
Léčivé látky z mořských organismů v klinických studiích a praxi
Active substances from marine organisms in clinical trials and practice
Oceans cover a large part of our planet and they are a home for an enormous amount of species. A lot of them are still waiting to be discovered by man, much like the chemicals they synthesize. Marine pharmacology concerns itself with the study of these chemicals and their potential use in medicine. Origin in marine species is for the most part the only thing this large and diverse group of substances have in common, so the spectrum of possible applications is quite wide. Many of these substances have a unique mechanism of action, offering new therapeutic possibilities. Although just a few of them are used in a clinical practice today (e.g. eribulin, cytarabine), the future looks quite promising. Current clinical trials focus mostly on the therapy of cancer, but trials for therapy of pain or Alzheimer’s disease and many others are also underway.
Key words:
marine pharmacology
Authors:
D. Mareček; PharmDr. Jana Rudá-Kučerová, PhD.
Published in:
Čes. slov. Farm., 2017; 66, 191-207
Category:
Review Articles
Overview
Oceány pokrývají velkou část povrchu naší planety a jsou domovem nepřeberného množství organismů. Řada z nich teprve čeká na své objevení, podobně jako chemické látky, které syntetizují. Odvětví farmakologie označované v zahraniční literatuře termínem „marine pharmacology“ se zabývá studiem právě těchto látek a jejich využitím v medicíně. Původ v mořských organismech je pro většinu z nich jediným pojítkem a tato různorodost se promítá i do širokého spektra možného využití. Mnohé se vyznačují zcela unikátním mechanismem účinku nabízejícím nové možnosti terapeutického působení. Ačkoliv jich do klinické praxe zatím proniklo jen několik (například eribulin či cytarabin), potenciál je obrovský. V klinických studiích v současnosti sice převažují farmaka pro terapii nádorových onemocnění, zkoumány jsou ale i látky s potenciálním využitím v léčbě bolesti či Alzheimerovy choroby a mnoha dalších.
Klíčová slova:
léčiva mořského původu
Úvod
Oceány pokrývají přes 70 % povrchu naší planety a jsou dle odhadů domovem pro více než 25 % všech rostlinných a živočišných druhů1). Je tedy logické, že ani tato oblast neušla zájmu vědy při hledání nových biologicky účinných látek a farmak. Objev cefalosporinů v plísni Cephalosporium acremonium, kultivované z mořské vody profesorem Giuseppem Brotzu (a následná práce výzkumného týmu oxfordské univerzity2)) během čtyřicátých let 20. století je jedním z velkých příběhů o „lovcích mikrobů“ a zároveň počátkem vývoje celé skupiny antibiotik, dnes tak běžných v klinické praxi3).
Podobně i počátek výzkumu potenciálního farmakologického využití metabolických produktů mořských bezobratlých sahá až do padesátých let 20. století4, 5). Jeho prvním cílem se stali především zástupci houbovců (Porifera), pláštěnců (Tunicata) a měkkýšů (Mollusca) obývající pobřežní oblasti oceánu, vzhledem k jejich relativně snadné dostupnosti4, 6). Později, s rozvojem technologie přístrojového potápění se začala oblast výzkumu rozšiřovat. V centru pozornosti nicméně stále zůstávají korálové útesy, porosty mangrovů a další ekosystémy s vysokou biodiverzitou, které tak poskytují největší množství potenciálně zajímavých látek6). Těmito látkami jsou nejčastěji sekundární metabolity, tedy produkty, které organismus nezbytně nepotřebuje ke svému přežití. Zdá se však, že situace není zdaleka tak jednoduchá a u řady látek dříve izolovaných se prokázalo, že tyto ve skutečnosti nejsou produktem daného organismu ale symbiotických mikroorganismů7, 8). Mikroorganismy mohou podle některých studií tvořit 40 až 60 % biomasy některých houbovců9), kteří jsou skutečnou „pokladnicí” potenciálních léčiv5, 10).
Mnohdy velmi komplexní struktura těchto látek představuje skutečný oříšek pro jejich průmyslovou syntézu11, 12). Ta je však často ekonomicky i časově výhodnější metodou zisku dostatečného množství účinné látky než lov volně žijících organismů, ať už se jedná o semisyntézu (tedy syntézu vycházející z určitého prekurzoru) či syntézu totální4, 13). Pro výrobu gramových množství je totiž u řady z těchto látek potřeba několika tun daného organismu13–15).
Chemická syntéza není jedinou alternativou ke zpracovávání volně žijících organismů. Některé z nich se daří pěstovat přímo v moři či laboratorně16, 17). Obě varianty však přináší řadu problémů. Při pěstování v moři můžeme jen těžko ovlivnit celou řadu environmentálních faktorů, v laboratorních podmínkách může představovat velkou výzvu vytvoření vhodného mikroprostředí (teplota, salinita, pH a mnohé další faktory)13). Ani v případě, že se podaří např. houbovce vypěstovat, není úspěch zaručen, jelikož i dobře rostoucí organismy mohou mít velmi nízký obsah účinné látky. Zdá se, že příčinou budou faktory prostředí, které sice mohou vyhovovat makroorganismu, zásadním způsobem však ovlivňují společenství mikroorganismů13, 17, 18).
Další výhodnou metodou může být biosyntéza účinných látek ve snadněji kultivovatelných organismech (formou heterelogní exprese)19).
Smyslem tohoto textu je postihnout informace o nejzajímavějších léčivech mořského původu nacházejících se v klinické fázi vývoje, jejichž shrnutí obsahuje tabulka 1. V dalším textu pak následují podrobné informace o jednotlivých látkách.
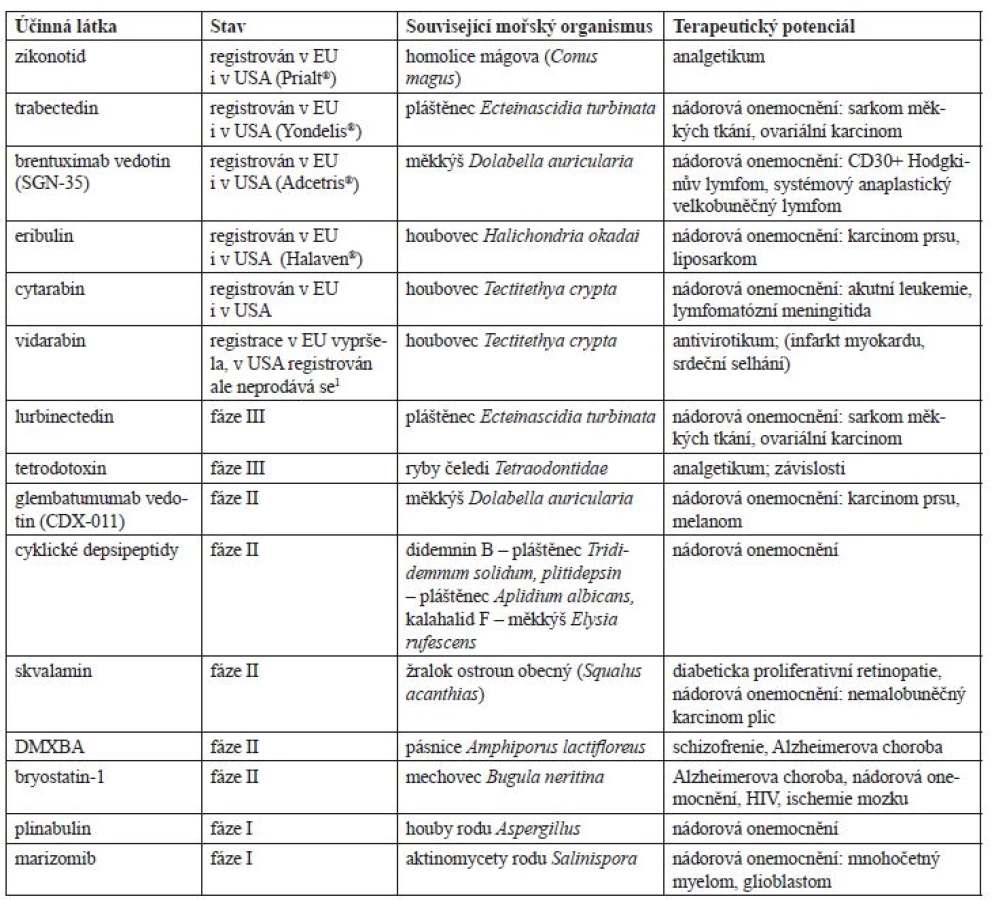
Zikonotid (SNX-111)
Zikonotid je syntetický analog ω-konopeptidu MVIIA, který je součástí jedu mořského plže Conus magus20). Působí jako selektivní blokátor napěťově řízených vápníkových kanálů typu N. Tento typ kanálů pro vápenaté ionty se nachází např. na neuronech21), v srdci22) či v ledvinách23), kde se účastní regulace řady kalcium-dependentních procesů, jako je uvolňování neurotransmiterů či aktivace systému druhých poslů. Lokalizace těchto kanálů na neuronech dorzálních míšních rohů a blok ascendentní dráhy bolesti jsou pravděpodobným vysvětlením analgetického působení zikonotidu20, 24).

