
Vliv EUDRAGITU® RS na disoluční chování pevných disperzí theofylinu
Effect of EUDRAGIT® RS on the release behaviour of theophylline solid dispersions
The purpose of this study was to extend the release of theophylline using Eudragit® RS 100 and Eudragit® RSPO as carriers. Solid dispersions of theophylline were prepared by the solvent evaporation technique using Eudragit® RS 100, Eudragit® RSPO and their blend in various drug : polymer ratios. The prepared solid dispersions were characterized with respect to entrapment efficiency, solubility and recovery yield. In vitro drug release of theophylline from the solid dispersions was evaluated in simulated gastric fluid (SGF) and simulated intestinal fluid (SIF) without enzymes. Solubility studies demonstrated a decrease in the solubility of the drug from the solid dispersions. The solubilities of pure drug and solid dispersions were lowered in SGF compared to SIF. Solid dispersions prepared with Eudragit® RS 100 entrapped a greater amount of theophylline in comparison to those with Eudragit® RSPO or the polymer blends and were able to extend the release of theophylline over 24 hrs. Formulation SD4 released 95.52% of the drug in SIF and 93.56% in SGF. Hence, it was selected as the optimized formulation because it was able to extend the release of theophylline over 24 hrs.
Key words:
solid dispersion • extended release • Eudragit® • drug release
Authors:
Oluwaseun Orugun 1; Avosuahi Oyi 1; Adeniji Olowosulu 1; Yonni Apeji 1; Olubunmi Olayemi 2
Authors‘ workplace:
Department of Pharmaceutics and Pharmaceutical Microbiology
Faculty of Pharmaceutical Sciences
Ahmadu Bello University, Zaria
1; Department of Pharmaceutical Technology and Raw Materials
Development, National Institute for Pharmaceutical Research and
Development (NIPRD), Idu, Abuja
2
Published in:
Čes. slov. Farm., 2016; 65, 226-231
Category:
Original Articles
Overview
Cílem této studie bylo prodloužení uvolňování theofylinu pomocí Eudragit® RS 100 a Eudragit® RSPO ve funkci nosičů. Pevné disperze theofylinu byly připraveny za použití techniky odpaření rozpouštědla ze směsi léčiva a polymerů Eudragit® RS 100, Eudragit® RSPO nebo jejich směsi v různých poměrech. Připravené pevné disperze byly charakterizovány se zaměřením na enkapsulační účinnost, rozpustnost a výtěžek procesu. In vitro uvolňování účinné látky theofylinu z pevné disperze bylo hodnoceno v simulované žaludeční šťávě (SGF) a střevní šťávě (SIF) bez enzymů. Disoluční studie prokázaly snížení uvolňování léčiva z pevných disperzí. Rozpustnost čistého léčiva a pevných disperzí byly nižší v SGF ve srovnání v SIF. Pevné disperze připravené z Eudragitu® RS 100 vykazovaly vyšší enkapsulační účinnost theofylinu v porovnání s pevnými disperzemi na bázi Eudragitu® RSPO nebo polymerních směsí a zajistily prodloužené uvolňování theofylinu v intervalu více než 24 hodin. Vzorek SD4 uvolnil 95,52 % léčiva v SIF a 93,56 % v SGF. Z toho důvodu byl vybrán jako optimální formulace.
Klíčová slova:
pevné disperze • prodloužené uvolňování • Eudragit® • uvolňování léčiva
Introduction
Drug delivery via the oral route remains the most acceptable route to patients for the administration of therapeutically active drug molecules. This has been attributed to the safety profile, ease of administration, patient acceptance and cost-effective manufacturing process associated with oral dosage forms1). Oral route of administration is considered more flexible in dosage form design than the parenteral route and drugs administered via this route are well-absorbed2).In the formulation of oral drug delivery systems, controlled release preparations are preferred because they have been able to address many problems associated with conventional multiple dosing regimen of drugs with rapid onset of action and short half-life3). Controlled drug delivery systems are designed to release the entrapped drug in its matrix over an extended period with better patient compliance.
Several approaches to sustain the release of drugs have been employed ranging from coated tablets and gels to biodegradable microparticles and osmotic systems. However, most of the controlled release formulations prepared in this manner produces a rapid initial release of drug in the release medium before a steady rate of release is attained. This phenomenon referred to as “burst release” leads to higher initial drug delivery and reduces the effective lifetime of the device3). Hence, an alternative to controlling drug release was found in the solid dispersion approach.
Solid dispersions have been used widely in various formulations to modulate dissolution rate and bioavailability of poorly water-soluble drugs. Many studies have reported the development of controlled release delivery systems using solid dispersion approach4–6). In the preparation of controlled release solid dispersions, water-insoluble carriers like ethylcellulose, hydroxypropyl cellulose, hydroxypropylmethyl cellulose and methacrylic acid polymers have been used. These carriers have exerted a major influence on the release profile of the dispersed drug.
