesium aluminometasilikátů44) a syntetický amorfní silikagel101).
Hodnocení systémů kapalina v pevné fázi
Kromě zkoušek používaných již k hodnocení vlastností LSS a jejich složek v rámci preformulačních studií jsou liquisolid systémy formulované do finální lékové formy obvykle podrobeny testům s cílem zajistit jejich vhodné fyzikálně-chemické vlastnosti a stabilitu a také popsat děje, k nimž může v soustavě kapalina-pevná látka v průběhu přípravy docházet.
Při hodnocení práškové směsi pro přípravu liquisolid systémů se provádí zkoušky lékopisné a několik zkoušek nelékopisných102).
Z lékopisných zkoušek se hodnotí především zkoušky tokových vlastností a stanovení hustot, z nichž se následně vypočítávají faktory související s možností dalšího zpracování do finální jednotkové lékové formy102):
- sypnost (hmotnostní rychlost sypání)
- sypný úhel
- sypná a setřesná hustota
- Hausnerův poměr a index stlačitelnosti
- pyknometrická hustota
K odhalení případných interakcí mezi léčivem a pomocnými látkami (nebo jen mezi pomocnými látkami) již před zpracováním do finální lékové formy se používá diferenciální skenovací kalorimetrie (DSC)35, 40). V některých případech je doporučeno provést rovněž rentgenovou difrakci a skenovací elektronovou mikroskopii pro zjištění krystalinity léčiva. Obecně platí, že nepřítomnost charakteristického píku nebo krystalů léčiva značí, že léčivo je převedeno na amorfní formu, nebo je v liquisolid systému přítomno v podobě roztoku56, 103).
Mezi další nelékopisné zkoušky se řadí úhel skluzu, stanovení rozpustnosti léčiva, viskozita disperze účinné látky, stanovení specifického měrného povrchu a porozity nosiče a obalovacího materiálu atd.
Hodnocení finální lékové formy formulované LSS technikou probíhá podobě jako u ostatních pevných lékových forem. Kromě lékopisných zkoušek se často hodnotí i specifické parametry související pouze s tímto typem formulací.
Lékopisné zkoušky u liquisolid tablet102):
- hmotnostní stejnoměrnost
- obsahová stejnoměrnost
- pyknometrická hustota
- pevnost
- oděr
- rozpad
- disoluce
Zvláštními nelékopisnými metodami jsou např. stanovení kontaktního úhlu nebo nasákavosti tablety.
Díky měření kontaktního úhlu je možné stanovit smáčivost liquisolid systémů disolučním médiem. Rychlost a kvalita smáčení má vliv na rychlost průniku gastrointestinální tekutiny do lékové formy a s tím související rychlost uvolňování léčiva. Kapka disolučního média se nanese na rovný povrch tablety a měří se výška a průměr kapky, z čehož se získává hodnota kontaktního úhlu [11] (obr. 4). LSS vykazují, díky hydrofilizaci povrchu tablety zapracováním hydrofilního rozpouštědla s dispergovaným léčivem, obvykle nižší hodnoty kontaktního úhlu, a tím pádem vyšší smáčivost tablet a rychlost uvolňování léčiva než běžné tablety29, 60).