V České republice je zikonotid dostupný v registrovaném léčivém přípravku PRIALT, který však v současnosti není na Seznamu cen a úhrad léčivých přípravků a potravin pro zvláštní lékařské účely, není tedy hrazen z veřejného zdravotního pojištění. Dle SPC je PRIALT indikován k léčbě silné, chronické bolesti u dospělých, kteří vyžadují intratekální analgezii. Výraznou výhodou zikonotidu oproti opiovým i jiným analgetikům je, že na jeho analgetický účinek nevzniká tolerance25). Mezi hlavní nevýhody patří nutnost intratekálního (IT) podání, časté nežádoucí účinky (závratě, nauzea, zmatenost či jiné kognitivní a neuropsychiatrické projevy a další) a úzký terapeutický index. Z klinických studií vyplývá i výhodnost kombinované IT analgezie s morfinem u obtížně kontrolovatelné bolesti, která umožňuje snížení dávky zikonotidu i opioidů26, 27).
Jedna případová studie (tři pacienti s různě pokročilým nádorovým onemocněním a obtížně zvladatelnou bolestí a jeden pacient s centrální neuropatickou bolestí) ukazuje na možnost využití pro intracerebroventrikulární terapii28).
Kromě analgetického působení by snad v budoucnu mohl zikonotid najít uplatnění i v terapii ischemického poškození mozku. Studie na potkanech a králících ukázaly, že zikonotid významně snižuje hromadění vápníku v ischemických neuronech a může tak snižovat míru jejich poškození29–31).
Trabectedin (Ecteinascidin-743, ET-743)
Trabectedin byl poprvé izolován z mořské sumky Ecteinascidia turbinata32). Na základě genomové analýzy se však zdá, že skutečným producentem je symbiotický mikroorganismus Candidatus endoecteinascidia frumentensis33, 34). Tento mikrob má velice redukovaný genom (ecteinascidin je dle genomové analýzy pravděpodobně jeho jediný sekundární metabolit) a zachování genu pro ecteinascidin zřejmě svědčí o významnosti produkce tohoto metabolitu pro vztah symbionta s hostitelem34).

Účinek trabectedinu v protinádorové terapii pravděpodobně vychází z několika mechanismů. Jedním vysvětlením je jeho vazba do oblasti malého žlábku (minor groove) DNA, kde působí alkylaci aminoskupiny guaninu na pozici 235). Vazba trabectedinu na DNA změní její strukturu a brání tak vazbě proteinů nutných pro replikaci DNA36) a naruší funkci nucleotide excision repair (NER) systému37). Narušení NER vede ke vzniku jednořetězcových zlomů DNA neúčinnou excizí alkylovaného nukleotidu38). Výsledky některých studií ukazují, že funkční NER je pro dobrý účinek trabectedinu zásadní. Porucha v NER systému výrazně snižuje jeho účinnost, naopak zvýšeně aktivní NER může vést k zesílení účinnosti trabectedinu38–40).
Jiné zdroje uvádějí, že alespoň část účinku trabectedinu v protinádorové léčbě vychází z jeho selektivního působení na makrofágy. Skupinou makrofágů, která nás v tomto případě zajímá, jsou tzv. tumor associated macrophages (TAM)41). TAM hrají významnou úlohu v nádorovém mikroprostředí a mohou přispívat k jeho progresi, např. proangiogeneticky42–44).
Dalším mechanismem může být kompetice trabectedinu o vazebné místo v malém žlábku DNA s high mobility group A (HMGA) proteiny, kde znemožňuje jejich vazbu a tím narušuje jejich vliv na regulaci transkripce řady genů a mimo jiné zvyšuje citlivost buněk k radioterapii45).
HMGA-1 jsou zvýšeně exprimovány v celé řadě malignit a hrají významnou roli v jejich progresi46).
Zajímavým účinkem trabectedinu je i snížení exprese P-glykoproteinu, které může zvýšit účinnost jak trabectedinu, tak i dalších cytotoxických látek47).
Trabectedin je v České republice registrován v přípravku YONDELIS, v současnosti není hrazen z veřejného zdravotního pojištění. Je indikován k léčbě pokročilého sarkomu měkkých tkání (poté, co selže léčba antracykliny a ifosfamidem nebo pokud tyto nejsou vhodné) a v kombinaci s pegylovaným liposomálním doxorubicinem k léčbě pacientek s relabujícím ovariálním karcinomem citlivým na platinu36).
Výsledky malé, multicentrické studie 2. fáze (52 pacientů) svědčí pro možné budoucí využití trabectedinu u pokročilého karcinomu prsu po terapii antracykliny a taxany (response rate 12 % u pacientů, jimž byl trabectedin podáván jednou za 3 týdny)48).
Brentuximab vedotin (SGN-35)
Brentuximab vedotin je konjugát protilátky anti-CD30 a cytotoxického agens monomethyl auristatinu E. Monomethyl auristatin E (MMAE) je syntetický analog inhibitoru polymerizace tubulinu, dolastatinu 1049), který byl izolován z mořského plže Dolabella auricularia50).
Na tubulinu se dolastatin váže do místa poblíž vazebného místa pro vinca alkaloidy a jeho vazba způsobuje nekompetitivní inhibici vazby vincristinu51).
Kromě inhibice polymerizace tubulinu působí dolastatin 10 i na dendritické buňky (mimo jiné zrychluje jejich maturaci) a jeho protinádorové působení tak vychází i z aktivace celulární imunity, dle výzkumu na myších vede porucha imunity k oslabení jeho účinku52).
Brentuximab vedotin je u nás registrován v léčivém přípravku ADCETRIS53), je indikován k léčbě dospělých pacientů s relabujícím CD30+ Hodgkinovým lymfomem po autologní transplantaci kmenových buněk (ASCT) nebo po nejméně dvou předchozích terapiích v případech, kdy ASCT nebo kombinovaná chemoterapie nepředstavuje léčebnou možnost. Dále je indikován k léčbě dospělých pacientů s relabujícím nebo refrakterním systémovým anaplastickým velkobuněčným lymfomem (sALCL)53).
Případové studie ukázaly na možnost využití brentuximab vedotinu v terapii CD30+ Sézaryho syndromu54–56) a pokročilé mycosis fungoides55–57), výsledky klinické studie fáze II pak potvrdily jeho účinnost u kožních T-buněčných lymfomů i lymfomatoidní papulózy58). Další klinická studie fáze II pak prokázala jeho účinnost v terapii difuzního velkobuněčného B lymfomu (DLBCL) s variabilní expresí CD3059).