Theophylline is the most effective bronchodilator of the methylxanthines used in the management of acute and chronic reversible airway obstruction7). Theophylline base is only slightly soluble in water therefore it is usually administered as the salt form containing various proportion of base. It is rapidly and completely absorbed after oral administration and plasma levels of 5–20 μg/mL are required after administration to achieve optimum therapeutic effects8). Conventional dosage forms of theophylline are administered 3–4 times daily to achieve effective plasma concentration7), thus requiring multiple dosing. This therefore, makes theophylline a suitable candidate for controlled release formulation.
In the present study, Eudragit® polymers were employed in the formulation of theophylline solid dispersions prepared by the solvent evaporation technique. The method aims to dissolve the drug and carrier simultaneously; followed by the removal of solvent by evaporation to produce co-evaporates of the drug and carrier. Eudragit® RS and RSPO were chosen for the study because they are less permeable compared to other Eudragit® carriers and drug release is controlled by diffusion mechanism.
Experimental part
Materials
The materials include theophylline (Sigma Aldrich laborchemikalien GmbH, Germany), Eudragit® RS 100 and RSPO (Evonik Pharma, Germany), Monobasic Potassium Phosphate (Sigma Chemical Co., USA), Sodium Hydroxide (Merck, Germany), Conc. Hydrochloric Acid, Potassium Chloride (BDH Chemicals Poole, England). All other chemicals used were of analytical grade.
Methods
Preparation of solid dispersions
Theophylline solid dispersions were prepared using Eudragit® RS 100 and Eudragit® RSPO by solvent evaporation method. Theophylline (2 g) was dissolved in a mixture of acetone and methanol (3 : 1), the carriers were dissolved in methanol (50 mL) and the two solutions were mixed. The resulting solution was stirred for 1 h and the solvent removed by evaporation at room temperature for 24 h. The residue (co-evaporate) obtained was pulverized and stored for further analysis. Physical mixtures containing the drug and carriers (Eudragit® RS 100 and RSPO) were prepared by simple trituration according to the ratios utilized for the dispersions (Table 1).
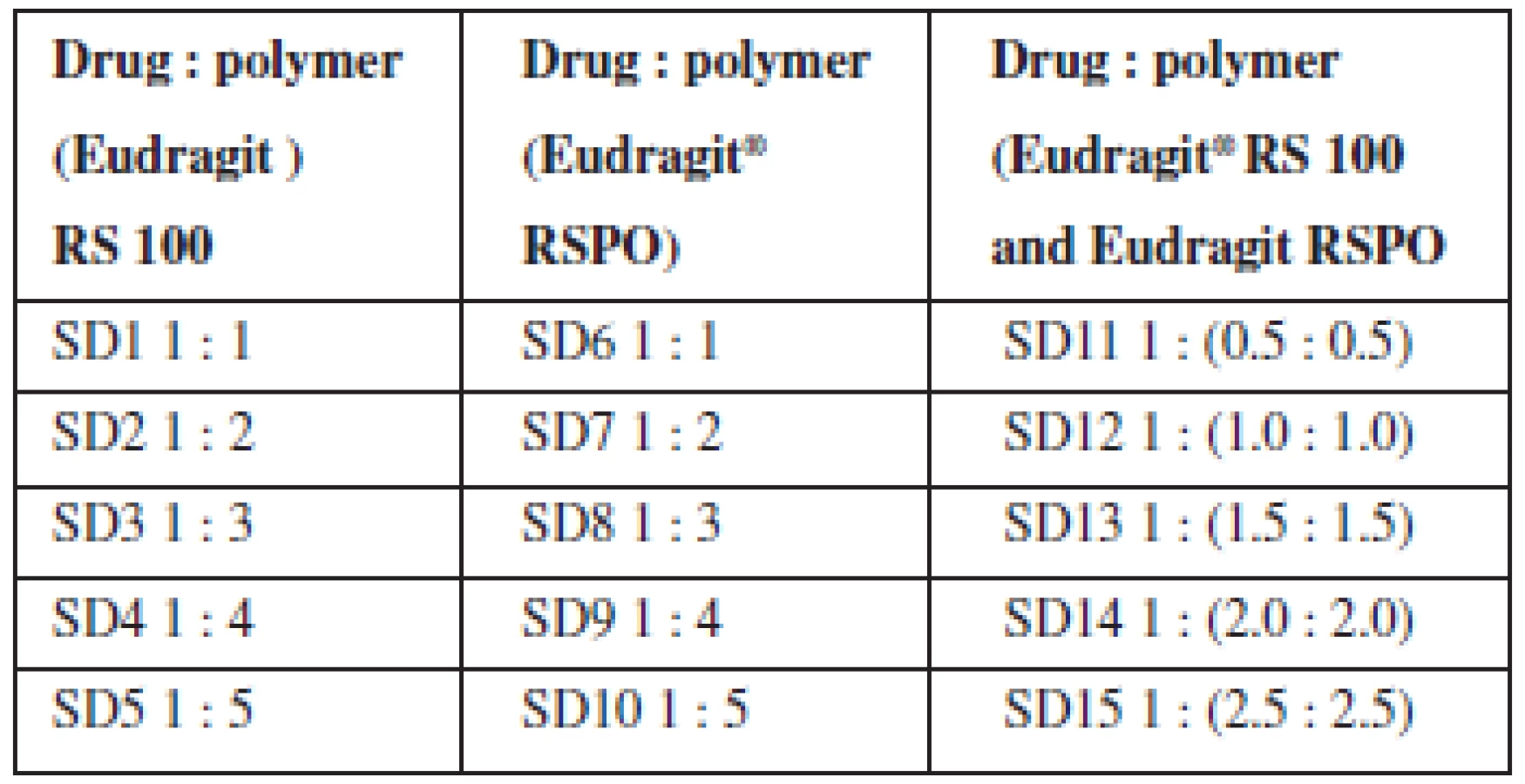
Determination of percentage yield
The percentage yield of theophylline solid dispersions was determined according to method described by Poovi et al9) using the following equation:

Entrapment efficiency