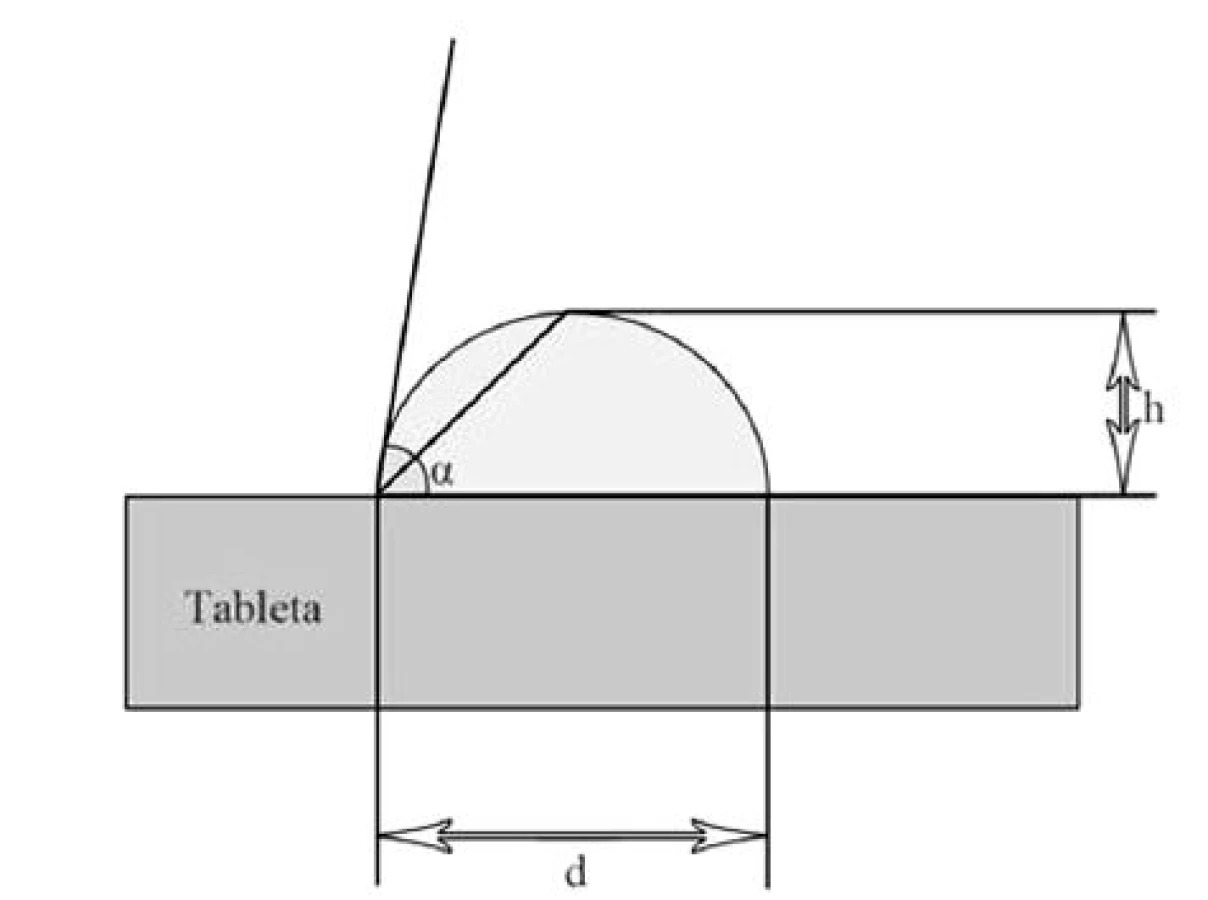
kde H je výška kapky (mm), D průměr kapky (mm), α velikost kontaktního úhlu (°).
Další možností k určení smáčivosti liquisolid přípravku je stanovení nasákavosti. Do Petriho misky se umístí houbička spolu s potřebným množstvím vodného roztoku barviva. Na houbičku se následně položí přesně zvážená tableta. Zaznamenává se čas, kdy dojde ke smáčení vrchního povrchu tablety a kdy je tableta smáčená kompletně. Poté se tableta opět zváží a vypočítá se nasákavost vyjádřená jako procentuální nárůst hmotnosti104).
Příklady léčiv formulovaných lss technikou
Příklady zlepšení rozpustnosti a optimalizace disolučního profilu:
Famotidin – rychlost disoluce zvýšena o 39 % oproti běžným tabletám98).
Piroxikam – výrazné zlepšení disolučního profilu díky přípravě LSS, rychlost disoluce zvýšena o 40 % oproti běžným tabletám105).
Indometacin – 60% zvýšení disoluční rychlosti106).
Příklady zlepšení farmakokinetických parametrů léčiv:
Hydrochlorothiazid – biologická dostupnost léčiva z liquisolid tablet zvýšena o 15 % než při aplikaci konvenčních tablet107).
Atorvastatin vápenatá sůl – léčivo uvolňované z liquisolid tablet při testech in vivo vykazovalo zlepšené hodnoty farmakokinetických parametrů, tj. AUC, tmax, cmax, než u konvenčních tablet připravovaných přímým lisováním41).
Formulace tablet s řízeným uvolňováním léčiva:
Propranolol hydrochlorid – polysorbát 80 jako rozpouštědlo a hydrofobní nosič Eudragit® RL hrály významnou roli v získání profilu prodlouženého uvolňování léčiva, dle kinetiky nultého řádu33).
Tramadol – potvrzen přínos použití HPMC K4M jako pomocné látky ve formulacích LSS pro řízený profil uvolňování léčiva53).
Theophylin – prokázán pozitivní vliv použití HPMC na získání požadovaného profilu uvolňování léčiva56).