Jedna případová studie, kdy byl brentuximab vedotin podán pacientovi s CD30+ plasmablastickým lymfomem60) a nadějné výsledky výzkumu na buněčných liniích mesotheliomu61) snad ukazují jen některé další budoucí cíle terapie brentuximab vedotinem.

Eribulin mesylát
Eribulin (E7389) je syntetický derivát halichondrinu B62), který působí jako inhibitor polymerizace tubulinu63). Na rozdíl od vinblastinu či paklitaxelu (dalších látek působících na dynamiku tubulinu) neinhibuje proces zkracování mikrotubulů, pouze jejich růst a dynamicitu62, 64).
Studie na potkanech dále ukázaly vliv eribulinu na prokrvení nádorové tkáně (eribulin zvyšuje prokrvení nádoru a ovlivňuje tak strukturu nádorového mikroprostředí a snad i umožňuje lepší průnik chemoterapeutik65)), další studie (na potkanech a nádorových buněčných liniích) ukázaly, že eribulin snižuje epiteliálně-mezenchymální tranzici (EMT) a naopak posiluje mezenchymálně-epitelialní tranzici (MET)66, 67). EMT je proces fenotypických změn buňky, kdy dochází např. ke změnám v mezibuněčné adhezi, ztrátě polarizace buňky či posílení její schopnosti migrovat. Tento proces zásadně ovlivňuje invazivitu a schopnost tvořit metastázy68).
Halichondrin B byl poprvé izolován z mořského houbovce Halichondria okadai69), později i z dalších houbovců (např. Phakellia carteri)70). Izolace ze zástupců dvou různých rodů hobovců by mohla svědčit pro jeho mikrobiální původ63).
Eribulin mesylát je u nás dostupný v registrovaném léčivém přípravku HALAVEN, je indikován k léčbě dospělých pacientů s lokálně pokročilým nebo metastazujícím karcinomem prsu, jejichž stav se dále zhoršil po nejméně jednom chemoterapeutickém režimu, dále je indikován k léčbě dospělých pacientů s neresekovatelným liposarkomem, kteří již podstoupili léčbu antracyklinem (nebo pro ně tato léčba nebyla vhodná)71). HALAVEN je hrazen z veřejného zdravotního pojištění.
Jeho účinnost v terapii metastazujícího karcinomu prsu byla potvrzena studií fáze III – EMBRACE72), srovnávací studie fáze III pro terapii pokročilého liposarkomu či leiomyosarkomu (eribulin vs dakarbazin) potvrdila účinnost eribulinu i v této indikaci73).
Studie fáze II pak ukázaly aktivitu u pokročilého nemalobuněčného karcinomu plic (NSCLC)74–76), metastatického na kastraci rezistentního karcinomu prostaty77), rekurentního karcinomu ovaria78) a několika podtypů sarkomu měkkých tkání79).
Studie na buňkách gemcitabin-rezistentního karcinomu pankreatu také vyšla s poměrně slibnými výsledky a eribulin by se tak snad v budoucnu mohl stát součástí terapie i tohoto onemocnění80).
Cytarabin (cytosin arabinosid, ara-C)
Cytarabin je derivát nukleosidu deoxycytidinu, v němž je deoxyribóza nahrazena arabinózou. V buňce dochází k jeho přeměně na cytosin arabinosid trifosfát, který soutěží s deoxycytidin trifosfátem o zabudování do struktury DNA. Pokud dojde k zabudování arabinosidu, DNA polymeráza je inhibována a další syntéza DNA se zastaví81). To vede k zástavě buněčného cyklu a později k apoptóze.
Historie cytarabinu je na svém začátku výrazně spojena s vidarabinem, vývoj obou totiž vycházel z objevu spongonukleosidů v houbovci Tectitethya crypta82).
V České republice je cytarabin dostupný v registrovaných přípravcích ALEXAN, CYTARABIN KABI, CYTARABINE ACCORD, CYTOSAR a DEPOCYTE. Je indikován zejména k dosažení remise při akutní myeloidní leukemii u dospělých a při jiných formách akutní leukemie u dospělých a dětí.
V přípravku DEPOCYTE (forma s prodlouženým uvolňováním) je pak indikován k intratekální léčbě lymfomatózní meningitidy. Ačkoliv lze u meningeálního postižení při leukemii využít i jiné přípravky s cytarabinem, forma s prodlouženým uvolňováním nabízí výhodnější dávkovací režim83–86).
Kombinace cytarabinu s daunorubicinem (označovaná jako DA režim či „7+3”; 7 dní kontinuální infuze cytarabinu + 3 dny infuze daunorubicinu) je už poměrně dlouhou dobu využívaná v první linii terapie akutní myeloidní leukemie87–89). Studie zkoumající jiné možné režimy spočívají obvykle ve změně antracyklinu (např. za idarubicin90, 91)), přidání dalšího léku (např. kladribinu92, 93)) či kombinaci obojího (např. CLA-Ida režim obsahující kladribin, cytarabin a idarubicin94, 95)). Ačkoliv se tyto kombinace stále zlepšují ve snaze maximalizovat terapeutickou odpověď a minimalizovat toxicitu, cytarabin zůstává jejich ústřední komponentou. To platí i v terapii řady lymfomů před autologní transplantací kmenových buněk, např. režim BEAM (zahrnující karmustin, etoposid, cytarabin a melphalan96)) či jeho varianty LEAM (místo karmustinu je použit lomustin97)) nebo NEAM (místo karmustinu je použit mitoxantron98)).
O významu cytarabinu v současnosti i do budoucna snad svědčí počet probíhajících studií: 136 v EU (22 s účastí v ČR)99) a 289 v USA100)).

Vidarabin (adenin arabinosid, ara-A)
Počátek historie využití vidarabinu (a cytarabinu) sahá až k přelomu čtyřicátých a padesátých let 20. století, kdy Werner Bergmann s kolegy poprvé izoloval spongonukleosidy – spongothymidin a spongouridin – z houbovce Tectitethya crypta82, 101). Vidarabin je syntetický analog spongouridinu101).
Jeho antivirotické působení pak zaznamenali Privat de Garilhe a De Rudder už v roce 1964101, 102). Mechanismem jeho účinku je kompetitivní inhibice DNA polymerázy, která zabrání viru v replikaci103).
V USA byl vidarabin registrován od roku 1976 a jeho prodej byl ukončen v červnu 2001 z ekonomických důvodů, jelikož byl postupně „vytlačován” novějšími antivirotiky11).
To ale pro vidarabin nemusí znamenat definitivní konec. Studie na myších ukázaly, že vidarabin funguje jako relativně selektivní inhibitor adenylyl cyklázy 5 (AC5) a zpomaluje progresi poinfarktového srdečního selhání104). AC5 je (spolu s AC6) hlavní izoformou adenylyl cyklázy v srdci. Myši s knockoutovaným genem pro AC5 žijí déle, mají nižší riziko rozvoje diabetes mellitus a obezity a lépe snášejí zátěž105). Vidarabin by snad mohl vyvolávat podobné účinky bez nutnosti zásahu do genomu či genové exprese. Ve studii na prasatech bylo dále zaznamenáno zmenšení infarktového ložiska po koronární obstrukci u skupiny, jíž byl podán vidarabin (oproti vehikulu). Navíc se ukázalo, že ke zmenšení ložiska došlo i v případě, že byl vidarabin podán až po reperfuzi koronárních arterií. To má velký význam pro kliniku, kdy nelze s obnovením průtoku otálet. Adenosin, se kterým byl vidarabin porovnáván, snižoval velikost ložiska, pokud byl podán před reperfuzí, ke zmenšení ložiska po reperfuzi bylo potřeba podat pětinásobnou dávku. Vysoké dávky adenosinu však v tomto případě nejsou vhodné, jelikož vedou ke snížení systolického tlaku. U vidarabinu nebyl významný vliv na tlak zaznamenán106).