Solid dispersion equivalent to 100 mg theophylline was dissolved in 100 mL simulated intestinal fluid (SIF) without pancreatin (pH 7.4). One (1) mL of the solution was diluted to 50 mL with the same fluid, shaken vigorously, filtered and then analyzed spectrophotometrically (Jenway Model 6405 UV-Vis spectrophotometer, England) for drug content at 271 nm. The entrapment efficiency (EE) was calculated using the formula below10):

Determination of solubility
Solid dispersions equivalent to 100 mg of theophylline was transferred to a flask containing 50 mL SIF without pancreatin (pH 7.4). The sample was agitated at 80 rpm in a thermostated water bath shaker (CS 200G) at 37 ± 0.5 °C for 24 h. The supernatant solution was filtered (0.45 ∝m Whatman filter paper), diluted and the absorbance was measured using Jenway Model 6405 UV-Vis spectrophotometer, England. The procedure was repeated using the simulated gastric fluid (SGF) without pepsin (pH 1.2). A mean of three determinations was obtained for each evaluation.
Dissolution studies
The dissolution test for the prepared solid dispersions and physical mixtures was determined as described in the USP11) using dissolution apparatus type II (Universal dissolution tester, UDT 804, United States). SGF without pepsin (pH 1.2, 900 mL) was prepared and thermostated at 37 ± 0.5 °C. Samples equivalent to 100 mg theophylline were placed in a polycarbonate dialysis membrane (8000 molecular weight cut off, Sigma Aldrich, Germany) already pretreated and immersed in the dissolution medium as illustrated in Figure 1. The dissolution apparatus was stirred at a rate of 50 rpm as described by Ofokansi et al.10). At predetermined time intervals, 2 mL aliquots of the dissolution medium were withdrawn and replaced with equivalent volume of fresh medium maintained at the same temperature to ensure sink conditions throughout the study period. The withdrawn samples were filtered and assayed spectrophotometrically at 271 nm. The dissolution of pure theophylline powder was also carried out to compare its release profile with the dispersions. The entire dissolution process was repeated in SIF without pancreatin (pH 7.4). The time taken to release 75% and 90% of the drug, respectively (T75 & T90), and the amount of drug released after 5 and 15 h, respectively (D5 & D15), were extrapolated from the dissolution plot.

Statistical analysis
The results obtained were subjected to statistical analysis and expressed as mean ± standard deviation. The differences between means were considered significant at p < 0.05 using one sample t-test. Relationship was established at p > 0.05 using Pearson’s correlation. All statistical analyses were carried out using SPSS (version 20).