Závěr
Systémy kapalina v pevné fázi jsou moderní formulace vznikající nasorbováním léčiva v kapalné podobě na vysoce porézní nosič, který je následně obalen materiálem s vysokým měrným povrchem částic za vzniku volně tekoucího, nepřilnavého, dobře stlačitelného prášku, vhodného pro další zpracování do finální lékové formy. Hlavní výhodnou těchto systémů je zlepšení rozpustnosti a následně zvýšení biologické dostupnosti špatně rozpustných léčivých látek, především díky přítomnosti již rozpuštěného léčiva, které tak obchází jeden z limitujících kroků systémové absorpce – rozpouštění. Metoda přípravy liquisolid systémů je v samotném počátku svého vývoje, přesto se však předpokládá, že by liquisolid systémy mohly hrát důležitou roli při přípravě moderních pevných lékových forem s ohledem na jejich výhody (např. nízké výrobní náklady, konečné zpracování podobné klasickým tabletám a tobolkám, možnost průmyslové výroby a přípravy lékových forem s řízeným uvolňováním léčiva).
Seznam symbolů a zkratek
α velikost kontaktního úhlu
θ úhel skluzu (angle of slide)
σi index pórovitosti
Φ tokový retenční potenciál (flowable liquid retention potential)
Ψ lisovací retenční potenciál (compressible liquid retention potential)
Ω maximální destrukční síla přepočtena na 1g tablety
Ω0 vnitřní kompaktnost
AUC plocha pod křivkou
Cm poměr hmotností kapalné/pevné fáze (obsah rozpouštědla)
cmax maximální koncentrace v plazmě po jednorázové aplikaci
D průměr kapky
H výška kapky
HMS duté mezoporézní silikát (hollow mesoporous silicas)
Lf absorpční faktor
LfO optimální retenční faktor
LSC test lisovatelnosti liquisolid systémů (liquisolid compresibility test)
LSS liquisolid systémy
m hmotnost kapalné fáze
MCC mikrokrystalická celulosa
N množství nosiče
NO optimální množství nosiče
O množství obalovacího materiálu
OO optimální množství obalovacího materiálu
PEG magrogoly, polyethylenglykoly
PG propylenglykol
R poměr mezi nosičem a obalovacím materiálem
SSA specifický měrný povrch (specific surface area)
TA tokoferol acetát
tmax doba dosažení maximální plazmatické koncentrace
Střet zájmů: žádný.
Došlo 9. února 2015
Přijato 10. dubna 2015
PharmDr. Barbora Vraníková • J. Gajdziok
Veterinární a farmaceutická univerzita Brno, Farmaceutická fakulta
Ústav technologie léků
Palackého třída 1/3, 612 42 Brno
e-mail: vranikovab@vfu.cz
Sources
1. Kawabata Y., Wada K., Nakatani M., Yamada S., Onoue S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: Basic approaches and practical applications. Int. J. Pharm. 2011; 420, 1–10.
2. Okáčová L., Vetchý D., Franc A., Rabišková M., Kratochvíl B. Zvýšení biodostupnosti těžce rozpustných léčivých látek jejich modifikací. Chem. Listy 2010; 104, 21–26.
3. Balakrishnan A., Rege B. D., Amidon G. L., Polli E. Surfactant-mediated dissolution: Contributions of solubility enhancement and relatively low micelle diffusivity. J. Pharm. Sci. 2004; 93, 2064–2075.
4. Bajaj H., Bisht S., Yadav M., Singh V. Bioavaolability enhancement: a review. International Journal of Pharma and Bio Sciences 2011; 2, 202–216.
5. Sikarra D., Shukla V., Kharia A. A., Chatterjee D. P. Techniques for solubility enhancement of poorly soluble drugs: an overview. Journal of Medical Pharmaceutical and Allied Sciences 2012; 1, 1–22.
6. Li P., Yhao L. Solubilization of flurbiprofen in pH-surfactant solutions. J. Pharm. Sci. 2003; 92, 951–956.
7. Rangel-Yagui C. O., Pessoa Jr. A., Taveres L. C. Micellar solubilization of drugs. J. Pharm. Pharm. Sci. 2005; 8, 147–163.
8. Zhu J., Bierwagen G. P. The surface chemistry of water-reducible polymer solutions/dispersions – 1. Surface tension behavior. Prog. Org. Coat. 1995; 26, 87–100.
9. Kawakami K., Oda N., Miyoshi K., Funaki T., Ida Y. Solubilization behavior of a poorly soluble drug under combined use of surfactants and cosolvents. Eur. J. Pharm. Sci. 2006; 28, 7–14.