Studie na psech s pacingem-indukovanou dilatační kardiomyopatií ukázala působení vidarabinu proti progresi systolické dysfunkce, apoptóze kardiomyocytů a srdeční intersticiální fibróze107).
Lurbinectedin (PM01183)
Lurbinectedin je syntetický alkaloid strukturně příbuzný trabectedinu. Podobně jako on se váže do malého žlábku DNA, působí vznik dvojřetězcových zlomů, blok buňky v S fázi a následnou apoptózu108, 109). Rovněž působí redukci počtu TAM (tumor associated macrophages – viz trabectedin)110). Preklinicky vykazoval protinádorovou aktivitu in vitro i in vivo108, 109, 111), klinická studie fáze I pro terapii pokročilých solidních nádorů pak prokázala jeho dobrou toleranci i aktivitu112). V současnosti probíhají např. studie fáze III pro terapii ovariálního karcinomu s rezistencí na platinu (studie113), studie fáze II pro terapii metastatických či neresekovatelných sarkomů114) a s BRCA 1/2 asociovaného nebo metastatického karcinomu prsu115).
Tetrodotoxin (TTX)
Tetrodotoxin je jeden z vůbec nejsilnějších neurotoxinů116), známý zejména díky přítomnosti v rybách čeledi Tetraodontidae117), ale izolován byl i z dalších mořských i suchozemských organismů116, 118–121).
Přinejmenším u ryb čeledi Tetraodontidae však bylo prokázáno, že skutečným producentem TTX jsou endosymbiotické bakterie obývající jejich trávicí trakt116, 122, 123).
I přes jeho poměrně složitou strukturu dnes existuje několik způsobů průmyslové syntézy124), což je dobrým východiskem pro budoucí využití.
V budoucnu by se tetrodotoxin mohl uplatnit jako analgetikum116) a snad i jako lék pro snížení relapsů drogových závislostí (studie s nízkými dávkami TTX ke snížení cravingu a úzkosti u pacientů léčících se ze závislosti na heroinu125, 126)).
Tetrodotoxin působí jako selektivní blokátor sodíkových kanálů117). Podle senzitivity těchto kanálů je dělíme na tetrodotoxin senzitivní (TTX-S) a tetrodotoxin rezistentní (TTX-R)127–129).
Zásadní otázkou pro účinnost TTX jako analgetika jsou role TTX-R a TTX-S kanálů při přenosu signálu v nocicepční dráze130, 131). Studie na potkanech ukázaly, že TTX-S jsou zcela klíčové pro propagaci signálu v C i Aδ vláknech (tenká vlákna vedoucí informace o bolesti a teplotě), zatímco TTX-R jsou významněji zastoupeny pouze na tělech neuronů a periferních zakončeních C vláken a jejich přítomnost není pro šíření akčního potenciálu nutná132, 133). Jiné studie ale přikládají TTX-R kanálům mnohem větší význam134). Zdá se, že z funkčního pohledu jsou TTX-R důležité např. pro vnímání bolesti při nízkých teplotách135).
V Kanadě proběhla studie III. fáze s tetrodotoxinem pro léčbu nádorové bolesti, data však zatím nebyla zveřejněna136). V USA byla předčasně ukončena II. fáze klinického testování (na základě průběžné analýzy dat, zdá se, že výzkum bude pokračovat studií III. fáze) pro terapii chemoterapií indukované neuropatické bolesti137).

Glembatumumab vedotin (CDX-011)
Glembatumumab vedotin je konjugát protilátky anti-gpNMB (glycoprotein nonmetastatic melanoma B, též osteoactivin, DC-HIL) a monomethyl auristatinu E138). GpNMB je zvýšeně exprimován v řadě malignit, např. některých typech melanomu139, 140), gliomu141), karcinomu prsu142), osteosarkomu143) či nemalobuněčném karcinomu plic144).
Exprese gpNMB v těchto tumorech zřejmě potencuje invazivní a metastatické chování141, 144).
Studie glembatumumabu vedotinu fáze I/II145) a 146) ukázaly poměrně dobrou snášenlivost a účinnost v terapii karcinomu prsu (30 % ORR (overall response rate) u pacientů se zvýšenou expresí gpNMB ve více než 25 % nádorových buněk – studie EMERGE). Další studie fáze II – METRIC (Metastatic triple-negative breast cancer) právě probíhá147).
Studie fáze I/II ukázaly možnost budoucího využití pro terapii pokročilého melanomu138).
Dále probíhají studie pro využití v terapii pokročilého malobuněčného karcinomu plic (NCT02713828), osteosarkomu (NCT02487979) a uveálního melanomu (NCT02363283).
Cyklické depsipeptidy
Jedná se o polypeptidy, ve kterých je nejméně jedna aminokyselina nahrazena hydroxykyselinou, což vede ke vzniku nejméně jedné esterové vazby v cyklu. Tato skupina látek, z nichž některé byly izolovány z mořských organismů, má velmi rozmanité spektrum účinků148).
Didemnin B je cyklický depsipeptid poprvé izolovaný ze sumky Trididemnum solidum149). V buňkách pravděpodobně působí inhibici syntézy DNA a proteinů, což vede k zástavě jejich růstu149, 150). Preklinicky vykazoval slibnou aktivitu proti řadě nádorů151), v několika klinických studiích však ztroskotal kvůli slabým výsledkům a závažné toxicitě152–155).
Plitidepsin (aplidin, dehydrodidemnin B) byl poprvé izolován ze sumky Aplidium albicans156). Mechanismus jeho působení je zřejmě komplexní. V buňkách vyvolává dlouhodobou aktivaci signálních kináz JNK a p38 MAPK, významnou úlohu zřejmě hraje abnormální aktivace EGFR, indukce oxidativního stresu a deplece glutathionu157–159). Účinně blokoval buňky v G1 fázi i v přítomnosti mutované TP53160). Dále má zřejmě vliv na angiogenezi zásahem do sekrece VEGF157, 161) či přímým působením na endoteliální buňky162).
V preklinických studiích vykazoval protinádorovu aktivitu např. na buněčných liniích AML161), karcinomu štítné žlázy160) či karcinomu prsu159). V klinických studiích fáze I a II se potvrdila jeho aktivita u pokročilého melanomu (v kombinaci s dakarbazinem163), refrakterního či relabujícího mnohočetného myelomu164) nebo neresekabilního pokročilého renálního karcinomu165), výsledky ale nejsou ohromující. Podobně plitidepsin neukázal nikterak „zázračné” účinky – např. u myelofibrózy166), pokročilého či metastatického karcinomu z buněk přechodního epitelu167) nebo u malobuněčného karcinomu plic168). Nicméně se jedná o velmi zajímavou látku z hlediska unikátního mechanismu působení na sekreci VEGF.
Kalahalid F je cyklický depsipeptid izolovaný z měkkýše Elysia rufescens a zelených řas, kterými se živí169). V preklinických studiích ukázal slibnou cytotoxickou aktivitu např. na buňkách karcinomu prsu či prostaty170, 171). Mechanismem jeho účinku je pravděpodobně narušení organel buňky, vznik osmotické dysbalance a smrt buněk onkózou170). Studie fáze II využití kalahalidu F v terapii pokročilého maligního melanomu sice ukázala dobrý bezpečnostní profil, nicméně byla ukončena pro nedostatečnou protinádorovou aktivitu172).