Results
Percentage yield of solid dispersions
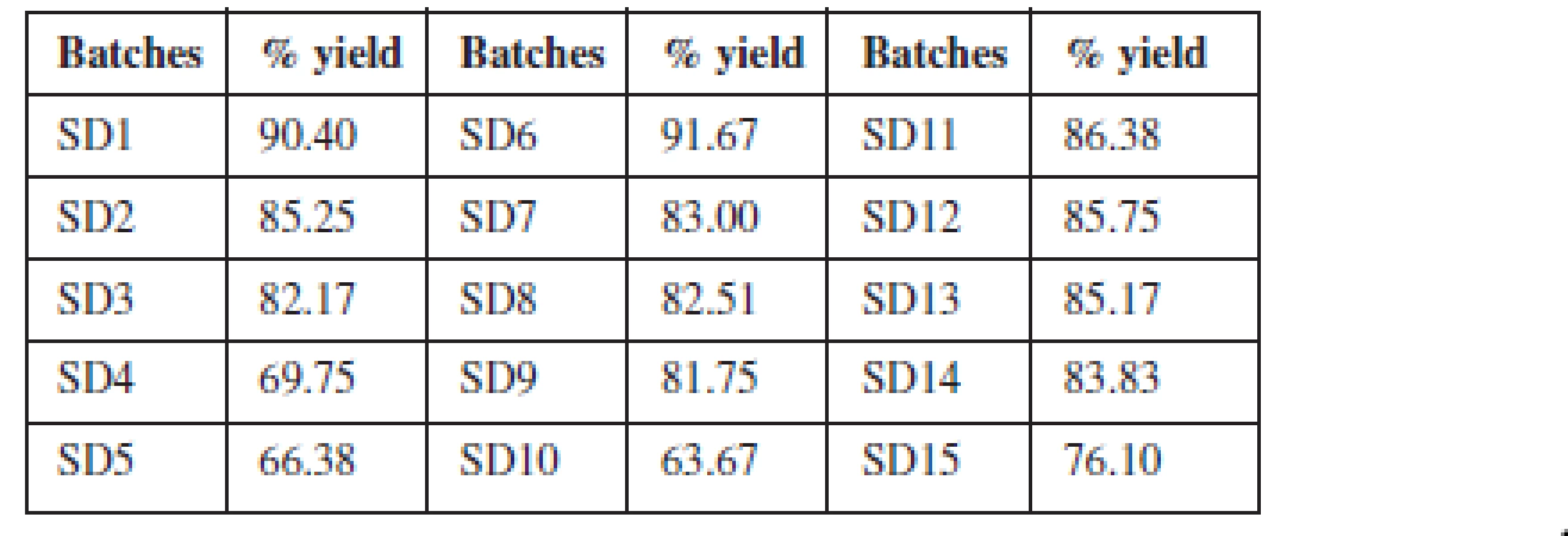
The % yield of solid dispersions prepared using the two polymers and their blends ranged between 63–91% as presented in Table 2. The yield decreased as the proportion of polymer in the dispersion increased. The blend of the two polymers in equal proportions as a carrier system in the formulation of solid dispersions did not significantly influence the yield.
Entrapment efficiency of solid dispersions
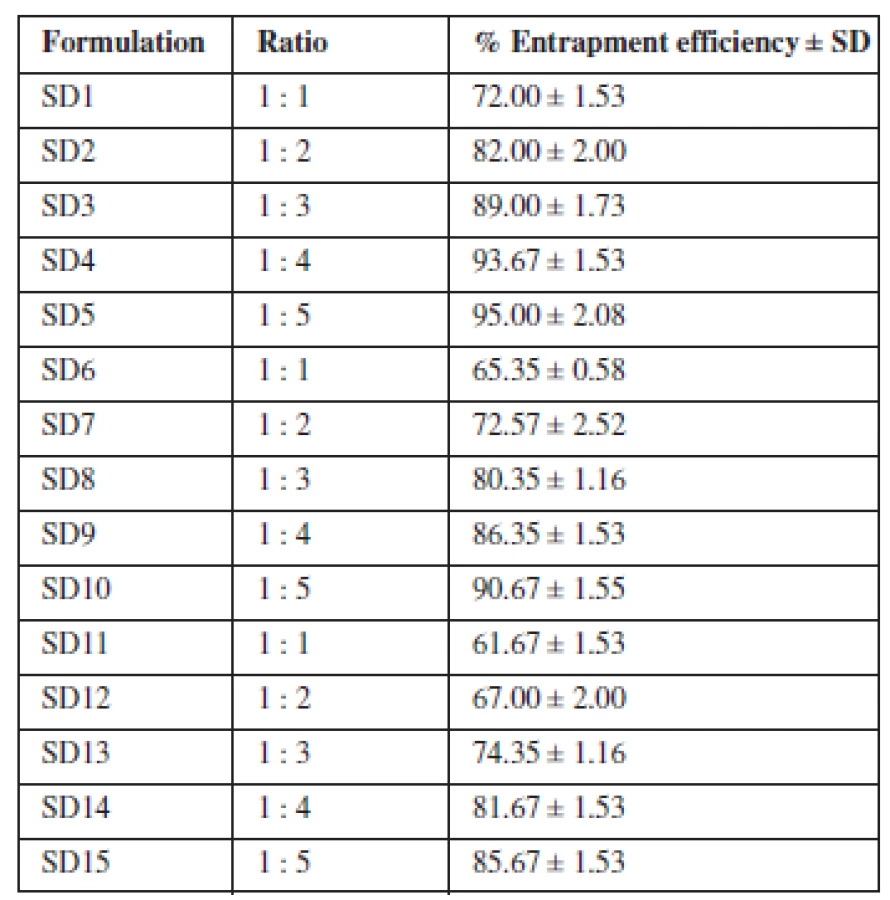
The amount of drug entrapped by the various formulations ranged between 61.67–95% (Table 3). There was a corresponding increase in the amount of drug entrapped as the proportion of the carrier increased in the dispersion. Maximum entrapment efficiency was attained with formulations containing Eudragit® RS 100 as carrier. Comparing the entrapment efficiency of the individual polymers and their blend, a greater degree of drug entrapment was achieved with the individual polymers relative to their blend. Hence, the combination of the two polymers did not demonstrate any synergy with respect to entrapment efficiency. The extent to which theophylline was entrapped by the polymers and their blend commends the suitability of the solvent evaporation technique in developing solid dispersions.