10. Vemula V. R., Lagishetty V., Lingala S. Solubility enhancement techniques. International Journal of Pharmaceutical Sciences Review and Research 2010; 5, 41–51.
11. Jangher A., Griffiths P. C., Paul A., King S. M., Heenan R. K., Schweins R. Polymeric micelle disruption by cosolvents and anionic surfactants. Colloid and Surface A 2011; 391, 88–94.
12. Patil A. E., Devtalu S. V., Bari M. M., Barthel S. D. A review on: novel solubility enhancement technique hydrotropy. Indo American Journal of Pharmaceutical Research 2013; 3, 4670–4679.
13. Kumar V. S., Raja C., Jayakumar C. A review on solubility enhancement using hydrotropic phenomena. International Journal of Pharmacy and Pharmaceutical Sciences 2014; 6, 1–7.
14. Terao K., Nakata D., Fukumi H., Schmid G., Arima H., Hirayama F., Uekama K. Enhancement of oral bioavailability of coenzyme Q10 by complexation with γγ-cyclodextrin in healthy adults. Nutr. Res. 2006; 26, 503–508.
15. Gowardhane A. P., Kadam N. V., Dutta S. Review on enhancement of solubilisation precess. Journal of Pharmacy and Phytotherapeutics 2013; 2, 28–38.
16. Kataria M. K., Bhandari A. Solubility and dissolution enhancement: technologies and research emerged. Journal of Biological and Scientific Opinion 2013; 1, 105–116.
17. Dvořáčková K. Principy uvolňování léčiv z perorálních matricových tablet obsahujících hypromelosu. Chem. Listy 2009; 103, 66–72.
18. Okáčová L., Vetchý D., Franc A, Rabišková M. Zvýšení biodostupnosti těžce rozpustných léčivých látek technologickými postupy usnadňujícími jejich rozpouštění. Chem. Listy 2011; 105, 34–40.
19. Sinha S., Ali M., Baboota S., Ahuja A., Kumar A., Ali J. Solid dispersion as an approach for bioavailability enhancement of poorly water-soluble drug ritonavir. AAPS PharmSciTech. 2010; 11, 518–527.
20. Baek H. H., Kim D. H., Kwon S. Y., Rho S. J., Kim D. W., Choi H. G., Kim Y. R., Yong C. S. Development of novel ibuprofen-loaded solid dispersion with enhanced bioavailability using cycloamylose. Arch. Pharm. Res. 2012; 35, 683–689.
21. Zatloukal Z. Interaktivní práškové směsi. Čes. slov. Farm. 2004; 53, 165–171.
22. Allahham A., Stewart P. J. Enhancement of the dissolution of indomethacin in interactive mixtures using added fine lactose. Eur. J. Pharm. Biopharm. 2007; 67, 732–742.
23. Watano S., Imada Y., Hamada K., Wakamatsu Y., Tanabe Y., Dave R. N., Pfeffer R. Microgranulation of fine powders by a novel rotating fluidized bed granulation. Powder Technol. 2003; 131, 250–255.
24. Planinšek O., Kovačič B., Vrečer F. Carvediol dissolution improvement by preparation of solid dispersions with porous silica. Int. J. Pharm. 2011; 406, 41–48.
25. Krupa A., Jachowicz R., Kurek M., Figiel W., Kwiecień M. Preparation of solid self-emulsifying drug delivery systems using magnesium aluminometasilicates and fluid-bed coating proces. Powder Technol. 2014; 266, 329–339.
26. Yang H., Teng F., Wang P., Tian B., Lin X., Hu X., Zhang L., Zhang K., Zhang Y., Tang Y. Investigation of a nanosuspension stabilized by Soluplus® to improve bioavailability. Int. J. Pharm. 2014; 477, 88–95.
27. Shaikh J., Ankola D. D., Beniwal V., Singh D., Kumer M. N. Nanoparticle encapsulation improves oral bioavailability of curcumin by at least 9-fold when compared to curcumin administered with piperine as absorption enhancer. Eur. J Pharm. Sci. 2009; 37, 223–230.
28. Chen Y., Lu Y., Chen J., Lai J., Sun J., Hu F., Wu W. Enhanced bioavailability of the poorly water-soluble drug fenofibrate by using liposomes containing a bile salt. Int. J. Pharm. 2009; 376, 153–160.
29. Kulkarni S. A., Aloorkar N. H., Mane M. S. Liquisolid systems: A review. International Journal of Pharmaceutical Sciences and Nanotechnology 2010; 3, 795–802.