Elisidepsin (PM02734) je syntetický cyklický peptid odvozený od kalahalidu F. I on vykazoval slibné výsledky preklinicky173), ve studii fáze Ib/II pro terapii metastatického či pokročilého gastroezofageálního karcinomu (studie IMAGE) měl však jen nízkou účinnost174). Mezi další farmakologicky zajímavé cyklické depsipeptidy izolované z mořských organismů dále patří např. neamphamid A izolovaný z houbovce Neamphius huxleyi, s anti-HIV aktivitou in vitro175), neamphamid B izolovaný z jiného houbovce Neamphius sp., s antimykobakteriální aktivitou in vitro176) a celá řada dalších.
Skvalamin laktát
Skvalamin byl poprvé izolován ze žraloka ostrouna obecného (Squalus acanthias)177). Ačkoliv jeho objevitelé popisují zejména jeho antibiotické působení, později zaujal spíše svým antiangiogenetickým a protinádorovým potenciálem178, 179). V klinické studii fáze I/IIA využití skvalamin laktátu v terapii pokročilého nemalobuněčného karcinomu plic (v kombinaci s karboplatinou a paklitaxelem) určitý potenciál ukázal180), výsledky studií fáze II zkoumající vliv skvalamin laktátu na proliferativní diabetickou retinopatii181) a neovaskulární věkem podmíněnou makulární degeneraci182) zatím nebyly zveřejněny.
DMXBA (GTS-21, 3-(2,4-dimethoxybenzyliden)-anabasein)
DMXBA je syntetický analog anabaseinu, nikotinového alkaloidu poprvé izolovaného z mořské pásnice Amphiporus lactifloreus183), přítomný je ale např. i u některých druhů mravenců rodu Aphaenogaster184) a dalších živočichů.
Anabasein je neselektivní agonista nikotinových receptorů pro acetylcholin, preferenčně ale stimuluje podtyp receptorů v kosterním svalstvu a α7 podtyp v mozku. DMXBA je selektivní agonista právě pro α7 podtyp nikotinových receptorů185). Tyto receptory se nachází na neuronech jak presynapticky, tak postsynapticky186, 187) a představují zajímavý terapeutický cíl zejména pro terapii schizofrenie188–191) a Alzheimerovy choroby192, 193). Tvorba acetylcholinu a exprese cholinergních receptorů však rozhodně není výsadou neuronů, acetylcholin i jeho receptory můžeme najít téměř ve všech buňkách lidského těla194). Příkladem za všechny budiž role acetylcholinu jako mediátoru cholinergní antiinflamatorní cesty/reflexu, komunikace nervového a imunitního systému, která má významný vliv na regulaci zánětlivých procesů195). Studie in vitro196) i na myších197) na toto možné antiinflamatorní působení DMXBA ukazují, v pilotní studii na lidech však prokázáno nebylo198).
Co se týče terapie Alzheimerovy choroby, preklinické studie ukazují, že samotná vazba agonisty na α7 cholinergní receptor chrání neurony před Aβ mediovanou cytotoxicitou199–201). DMXBA navíc v pilotní studii na zdravých dobrovolnících ukázal vliv na zlepšení kognitivních funkcí (pozornosti, pracovní paměti a sekundární epizodické paměti)202).
K prokazatelnému zlepšení kognitivních funkcí došlo i v pilotní studii využití DMXBA pro terapii schizofrenie203). Ve studii fáze II tento vliv na kognici pozorován nebyl, došlo ale ke zlepšení negativních symptomů schizofrenie (hodnoceno na stupnici SANS, zohledňující mimo jiné anhedonii, apatii či poruchy pozornosti)204).
Klinické studie pro terapii Alzheimerovy choroby205) a ADHD206) zůstávají zatím bez publikovaných výsledků.

Bryostatin-1
Je látka (chemicky se jedná o makrocyklický lakton) poprvé izolovaná z Bugula neritina, mořského živočicha z kmene mechovců207). Analýza genových clusterů a další indicie ale svědčí pro mikrobiální původ, a to ze symbiotického mikroorganismu Candidatus Endobugula sertula208, 209). Bryostatin vykazuje velmi rozmanité účinky210, 211). Hlavním mechanismem jeho působení (pravděpodobně ne jediným) je vazba do vazebného místa pro diacylglycerol (DAG) na proteinkinase C (PKC), která vyvolá její aktivaci212). Na rozdíl od např. forbol esterů (které také fungují jako aktivátory PKC) ale nepůsobí jako tumor promoter213).
PKC funguje jako ústřední regulátor růstu a diferenciace buňky213, 214) a protinádorové působení je jedním z účinků bryostatinu. Přesný mechanismus nebyl zatím objasněn, zřejmě však bude vícesložkový215), uplatňuje se např. defosforylace cyklin-dependentní kinázy 2216), uvolnění TNF-α217) či modulace imunitního systému218).
Izoformy PKC mají dále zásadní vliv na správné fungování paměťových funkcí a jejich poruchy patří mezi první abnormality objevující se v mozku pacientů trpících Alzheimerovou chorobou219, 220). Snížená aktivita PKC pravděpodobně souvisí i s depresí221, 222). Bryostatin prokázal antidepresivní účinek a vliv na zlepšení paměti u potkanů223). Klinická studie fáze II využití bryostatinu pro terapii středně těžké a těžké Alzheimerovy choroby právě probíhá224).
Působení bryostatinu na PKC vede i k reaktivaci latentního HIV-1225, 226), tento účinek by snad v budoucnu mohl najít využití v terapii HIV, kde tyto rezervoáry latentního viru představují závažný terapeutický problém. Bryostatin navíc působí dočasné snížení exprese CD4 a CXCR4 na T buňkách 225, 227). Klinická studie fáze I zkoumající využití bryostatinu pro reaktivaci latentního viru u pacientů léčených antiretrovirovou terapií sice neprokázala vliv bryostatinu na aktivitu PKC ani reaktivaci latentního viru, problémem však mohly být příliš nízké plazmatické koncentrace228).
Co se týče protinádorového působení bryostatinu, proběhla od devadesátých let 20. století celá řada studií. Zdá se, že protinádorový účinek samotného bryostatinu je příliš slabý, ukázal však účinnost v kombinaci s jinými protinádorovými léky210). Účinná byla např. kombinace s paklitaxelem v terapii pokročilého gastrického adenokarcinomu nebo karcinomu gastroezofagealní junkce (lepší výsledek než monoterapie paklitaxelem229)) či kombinace s vinkristinem pro terapii relapsu agresivního non-Hodgkinova lymfomu po autologní transplantaci kmenových buněk230). Naopak úspěch nepřinesly např. kombinace s paklitaxelem pro terapii pokročilého karcinomu pankreatu231) či kombinace s cisplatinou pro terapii pokročilého či rekurentního karcinomu děložního hrdla232). Větší naději na klinické využití v terapii nádorů snad budou mít analoga bryostatinu (bryologa), např. picolog233).
Dle výzkumu na potkanech by bryostatin mohl snižovat poškození mozku při ischemii a prodlužovat okno pro podání trombolytické terapie234, 235). Navíc v této situaci zřejmě působí protektivně na paměťové funkce (retrográdní i anterográdní)236).

Plinabulin (NPI-2358)
Plinabulin je syntetický analog diketopiperazinu fenylahistinu (rovněž halimidu)237), izolovaného z mořských238) i suchozemských239) zástupců hub rodu Aspergillus. V in vitro studiích vykazoval protinádorovou aktivitu spojenou s disruptivním vlivem na cévní stěnu237). Klinické studie fáze I potvrdily toleranci a ukázaly i slibnou aktivitu240, 241).