Solubility determination
The solubilities of theophylline and its various solid dispersions in SIF and SGF media is represented by Figures 2a, b, respectively. The solubilities of the solid dispersions in both media were found to be lower than that of the pure drug. It was observed that the solubility of theophylline in the solid dispersion decreased as the content of Eudragit® polymer (RS 100 & RSPO) increased. Hence, four solid dispersion formulations (SD4, SD5, SD9 and SD10) containing maximum concentrations of the two polymers were selected for further studies. The remaining formulations were discarded at this point.

Dissolution studies
The drug release from the various SDs formulated with Eudragit® RS 100 and RSPO in SIF and SGF media is depicted in Fig. 3a, b, respectively. The graph shows that the maximum release of SDs was found to be 95.52% in 24 h (SIF) and 93.56% in 24 h (SGF). The parameters generated from the plots are presented in Tables 4 and 5 for SIF and SGF media, respectively. All the SDs were able to extend the release of theophylline over a period of 24 h relative to the pure drug that achieved maximum release after 8 h and was not sustained afterwards. The drug release profile of the two polymers did not differ significantly from each other.

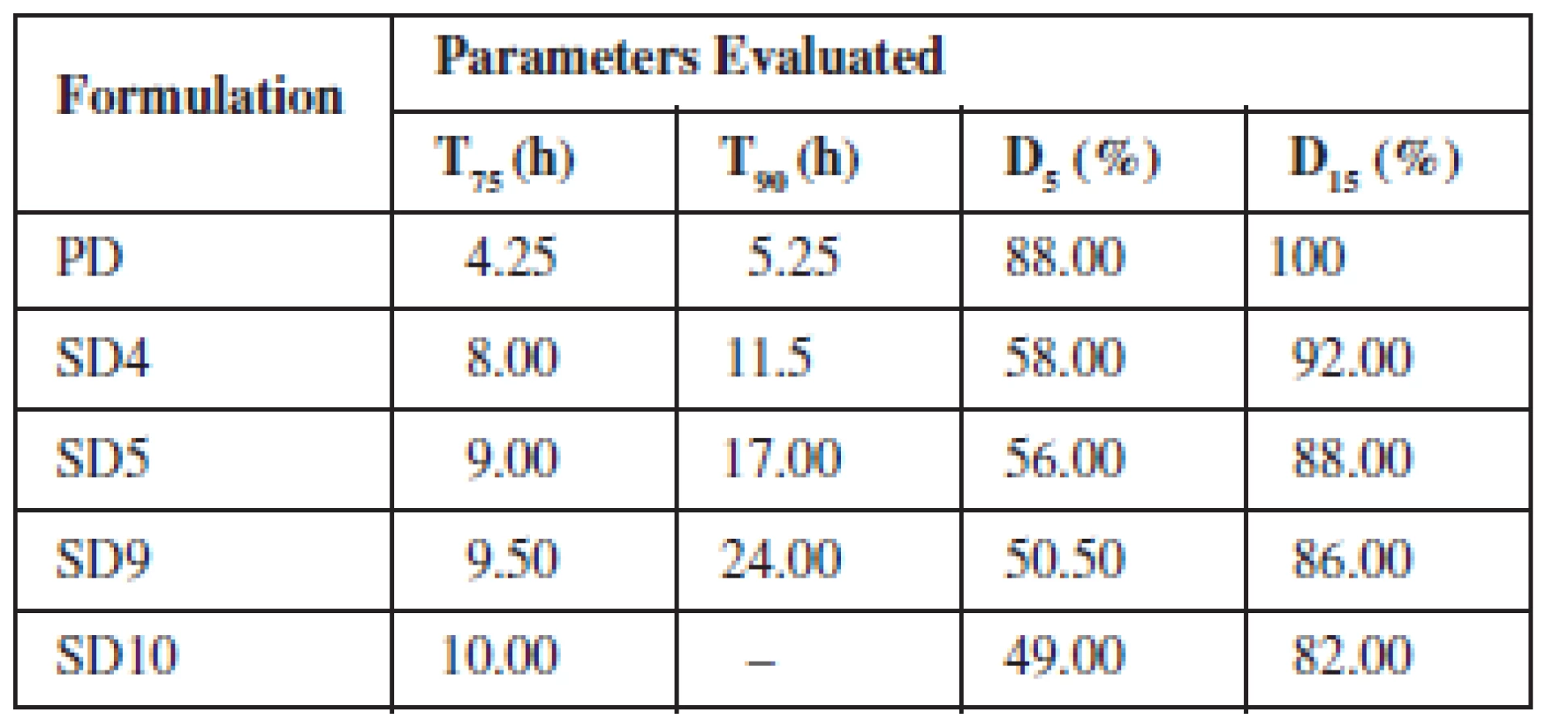
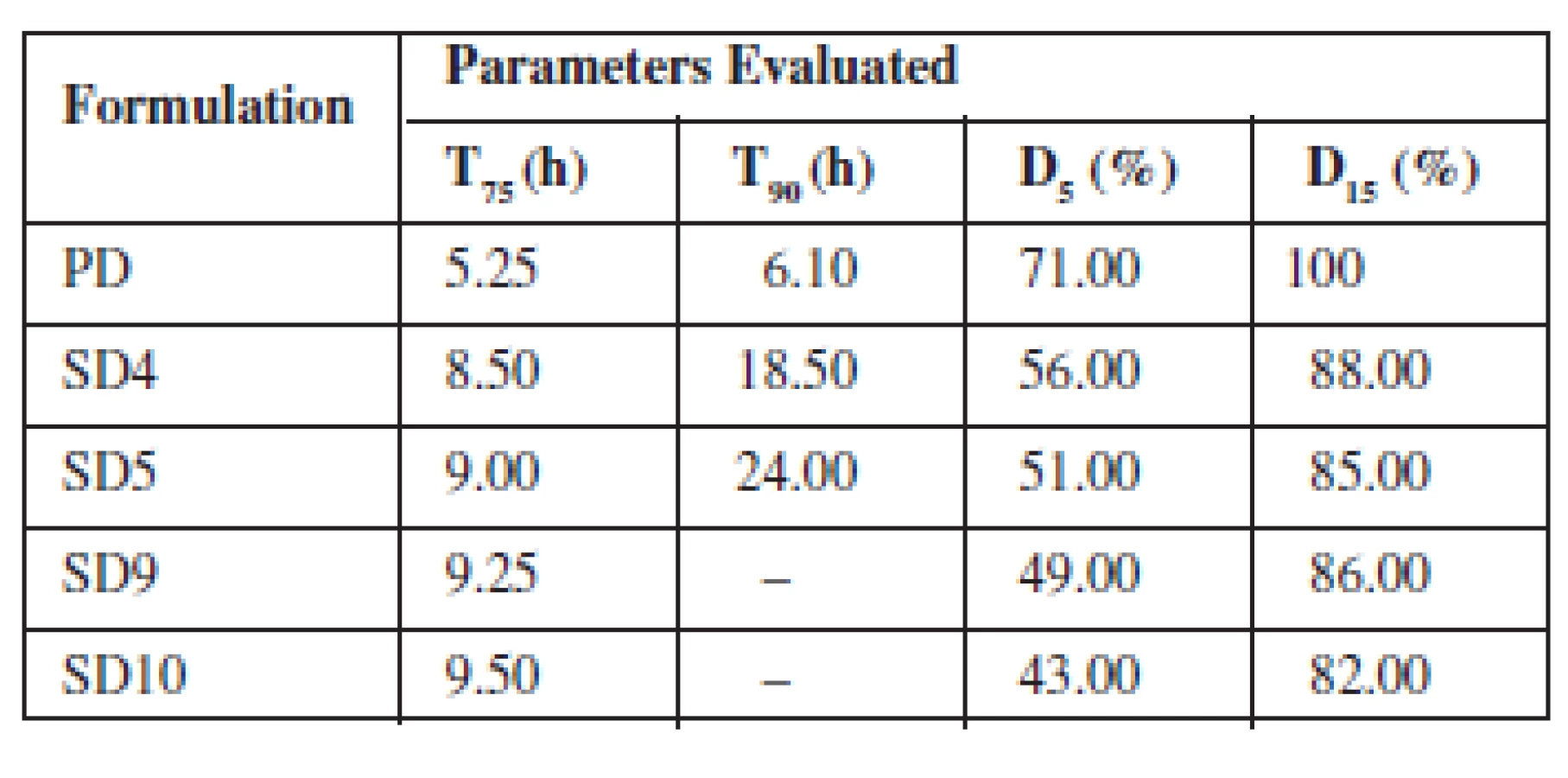
The rate at which drug was released from the SDs was much slower and in a sustained manner compared to the pure drug. A two-fold increase was observed in the time taken to achieve 75% and 90% release (T75 & T90) from the SDs when compared to the time taken for the same amount of pure drug to be released. There was a cumulative increase in the amount of drug released over a period of 24 h by the SDs compared to the pure drug whose release peaked after 8 h.
Discussion
Preliminary investigations
The percentage yield of theophylline SDs was determined to ascertain the losses incurred during formulation. A relatively low yield of the solid dispersions was obtained as the concentration of the polymer increased. For dispersions formulated with the individual polymers, the batches formulated with Eudragit® RSPO had a higher yield than the batches formulated with Eudragit® RS 100. The lower yield of solid dispersions with Eudragit RS 100 may be due to the fact that at high concentrations, it forms a rubbery mass which is very sticky and attaches to the container. Hence, some of the dispersion formed may be lost in the process of trying to recover the entire mass.