30. Kavitha K., Lova Raju K. N. S., Ganesh N. S., Ramesh B. Effect of dissolution rate by liquisolid compacts approach: An Overview. Der Pharmacia Lettre 2011; 3, 71–83.
31. Nokhodchi A., Hentzschel C. M., Leopold C. S. Drug release from liquisolid systems: speed it up, slow it down. Expert Opin. Drug Del. 2011; 8, 191–205.
32. Spireas S., Bolton S. M. Liquisolid systems and methods of preparing same. US 2002; 6, 339, 423.
33. Javadzadeh Y., Musaalrezaei L., Nokhodchi A. Liquisolid technique as a new approach to sustain propranolol hydrochloride release from tablet matrices. Int. J. Pharm. 2008; 362, 102–108.
34. Tang B., Cheng G., Gu J. C., Xu C. H. Development of solid self-emulsifying drug delivery systems: preparation techniques and dosage forms. Drug Discov. Today 2008; 13, 616–612.
35. Karmarkar A. B., Gonjari I. D., Hosmani A. H., Dhabale P. N., Bhise S. B. Liquisolid tablets: a novel approach for drug delivery. Int. J. Health. Res. 2009; 2, 45–50.
36. Nagabandi V. K., Ramarao T., Jayaveera K. N. Liquisolid compacts: A novel approach 30. to enhance bioavailability of poorly soluble drugs. International Journal of Pharmacy and Biological Sciences 2011; 1, 89–102.
37. El-Hammadi M., Awad N. Investigating the use of liquisolid compacts technique to minimize the influence of pH variations on loratadine release. AAPS PharmSciTech. 2012; 13, 53–58.
38. Spireas S., Bolton S. M. Liquisolid systems and methods of preparing same. US 1998; 5, 800, 834.
39. Javadzadeh Y., Siahi M. R., Asnaashari S., Nokhodchi A. Liquisolid technique as a tool for enhancement of poorly water-soluble drugs and evaluation of their physicochemical properties. Acta Pharm. 2007; 57, 99–109.
40. Gavali S. M., Pacharane S. S., Sankpal S. V., Jadhav K. R., Kadam V. J. Liquisolid compact: A new technique for enhancement of drug dissolution. International Journal of Research in Pharmacy and Chemistry 2011; 1, 705–713.
41. Gubbi S., Jarag R. Formulation and characterization of atorvastatin calcium liquisolid compacts. Asian Journal of Pharmaceutical Sciences 2010; 5, 50–60.
42. Hentzschel C. M., Alnaief M., Smirnova I., Sakmann A., Leopold C. S. Enhancement of griseofulvin release from liquisolid compacts. Eur. J. Pharm. Biopharm. 2012; 80, 130–135.
43. Tayel S. A., Soliman I. I., Louis, D. Improvement of dissolution properties of carbamazepine through application of the liquisolid tablet technique. Eur. J. Pharm. Biopharm. 2008; 69, 342–347.
44. Zhao X., Zhou Y. Q., Potharaju S., Lou H., Sun H. M., Bruson E., Almoazen H., Johnson J. Development of a self micro-emulsifying tablet of cyclosporine A by the liquisolid compact technique. International Journal of Pharmaceutical Sciences and Research 2011; 2, 2299–2308.
45. Yada, A. V., Shete S. A., Dabke A. P. Formulation and evaluation of orodispersible liquisolid compacts of aceclofenac. Indian J. Pharm. Educ. 2010; 44, 227–235.
46. Gonjari I. D., Karmarkar A. B., Hosmani A. H. Evaluation of in vitro dissolution profile comparison methods of sustained release tramadol hydrochloride liquisolid compact formulations with marketed sustained release tablets. Dig. J. Nanomater. Bios. 2009; 4, 651–661.
47. Nokhodchi A., Aliakbar R., Desai S., Javadzadeh Y. Liquisolid compacts: The effect of cosolvent and HPMC in theophylline release. Colloid Surface B 2010; 79, 262–269.
48. Vraníková B., Gajdziok J. Liquisolid systems and aspects influencing their research and development. Acta Pharm. 2013; 63, 447–465.
49. El-Say K. M., Samy A. M., Fetouh M. I. Formulation and evaluation of rofecoxib liquisolid tablets. International Journal of Pharmaceutical Sciences Review and Research 2010; 3, 135–142.
50. Hentzschel C. M., Sakmann A., Leopold C. S. Suitability of various excipients as carrier and coating materials for liquisolid compacts. Drug. Dev. Ind. Pharm. 2011; 37, 1200–1207.