Marizomib (salinosporamid A, NPI-0052)
Marizomib je proteasomový inhibitor, poprvé izolovaný z mořské aktinomycety rodu Salinispora242). Inhibice proteasomu působí protinádorově a proapoptoticky komplexním mechanismem. Proteolýza hraje nezastupitelnou roli v řízení buněčného cyklu, např. regulací množství aktivátorů či inhibitorů CDK243). Dalším aspektem je hromadění poškozených proteinů, které sami o sobě působí proapoptoticky a ke kterému jsou nádorové buňky náchylnější díky nižší genetické stabilitě a rychlejší proliferaci244).
Na rozdíl od bortezomibu (dalšího proteasomového inhibitoru, v současnosti registrovaného např. pro terapii mnohočetného myelomu), marizomib inhibuje všechny tři podjednotky proteasomu: chymotrypsinu-podobnou (CT-L), trypsinu podobnou (T-L) a kaspáze-podobnou (C-L)245).
Marizomib působí i inhibici aktivace NF-κB245, 246), který hraje významnou úlohu v iniciaci a progresi nádorového růstu247). K inhibici aktivace NF-κB zřejmě dochází inhibicí degradace jeho regulátoru IκB247). I bortezomib vyvolává inhibici dráhy NF-κB, ale zdá se, že jen v některých buňkách (např. ve stromálních buňkách kostní dřeně), v buňkách mnohočetného myelomu naopak vyvolává jeho aktivaci. Cytotoxicita bortezomibu tedy zřejmě není závislá na inaktivaci NF-κB248). Marizomib a bortezomib působily ve studiích na nádorových liniích synergicky245, 246).
In vitro vykazoval marizomib účinnost proti nádorovým buňkám mnohočetného myelomu245), karcinomu tlustého střeva249) či chronické250) a akutní leukemie251).
V klinické studii fáze I v kombinaci s vorinostatem (inhibitor histon-deacetylasy) u pacientů s melanomem, karcinomem slinivky či plic byla zjištěna dobrá snášenlivost a synergická protinádorová aktivita s vorinostatem252). I studie fáze I pro terapii relabovaného či relabovaného a refrakterního mnohočetného myelomu došly k nadějným výsledkům253, 254) a další studie fáze I/II pro tuto indikaci měla proběhnout, její stav je však nejasný255). V počátcích jsou i studie fáze I využití marizomibu v terapii maligního gliomu256, 257).
Závěr
Látky, o kterých tento článek pojednává, jsou rozmanité jak chemicky, tak i svým působením na lidský organismus. Jejich jediným pojítkem je původ organismů, ze kterých byly poprvé izolovány. Hodnota této informace pro klinickou praxi je zřejmě zanedbatelná, ale inspirace přírodními látkami hraje bezpochyby zcela zásadní úlohu ve vývoji nových léčiv. Moře a jeho obyvatelé představují bohatý zdroj nových molekul s biologickou aktivitou, jsou ovšem ohrožováni celou řadou vlivů, ať už mluvíme o oteplování, znečištění, nevhodných způsobech těžby a lovu či mnoha dalších faktorech. Opomenout nelze ani význam působení samotných farmak, jejich metabolitů a odpadních látek vznikajících při jejich výrobě na životní prostředí. Poškozování křehkých ekosystémů, úbytek druhů a pokles biodiverzity, vznikající jako možný následek těchto jevů, znamená i úbytek potenciálně zajímavých látek, jež tyto organismy vytvářejí. Blednutí korálových útesů, představující jeden z nejznámějších projevů tohoto fenoménu, tak není jen ztrátou plejády pestrobarevných korálů, ryb či korýšů, ale i celé řady potenciálních léčiv. A je to právě tato vzájemná blízkost farmakologie a ekologie, zásadní vztah ochrany přírody a vývoje budoucích léčiv, jež dává tomuto tématu význam. Jde samozřejmě jen o jeden ze střípků v rozsáhlé a mnohem složitější mozaice vzájemných souvislostí, nicméně jde o střípek, který by neměl být přehlédnutý.
Použité zkratky
ADHD - attention deficit hyperactivity disorder
DMXBA - 3-(2,4-dimethoxybenzyliden)-anabasein
EMT - epithelial-mesenchymal transition
FDA - Food and Drug Administration
IUCN - International Union for Conservation of Nature
MET - mesenchymal-epithelial transition
MMAE - monomethyl auristatin E
NER - nucleotide Excision Repair
NF-κB - nukleární faktor kappa B
SPC - summary of product characteristics
TAM - tumor associated macrophages
TTX - tetrodotoxin
VEGF - vascular endothelial growth factor
Studie vznikla na Masarykově Univerzitě v rámci projektu „Experimentální a translační farmakologický výzkum a vývoj“ číslo MUNI/A/1063/2016 podpořeného z prostředků účelové podpory na specifický vysokoškolský výzkum, kterou poskytlo MŠMT v roce 2017.
Střet zájmů: žádný.
Došlo 8. června 2017
Přijato 2. září 2017
PharmDr. Jana Rudá-Kučerová, PhD.
D. Mareček
Farmakologický ústav LF MU
Kamenice 5, 625 00 Brno
e-mail: jkucer@med.muni.cz
Sources
1. Mora C., Tittensor D. P., Adl S., Simpson A. G. B., Worm B. How Many Species Are There on Earth and in the Ocean? PLoS Biol. 2011; 9, e1001127.
2. Newton G. G., Abraham E. P. Cephalosporin C, a new antibiotic containing sulphur and D-alpha-aminoadipic acid. Nature 1955; 175, 548.
3. Bo G. Giuseppe Brotzu and the discovery of cephalosporins. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2000; 6 (Suppl 3), 6–9.
4. Radjasa O. K., Vaske Y. M., Navarro G., Vervoort H. C., Tenney K., Linington R. G., Crews P. Highlights of marine invertebrate-derived biosynthetic products: their biomedical potential and possible production by microbial associants. Bioorg. Med. Chem. 2011; 19, 6658–6674.
5. Anjum K., Abbas S. Q., Shah S. A. A., Akhter N., Batool S., Hassan S. S. ul. Marine Sponges as a Drug Treasure. Biomol. Ther. 2016; 24, 347–362.
6. Thomas T. R. A., Kavlekar D. P., LokaBharathi P. A. Marine drugs from sponge-microbe association – a review. Mar. Drugs 2010; 8, 1417–1468.
7. Faulkner D. J., He H. Y., Unson M. D., Bewley C. A., Garson M. J. New metabolites from marine sponges: Are symbionts important? Gazz Chem Ital 1993; 123, 301–307.
8. Haygood M. G., Schmidt E. W., Davidson S. K., Faulkner D. J. Microbial symbionts of marine invertebrates: opportunities for microbial biotechnology. J. Mol. Microbiol. Biotechnol. 1999; 1, 33–43.
9. Vacelet J., Donadey C. Electron microscope study of the association between some sponges and bacteria. J. Exp. Mar. Biol. Ecol. 1977; 301–314.
10. Anand T. P., Bhat A. W., Shouche Y. S., Roy U., Siddharth J., Sarma S. P. Antimicrobial activity of marine bacteria associated with sponges from the waters off the coast of South East India. Microbiol. Res. 2006; 161, 252–262.
11. Mayer A. M. S., Glaser K. B., Cuevas C., Jacobs R. S., Kem W., Little R. D., McIntosh J. M., Newman D. J., Potts B. C., Shuster D. E. The odyssey of marine pharmaceuticals: a current pipeline perspective. Trends Pharmacol. Sci. 2010; 31, 255–265.
12. Gerwick W. H., Fenner A. M. Drug Discovery from Marine Microbes. Microb. Ecol. 2013; 65, 800–806.
13. Gomes N. G. M., Dasari R., Chandra S., Kiss R., Kornienko A. Marine Invertebrate Metabolites with Anticancer Activities: Solutions to the “Supply Problem”. Mar. Drugs 2016; 14(5), 98.