The extent to which the formulated SDs were able to entrap the loaded drug may have been related to the particle properties of the polymers as they did not differ essentially in their composition. Overall, the entrapment efficiency increased as the concentration of the polymers increased. This is consistent with the findings of Aenugu and Abbaraju12) who prepared aspirin loaded ethyl cellulose by solvent evaporation method. The entrapment efficiency improved with increase in polymer concentration because more of the polymer was available to incorporate the drug. Entrapment efficiency was highest with Eudragit® RS 100 probably due to the availability of more spaces to take up the drug due to the porosity of the granules. More spaces for drug uptake are created with low bulk density materials (High porosity) as evidenced by the superior loading capacity of microcrystalline cellulose for direct compression13, 14). Entrapment efficiency is an important parameter for assessing the drug loading capacity of solid dispersions and their drug release profiles, thus suggesting the amount of the drug that would be available at the absorption site. The choice of the polymer and the method of formulation of the solid dispersion are two critical factors that combined to yield the high output of entrapment efficiency. In a related study, solid dispersion with high entrapment efficiency has been linked to the polymer combination and the reproducibility of the solvent evaporation method15).
Solubility studies
The rationale behind the development of solid dispersions is to modulate the aqueous solubility of poorly soluble drugs for enhanced drug delivery. In this study, solid dispersions were designed to extend the release of theophylline by lowering its solubility. The solid dispersions were developed with water insoluble carriers (methacrylic polymers) hence the decrease in solubility compared to the pure drug. Higher solubility was obtained in SIF relative to SGF. This observation was in contrast to the findings of Aiman16) who reported greater solubility of theophylline and the solid dispersions in SGF than SIF. The extent to which solubility decreased was a function of the concentration of the polymer utilized as higher concentrations resulted in the least solubility. The presence of low content of charged groups in the polymers prevents water permeability and swellability thereby limiting the solubility of the drug17). The difference in solubilities of the pure drug and solid dispersions in the two media was statistically significant at p < 0.05. Pearson’s correlation demonstrated a significant relationship between the solubility of the solid dispersions and the physical mixtures in each of the medium.
Dissolution studies
The dispersion of the drug in the polymer matrices strongly influenced their dissolution rate which appeared slower and more gradual than that of the pure drug. The presence of the polymer reduced the massive initial drug dissolution observed with pure theophylline. As the proportion of the polymer increased, the permeability of water in the formulation decreased and hence a better sustained release was achieved. Eudragit RS 100 and Eudragit RSPO are methacrylic acid polymers (insoluble polymers) and they exhibit pH independent swelling. They are water insoluble polymers that are widely used as wall material for sustained release formulations due to their biocompatibility, good stability and easy fabrication18). They both contain low content of quaternary ammonium groups and they are considered to have low permeability to water. The slow drug release from solid dispersions with Eudragit RS polymers can be attributed to the low permeability of the polymers which pose a significant hindrance to fluid penetration and passive drug diffusion19).
Comparing the drug release profile of the two polymers, it was observed that Eudragit RSPO extended the release more than Eudragit RS 100. It took a longer time for dispersions with Eudragit RSPO to attain 75% and 90% release and the amount of drug released in 5 and 15 h was seen to be higher in Eudragit RS 100 than Eudragit RSPO. The difference in particle properties of the two polymers may have been responsible for the slight variation in drug release. In solid dispersions, the drug forms a complex with an inert soluble carrier in solid state. The availability of the drug depends on the solubility of the polymer and the absorption rate of the drug. Therefore, for a drug to exhibit extended release the use of a water insoluble polymer is required. Eudragit RS polymers are neutral co-poly (Ethylacrylate, methyl methacrylate) and trimethyl aminoethyl methacrylate that do not dissolve readily in water and digestive juices but swells and demonstrates low permeability leading to drug release principally by diffusion20). Therefore the permeability of the drug through Eudragit RS polymers is independent of the pH of the digestive tract21).
Conclusion
Solid dispersions of theophylline prepared by solvent evaporation technique using Eudragit RS 100 and Eudragit RSPO extended the release of theophylline. Formulation SD4 containing drug: Eudragit RS 100 in ratio 1 : 4 gave the highest extended release in the simulated fluids of 93.56% in 24 h for SGF and 95.52% in 24 h for SIF while the pure drug completely dissolved in 6 h. A once daily dosage can therefore be formulated with these two polymers.
Conflict of interest: none.
Received November 11, 2016 / Accepted December 1, 2016
Dr. Yonni Apeji (PhD)
Department of Pharmaceutics and Pharmaceutical Microbiology
Faculty of Pharmaceutical Sciences
Ahmadu Bello University, Zaria
Kaduna State, Nigeria
e-mail: yehonathanapeji@gmail.com
Sources
1. Chalo C. S. L., Robinson J. R., Lee V. H. C. Sustained release drug delivery systems. In: Osol A., Gennaro A. R., Gibson M. R., et al. (eds.) Remington’s Pharmaceutical Sciences, 17th ed. Pennsylvania: Mack Publishing Co. 1985.
2. Lachman L., Lieberman H. A., Kanig J. I. The theory and practice of industrial pharmacy, 2nd ed. Bombay: Varghese Publishing House 1996.