51. Tiong N., Elkordy A. A. Effects of liquisolid formulations on dissolution of naproxen. Eur. J. Pharm. Biopharm. 2009; 73, 373–384.
52. Spireas S. S., Jarowski C. I., Rohera B. D. Powdered solution technology: Principles and mechanism. Pharm. Res. 1992; 9, 1351–1358.
53. Karmarkar A. B., Gonjari I. D., Hosmani A. H. Liquisolid technology for dissolution rate enhancement or sustained release. Expert Opi. Drug. Deliv. 2010; 7, 1227–1234.
54. Khanfar M., Sheikh S. M., Hawari R. Formulation factors affecting the release of ezetimibe from different liquisolid compacts. Pharm. Dev. Technol. 2013; 18, 417–427.
55. Spireas S., Bolton S. M. Liquisolid systems and methods of preparing same, US 5,968,550, 1999.
56. Rajesh K., Rajalakshmi R., Umamaheswari J., Ashok Kumar C. K. Liquisolid technique a novel approach to enhance solubility and bioavailability. International Journal of Biopharmaceutics 2011; 2, 8–13.
57. Elkordy A. A., Essa E. A., Dhuppad S., Jammiqumpula P. Liquisolid technique to enhance and to sustain griseofulvin dissolution: effect pf choice of non-volatile liquid vehicles. Int. J. Phar. 2012; 434, 122–132.
58. Grover R., Spireas S., Lau-Cam C. Development of a simple spectrophotometric method for propylene glycol detection in tablets. J. Pharm. Biomed. Anal. 1998; 16, 931–938.
59. Suliman A. S., Anderson R. J., Elkordy A. A. Norfloxacin as a model hydrophobic drug with unique release from liquisolid formulations prepared with PEG200 and Synperonic PE/L-61 non-volatile liquid vehicles. Powder Technol. 2014; 257, 156–167.
60. Sirisha V. N. L., Sruthi B., Namrata M., Bhavani Harika I., Kirankumar P., Kiran Kumar Rao Y., Pravani K., Sindhura S., Camsi Krishna N., Uma Maheshwar Rao O. A Review on liquid solid compacts. International Journal of Pharmaceutical and Phytopharmacological Research 2012; 2, 116–121.
61. Adibkia K., Shokri J., Barzegar-Jalali M., Solduzian M., Javadzadeh Y. Effect of solvent type on retardation properties of diltiazem HCl form liquisolid tablets. Colloids Surf. B: Biointerfaces 2014; 113, 10–14.
62. Elkordy A. A., Tan X. N., Essa E. A. Spironolactone release from liquisolid formulations prepared with CapryolTM 90, Solutol® HS-15 and Kollicoat® SR 30 D as a non-volatile liquid vehicles. Eur. J. Pharm. Biopharm. 2013; 83, 203–223.
63. Thakur N., Khokra S. L., Sharma D., Thakur N. S., Purohit R., Arya V. A review on pharmaceutical applications of liquisolid technique. American Journal of PharmTech Research 2011; 1, 1–18.
64. NTP-CERHR Expert Panel report on the reproductive and developmental toxicity of propylene glycol. Reprod Toxicol 2004; 18, 533–579.
65. Propylene Glycol – Pharmaceutical. http://www.propylene-glycol.com/index.php/propylene-glycol-ups-ep-pharmaceutical-grade/pharmaceuticals. (17. 7. 2014).
66. Chella N., Shastri N., Tadikonda R. R. Use of the liquisolid compact technique for improvement of the dissolution rate of valsartan. Acta Pharm. Sinica B 2012; 2, 502–508.
67. Jabbar A. S. A., Hussein A. A. Formulation and evaluation of piroxicam liquisolid compacts. International Journal of Pharmacy and Pharmaceutical Sciences 2013; 5, 132–141.
68. Pavani E., Noman S., Syed I. A. Liquisolid technique based sustained release tablet of trimetazidine dihydrochloride. Drug Invention Today 2013; 5, 302–310.
69. Mahajan H. S., Dhamne M. R., Gattani S. G., Rasal A. D., Shaikh H. T. Enhanced disolution rate of glipizide by a liquisolid technoque. Int. J. Pharm. Sci. Nanotech. 2011; 3, 1205–1213.
70. Saeedi M., Akbari J., Morteza-Semnani K., Enayati-Fard R., Sar-Reshteh-dar S., Soleymani A. Enhancement of dissolution rate of indomethacin using liquisolid compacts. Iran. J. Pharm. Res. 2011; 10, 25–34.