14. Schaufelberger D. E., Koleck M. P., Beutler J. A., Vatakis A. M., Alvarado A. B., Andrews P., Marzo L. V., Muschik G. M., Roach J., Ross J. T. The large-scale isolation of bryostatin 1 from Bugula neritina following current good manufacturing practices. J. Nat. Prod. 1991; 54, 1265–1270.
15. Cuevas C., Francesch A. Development of Yondelis (trabectedin, ET-743). A semisynthetic process solves the supply problem. Nat. Prod. Rep. 2009; 26, 322–337.
16. Mendola D. Aquaculture of three phyla of marine invertebrates to yield bioactive metabolites: process developments and economics. Biomol. Eng. 2003; 20, 441–458.
17. Duckworth A. Farming sponges to supply bioactive metabolites and bath sponges: a review. Mar. Biotechnol. N. Y. N 2009; 11, 669–679.
18. Belarbi E. H., Goméz A. C., Chisti Y., Camacho F. C., Grima E. M. Producing drugs from marine sponges. Biotechnol. Adv. 2003; 585–598.
19. Dunlap W. C., Battershill C. N., Liptrot C. H., Cobb R. E., Bourne D. G., Jaspars M., Long P. F., Newman D. J. Biomedicinals from the phytosymbionts of marine invertebrates: a molecular approach. Methods San Diego Calif 2007; 42, 358–376.
20. Prialt SPC. http://www.ema.europa.eu/docs/cs_CZ/document_library/EPAR_-_Product_Information/human/000551/WC500041929.pdf (30. 5. 2017).
21. Bowersox S. S., Luther R. Pharmacotherapeutic potential of omega-conotoxin MVIIA (SNX-111), an N-type neuronal calcium channel blocker found in the venom of Conus magus. Toxicon Off. J. Int. Soc. Toxinology 1998; 36, 1651–1658.
22. Molderings G. J., Likungu J., Göthert M. N-Type calcium channels control sympathetic neurotransmission in human heart atrium. Circulation 2000; 101, 403–407.
23. Hayashi K., Wakino S., Sugano N., Ozawa Y., Homma K., Saruta T. Ca2+ channel subtypes and pharmacology in the kidney. Circ. Res. 2007; 100, 342–353.
24. Gohil K., Bell J. R., Ramachandran J., Miljanich G. P. Neuroanatomical distribution of receptors for a novel voltage-sensitive calcium-channel antagonist, SNX-230 (omega-conopeptide MVIIC). Brain Res. 1994; 653, 258–266.
25. Malmberg A. B., Yaksh T. L. Effect of continuous intrathecal infusion of omega-conopeptides, N-type calcium-channel blockers, on behavior and antinociception in the formalin and hot-plate tests in rats. Pain 1995; 60, 83–90.
26. Webster L. R., Fakata K. L., Charapata S., Fisher R., MineHart M. Open-label, multicenter study of combined intrathecal morphine and ziconotide: addition of morphine in patients receiving ziconotide for severe chronic pain. Pain Med. Malden Mass 2008; 9, 282–290.
27. de la Calle Gil A. B., Peña Vergara I., Cormane Bornacelly M. A., Pajuelo Gallego A. Intrathecal Ziconotide and Morphine for Pain Relief: A Case Series of Eight Patients with Refractory Cancer Pain, Including Five Cases of Neuropathic Pain. Neurol. Ther. 2015; 4, 159–168.
28. Staquet H., Dupoiron D., Nader E., Menei P. Intracerebroventricular Pain Treatment with Analgesic Mixtures including Ziconotide for Intractable Pain. Pain Physician 2016; 19, E905–915.
29. Bowersox S. S., Singh T., Luther R. R. Selective blockade of N-type voltage-sensitive calcium channels protects against brain injury after transient focal cerebral ischemia in rats. Brain Res. 1997; 747, 343–347.
30. Perez-Pinzon M. A., Yenari M. A., Sun G. H., Kunis D. M., Steinberg G. K. SNX-111, a novel, presynaptic N-type calcium channel antagonist, is neuroprotective against focal cerebral ischemia in rabbits. J. Neurol. Sci. 1997; 153, 25–31.
31. Berman R. F., Verweij B. H., Muizelaar J. P. Neurobehavioral protection by the neuronal calcium channel blocker ziconotide in a model of traumatic diffuse brain injury in rats. J. Neurosurg. 2000; 93, 821–828.
32. Rinehart K. L., Holt T. G., Fregeau H. L., Stroh J. G., Keifer P. A., Sun F., Li L. H., Martin D. G. Ecteinascidins 729, 743, 745, 759A, 759B, and 770: potent antitumor agents from the Caribbean tunicate Ecteinascidia turbinata. J. Org. Chem. 1990; 4512–4515.
33. Rath C. M., Janto B., Earl J., Ahmed A., Hu F. Z., Hiller L., Dahlgren M., Kreft R., Yu F., Wolff J. J., Kweon H. K., Christiansen M. A., Håkansson K., Williams R. M., Ehrlich G. D., Sherman D. H. Meta-omic characterization of the marine invertebrate microbial consortium that produces the chemotherapeutic natural product ET-743. ACS Chem. Biol. 2011; 6, 1244–1256.
34. Schofield M. M., Jain S., Porat D., Dick G. J., Sherman D. H. Identification and analysis of the bacterial endosymbiont specialized for production of the chemotherapeutic natural product ET-743. Environ. Microbiol. 2015; 17, 3964–3975.
35. Pommier Y., Kohlhagen G., Bailly C., Waring M., Mazumder A., Kohn K. W. DNA sequence - and structure-selective alkylation of guanine N2 in the DNA minor groove by ecteinascidin 743, a potent antitumor compound from the Caribbean tunicate Ecteinascidia turbinata. Biochemistry (Mosc.) 1996; 35, 13303–13309.
36. Yondelis SPC. http://www.ema.europa.eu/docs/cs_CZ/document_library/EPAR_-_Product_Information/human/000773/WC500045832.pdf (30. 5. 2017).
37. Dubois E. A., Cohen A. F. Trabectedin. Br. J. Clin. Pharmacol. 2009; 68, 320–321.
38. Takebayashi Y., Pourquier P., Zimonjic D. B., Nakayama K., Emmert S., Ueda T., Urasaki Y., Kanzaki A., Akiyama S. I., Popescu N., Kraemer K. H., Pommier Y. Antiproliferative activity of ecteinascidin 743 is dependent upon transcription-coupled nucleotide-excision repair. Nat. Med. 2001; 7, 961–966.
39. Zewail-Foote M., Li V. S., Kohn H., Bearss D., Guzman M., Hurley L. H. The inefficiency of incisions of ecteinascidin 743-DNA adducts by the UvrABC nuclease and the unique structural feature of the DNA adducts can be used to explain the repair-dependent toxicities of this antitumor agent. Chem. Biol. 2001; 8, 1033–1049.
40. Soares D. G., Machado M. S., Rocca C. J., Poindessous V., Ouaret D., Sarasin A., Galmarini C. M., Henriques J. A. P., Escargueil A. E., Larsen A. K. Trabectedin and its C subunit modified analogue PM01183 attenuate nucleotide excision repair and show activity toward platinum-resistant cells. Mol. Cancer Ther. 2011; 10, 1481–1489.
41. Germano G., Frapolli R., Belgiovine C., Anselmo A., Pesce S., Liguori M., Erba E., Uboldi S., Zucchetti M., Pasqualini F., Nebuloni M., van Rooijen N., Mortarini R., Beltrame L., Marchini S., Fuso Nerini I., Sanfilippo R., Casali P. G., Pilotti S., Galmarini C. M., Anichini A., Mantovani A., D’Incalci M., Allavena P. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013; 23, 249–262.