3. Giri T. K., Kumar K., Alexander A., Badwaik H., Tripathi D. K. A novel and alternative approach to controlled release drug delivery system based on solid dispersion technique. Bulletin of Faculty of Pharmacy, Cairo University 2012; 50(2), 147–159.
4. Verma S., Rudraraju V. S. Disintegration mediated controlled release supersaturating solid dispersion formulation of an insoluble drug: design, development, optimization, and in vitro evaluation. AAPS PharmSciTech 2015; 16(1), 85–97.
5. Perge L., Robitzer M., Guillemot C., Devoisselle J. M., Quignard F., Legrand P. New solid lipid microparticles for controlled ibuprofenrelease: formulation and characterization study. International Journal of Pharmacutics 2012; 422(1–2), 59–67.
6. Kim H. J., Lee S. H., Lim E. A., Kim J. S. Formulation optimization of solid dispersion of mosapride hydrochloride. Archives of Pharmaceutical Research 2011; 34(9), 1467–1475.
7. Hanan M. E. Optimzation of Eudragit RS microspheres for controlled release of theophylline using response surface methodology. Journal of Pharmaceutical Sciences and Research 2010; 2(10), 663–671.
8. Katzung, B. G., Masters, S. B., Trevor, A. J. (eds.) Basic and Clinical Pharmacology, 11th ed. New York, NY: The McGraw-Hill Companies Inc. 2009.
9. Ofokansi K. C., Kenechukwu F. C., Isah A. B., Allagh T. S., Anumeka O. O. Formulation and evaluation of solid dispersions based on Eudragit RS 100 and PEG8000 for improved delivery of trandolapril. Africa Journal of Pharmaceutical Research and Development 2012; 4(1), 38–42.
10. Poovi G., Umamahaswari M., Sharmila S., Kumar S., Rajalakshimi N. Development of domperidone solid dispersions powders using sodium alginate as carrier. European Journal of Applied Sciences 2013; 5(2), 36–42.
11. United States Pharmacopeia. Published by the United States Pharmacopoeia convention 2011.
12. Aenugu S. R, Abbaraju K. S. Preparation and characterization of Aspirin loaded ethylcellulose nano particles by solvent evaporation technique. World Journal of Pharmacy and Pharmaceutical sciences 2014; 3(6), 1781–1793.
13. Kothari S. H., Umar V., Banker G. S. Comparative evaluations of powder and mechanical properties of low crystallinity celluloses, microcrystalline celluloses, and powdered celluloses. International Journal of Pharmaceutics 2002; 232, 69–80.
14. Bhimte N. A., Tayade P. T. Evaluation of microcrystalline cellulose prepared from sisal fibers as a tablet excipient: a technical note. AAPS PharmSciTech 2007; 8(1), E1–E7.
15. Rathinarag B. S., Rajeer C., Choudhury P. K., Gannesh S. B., Shinde G. V. Studies on the dissolution behaviour of sustained release solid dispersions of nimodipine. International Journal of Pharmaceutical Sciences Review Research 2010; 3(1), 77–82.
16. Aiman A. O. Modulation of the micro-environmental pH and its influence on the gel layer behaviour and release of theophylline from hydrophilic matrices. International Journal of Pharma and Bio Sciences 2013; 4(2), 794–802.
17. Jenquin M., Liebowitz S. M., Sarabia R. E., McGinity J. W. Physical and chemical factors influencing the release of drugs from acrylic films. Journal of Pharmaceutical Sciences, 1990; 79, 811–816.
18. Marwa H. A., Omaina A. S., Hanaa A. E., Hanan M. E., Waleed B. Development and characterization of controlled release ketoprofen microspheres. Journal of Applied Pharmaceutical Science 2012; 2(3), 60–67.
19. Shivkumar H. N., Desai B.G., Deshmukh G. Design and optimization of diclofenac sodium controlled release solid dispersion by response surface methodology. Indian Journal of Pharmaceutical Sciences 2008; 70, 22–30.
20. Kibbe, A. H. (ed.) Handbook of pharmaceutical excipients. Washington D.C., U.S.A: American Pharmaceutical Association 2000.
21. Apurba S. A., Atiqul H. P., Golam K. Reza U. J. In vitro release kinetic study of theophylline from Eudragit RSPO and Eudragit RLPO matrix tablets.
Labels
Pharmacy Clinical pharmacologyArticle was published in
Czech and Slovak Pharmacy

2016 Issue 6
Most read in this issue
- Hodnocení vlivu koncentrace účinné látky a technologie mísení na obsahovou stejnoměrnost směsi pro přípravu tablet s nízkým obsahem warfarinu
- Fraktální aspekty sypného a smykového chování volně sypných velikostních frakcí přímo lisovatelné farmaceutické pomocné látky sorbitolu
- Vliv EUDRAGITU® RS na disoluční chování pevných disperzí theofylinu
- Míra porozumění odborným lékařským termínům u italských studentů