71. Kutza C., Metz H., Kutza J. Toward a detailed characterization of oil adsorbates as “solid liquids”. Eur. J. Pharm. Biopharm. 2013; 84, 72–182.
72. Yadav V. B., Nighute A. B., Yadav A. V., Bhise S. B. Aceclofenac size enlargement by non aqueous granulation with improved solubility and dissolution. Arch. Pharm. Sci. Res. 2009; 1, 115–122.
73. Manogar P. G., Hari B. N. V., Devi D. R. Emerging liquisolid compact technology for solubility enhancement of BCSclass-II drug. J. Pharm. Sci. Res. 2011; 3, 1604–1611.
74. Akinlade B., Elkordy A. A., Essa E. A., Elhagar S. Liquisolid systems to improve the dissolution of furosemide. Sci. Pharm. 2010; 78, 325–344.
75. Kapure V. J., Pande V. V., Deshmukh K. P. Dissolution enhancement of rosuvastatin calcium by liquisolid compact technique. Journal of Pharmaceutics 2013; 2013, 1–9.
76. Lakshmi P. K., Srinivas C., Kalpana B. Preparation and comparative evaluation of liquisolid compacts and solid dispersions of valsartan. Stamford Journal of Pharmaceutical Sciences 2012; 4, 48 – 57.
77. Ceolus. http://www.ceolus.com/en/ceolus_func.html. (9. 8. 2014).
78. van Eerdenbrugh B., Froyen, L., van Humbeeck J., Martens J., Augustijns P., van Den Mooter G. Alternative matrix formers for nanosuspension solidification: Dissolution performance and X-ray microanalysis as an evaluation tool for powder dispersion. Eur. J. Pharm. Sci. 2008; 35, 344–353.
79. Schlack H., Bauer-Brandl A., Schubert R., Becker D. Properties of Fujicalin®, a new modified anhydrous dibasic calcium phosphate for direct compression: comparison with dicalcium phosphate dihydrate. Drug. Dev. Ind. Pharm. 2001; 27, 789–801.
80. Fuji Chemical Industry Co., LTD. Fujicalin – product info. http://www.fujicalin.com/product/index.php. (5. 1. 2015).
81. Kang M. J., Jung S. Y., Song W. H., Park J. S., Choi S. U., Oh K. T., Choi H. K., Choi Y. W., Lee J., Lee B. J., Chi S. C. Immediate release of ibuprofen from Fujicalin®-based fast-dissolving self-emulsifying tablets. Dug. Dev. Ind. Pharm. 2011; 37, 1298–1305.
82. Prajapati S. T., Bulchandani H. H., Patel D. M., Dumaniya S. K., Patel C. N. Formulation and evaluation of liquisolid compacts for olmesartan medoxomil. J. Drug. Deliv. 2013; 2013, 1–9.
83. Luštická I., Vyskočilová-Leirmannová E., Červený L. Funkcionalizace mezoporézních silikátových materiálů. Chem. Listy 2013; 107, 114–120.
84. Chen B., Wang Z., Quan G., Peng X., Pan X., Wang R., Xu Y., Li G., Wu C. In vitro and in vivo evaluation of ordered mesoporous silica as a nocel adsorbent in liquisolid formulation. Int. J. Nanomed. 2012; 7, 199–209.
85. Zhu W., Wan L., Zhang C., Gao Y., Zheng X., Jiang T., Wang S. Exploitation of 3D face-centered cubic mesoporous silica as a carrier for a poorly water soluble drug: influence of pore size on release rate. Mater. Sci. Eng. C. Mater. Biol. Appl. 2014; 34, 78–85.
86. Vialpando M., Backhuijs F., Martens J. A., van den Mooter G. Risk assessment of premature drug release during wet granulation of ordered mesoporous silica loaded with poorly soluble compounds itraconazole, fenofibrate, naproxen, and ibuprofen. Eur. J. Pharm. Biopharm. 2012; 81, 190–198.
87. Wu Z., Jiang Y., Kim T., Lee K. Effects of surface coating on the controlled release of vitamin B1 from mesoporous silica tablets. J. Control. Release 2007; 119, 215–221.
88. Mellaerts R., Mols R., Jammaer J. A. G., Aerts C. A., Annaert P., van Humbeeck J., van den Mooter G., Augustijns P., Martens J. A. Increasing the oral bioavailability of the poorly water soluble drug itraconazole with ordered mesoporous silica. Eur. J. Pharm. Biopharm. 2008; 69, 223–230.
89. van Speybroeck M., Mellaerts R., Mols R., Thi T. D., Martens J. A., van Humbeeck J., Annaert P., van den Mooter G., Augistijns P. Enhanced absorption of the poorly soluble drug fenofibrate by tuning its release rate from ordered mesoporous silica. Eur J Pharm Sci. 2010; 41, 623–630.
90. Mesoporous silica. http://news.easybranches.com/ wp–content/ uploads/2014/07/Mesoporous_silica_SEM-1-580x352.jpg (10. 4. 2015).
91. Chen B., Quan G., Wang Z., Chen J., Wu L., Xu Y., Li G., Wu C. Hollow mesoporous silicas as a drug solution delivery system for insoluble drugs. Powder Technol. 2013; 240, 48–53.
92. Fuji Chemical Industry Co., LTD. Neusilin – product info. http://www.neusilin.com/product/index.php. (6. 1. 2015).
93. Taniguchi H. Tabacco filter containing magnesium aluminometasilicate. EP 2,540,174 A1; 2013.
94. Krupa A., Majda D., Jachowicz R., Mozgawa W. Solid-state interaction of ibuprofen and Neusilin US2. Thermochim. Acta. 2010; 509, 12–17.
95. Sander C., Holm P. Porous magnesium aluminometasilicate tablets as carrier of a cyclosporine self-emulsifying formulation. AAPS PharmSciTech. 2009; 10, 1388–1395.
96. Hentzschel C. M., Alnaief M., Smirnova I., Sakmann A., Leopold C. S. Tableting properties of silica aerogels and other silicates. Drug. Dev. Ind. Pharm. 2012; 38,462–467.
97. Aerosil® fumed silica. https://www.aerosil.com/product/aerosil/en/ products/pages/default.aspx. (12. 8. 2014).
98. Fahmy R. H., Kassem M. A. Enhancement of famotidine dissolution rate through liquisolid tablets formulation: In vitro and in vivo evaluation. Eur. J. Pharm. Biopharm. 2008; 69, 993–1003.
99. Sharma S., Sher P., Badve S., Pawar A. P. Adsorption of Meloxicam on Porous Calcium Silicate: Characterization and Tablet Formulation. AAPS PharmSciTech. 2005; 6, 618–625.
100. El-Houssieny B. M., Wahman L. F., Arafa N. M. S. Bioavailability and biological activity of liquisolid compact formula of repaglinide and its effect on glucose tolerance in rabbits. Biosci. Trends. 2010; 4, 17–24.
101. Sheth A., Jarowski C. I. Use of powdered solutions to improve the dissolution rate of polythiazide tablets. Drug. Dev. Ind. Pharm. 1990; 16, 769–777.
102. Český lékopis 2009. 1. vydání. Praha: Grada Publishing 2009.
103. Syed I. A., Pavani E. The liquisolid technique: based drug delivery system. International Journal of Pharmaceutical Sciences and Drug Research 2012; 4, 88–96.
104. Kaur M., Bala R., Arora S. Formulation and evaluation of liquisolid compacts of amlodipine besylate. Intermational Research Journal of Pharma 2013; 4, 156–160.
105. Javadzadeh Y., Siahi-Shadbad M. R., Barzegar-Jalali M., Nokhodchi A. Enhancement of dissolution rate of piroxicam using liquisolid compacts. Farmaco 2005; 60, 361–365.
106. Nokhodchi A., Javadyadeh Y., Siahi-Shadbad M. R., Barzegar-Jalali M. The effect of type and concentration of vehicles on the dissolution rate of a poorly soluble drug (indomethacin) from liquisolid compacts. J. Pharm. Pharm. Sci. 2005; 8, 18–25.
107. Khaled A., Asiri Y. A., El-Sayed Y. M. In vivo evaluation of hydrochlorothiazide liquisolid tablets in beagle dogs. Int. J Pharm. 2001; 222, 1–6.
Labels
Pharmacy Clinical pharmacologyArticle was published in
Czech and Slovak Pharmacy

2015 Issue 3
Most read in this issue
- Enhancing of drug bioavailability using liquisolid system formulation
- The quality of life of patients suffering from the osteoporosis
-
Pharmaceutical history of capuchin monastery in Prague-Hradčan
Part II. Capuchin balsam (Balsamum capucinorum) -
Effectiveness of phytotherapy in supportive treatment of type 2 diabetes mellitus
II. Fenugreek (Trigonella foenum-graecum)