42. Bingle L., Brown N. J., Lewis C. E. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J. Pathol. 2002; 196, 254–265.
43. Comito G., Giannoni E., Segura C. P., Barcellos-de-Souza P., Raspollini M. R., Baroni G., Lanciotti M., Serni S., Chiarugi P. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene 2014; 33, 2423–2431.
44. Komohara Y., Takeya M. CAFs and TAMs: maestros of the tumour microenvironment. J. Pathol. 2017; 241, 313–315.
45. D’Angelo D., Borbone E., Palmieri D., Uboldi S., Esposito F., Frapolli R., Pacelli R., D’Incalci M., Fusco A. The impairment of the High Mobility Group A (HMGA) protein function contributes to the anticancer activity of trabectedin. Eur. J. Cancer Oxf. Engl. 2013; 49, 1142–1151.
46. De Martino M., Forzati F., Arra C., Fusco A., Esposito F. HMGA1-pseudogenes and cancer. Oncotarget 2016; 7, 28724–28735.
47. Kanzaki A., Takebayashi Y., Ren X.-Q., Miyashita H., Mori S., Akiyama S., Pommier Y. Overcoming multidrug drug resistance in P-glycoprotein/MDR1-overexpressing cell lines by ecteinascidin 743. Mol. Cancer Ther. 2002; 1, 1327–1334.
48. Goldstein L. J., Gurtler J., Del Prete S. A., Tjulandin S., Semiglazov V. F., Bayever E., Michiels B. Trabectedin as a single-agent treatment of advanced breast cancer after anthracycline and taxane treatment: a multicenter, randomized, phase II study comparing 2 administration regimens. Clin. Breast Cancer 2014; 14, 396–404.
49. Francisco J. A., Cerveny C. G., Meyer D. L., Mixan B. J., Klussman K., Chace D. F., Rejniak S. X., Gordon K. A., DeBlanc R., Toki B. E., Law C.-L., Doronina S. O., Siegall C. B., Senter P. D., Wahl A. F. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003; 102, 1458–1465.
50. Pettit G. R., Kamano Y., Fujii Y., Herald C. L., Inoue M., Brown P., Gust D., Kitahara K., Schmidt J. M., Doubek D. L., Michel C. Marine animal biosynthetic constituents for cancer chemotherapy. J. Nat. Prod. 1981; 44, 482–485.
51. Bai R. L., Pettit G. R., Hamel E. Binding of dolastatin 10 to tubulin at a distinct site for peptide antimitotic agents near the exchangeable nucleotide and vinca alkaloid sites. J. Biol. Chem. 1990; 265, 17141–17149.
52. Müller P., Martin K., Theurich S., Schreiner J., Savic S., Terszowski G., Lardinois D., Heinzelmann-Schwarz V. A., Schlaak M., Kvasnicka H-M., Spagnoli G., Dirnhofer S., Speiser D. E., von Bergwelt-Baildon M., Zippelius A. Microtubule-depolymerizing agents used in antibody-drug conjugates induce antitumor immunity by stimulation of dendritic cells. Cancer Immunol. Res. 2014; 2, 741–755.
53. Adcetris SPC. http://www.ema.europa.eu/docs/cs_CZ/document_ library/EPAR_-_Product_Information/human/002455/WC500135055.pdf (30. 5. 2017).
54. Corey K., Cook D., Bekker J., Mugnaini E., Lin J. H. A case of refractory Sézary syndrome with large-cell transformation responsive to brentuximab vedotin. JAMA Dermatol. 2014; 150, 210–212.
55. Mehra T., Ikenberg K., Moos R. M., Benz R., Nair G., Schanz U., Haralambieva E., Hoetzenecker W., Dummer R., French L. E., Guenova E., Cozzio A. Brentuximab as a treatment for CD30+ mycosis fungoides and Sézary syndrome. JAMA Dermatol. 2015; 151, 73–77.
56. Saintes C., Saint-Jean M., Renaut J. J., Dréno B., Quéreux G. Dramatic efficacy of brentuximab vedotin in two patients with epidermotropic cutaneous T-cell lymphomas after treatment failure despite variable CD30 expression. Br. J. Dermatol. 2015; 172, 819–821.
57. Criscuolo M., Fianchi L., Voso M. T., Pagano L. Rapid response of nodular CD30-positive mycosis fungoides to brentuximab vedotin. Br. J. Haematol. 2015; 168, 617.
58. Duvic M., Tetzlaff M. T., Gangar P., Clos A. L., Sui D., Talpur R. Results of a Phase II Trial of Brentuximab Vedotin for CD30+ Cutaneous T-Cell Lymphoma and Lymphomatoid Papulosis. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015; 33, 3759–3765.
59. Jacobsen E. D., Sharman J. P., Oki Y., Advani R. H., Winter J. N., Bello C. M., Spitzer G., Palanca-Wessels M. C., Kennedy D. A., Levine P., Yang J., Bartlett N. L. Brentuximab vedotin demonstrates objective responses in a phase 2 study of relapsed/refractory DLBCL with variable CD30 expression. Blood 2015; 125, 1394–1402.
60. Holderness B. M., Malhotra S., Levy N. B., Danilov A. V. Brentuximab vedotin demonstrates activity in a patient with plasmablastic lymphoma arising from a background of chronic lymphocytic leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013; 31, e197–199.
61. Dabir S., Kresak A., Yang M., Fu P., Wildey G., Dowlati A. CD30 is a potential therapeutic target in malignant mesothelioma. Mol. Cancer Ther. 2015; 14, 740–746.
62. Okouneva T., Azarenko O., Wilson L., Littlefield B. A., Jordan M. A. Inhibition of centromere dynamics by eribulin (E7389) during mitotic metaphase. Mol. Cancer Ther. 2008; 7, 2003–2011.
63. Bai R. L., Paull K. D., Herald C. L., Malspeis L., Pettit G. R., Hamel E. Halichondrin B and homohalichondrin B, marine natural products binding in the vinca domain of tubulin. Discovery of tubulin-based mechanism of action by analysis of differential cytotoxicity data. J. Biol. Chem. 1991; 266, 15882–15889.
64. Jordan M. A., Kamath K., Manna T., Okouneva T., Miller H. P., Davis C., Littlefield B. A., Wilson L. The primary antimitotic mechanism of action of the synthetic halichondrin E7389 is suppression of microtubule growth. Mol. Cancer Ther. 2005; 4, 1086–1095.
65. Funahashi Y., Okamoto K., Adachi Y., Semba T., Uesugi M., Ozawa Y., Tohyama O., Uehara T., Kimura T., Watanabe H., Asano M., Kawano S., Tizon X., McCracken P. J., Matsui J., Aoshima K., Nomoto K., Oda Y. Eribulin mesylate reduces tumor microenvironment abnormality by vascular remodeling in preclinical human breast cancer models. Cancer Sci. 2014; 105, 1334–1342.
66. Yoshida T., Ozawa Y., Kimura T., Sato Y., Kuznetsov G., Xu S., Uesugi M., Agoulnik S., Taylor N., Funahashi Y., Matsui J. Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial-mesenchymal transition (EMT) to mesenchymal-epithelial transition (MET) states. Br. J. Cancer 2014; 110, 1497–1505.
67. Kitahara H., Hirai M., Kato K., Bou-Gharios G., Nakamura H., Kawashiri S. Eribulin sensitizes oral squamous cell carcinoma cells to cetuximab via induction of mesenchymal-to-epithelial transition. Oncol. Rep. 2016.
68. Gavert N., Ben-Ze’ev A. Epithelial-mesenchymal transition and the invasive potential of tumors. Trends Mol. Med. 2008; 14, 199–209.
69. Hirata Y., Uemura D. Halichondrins – antitumor polyether macrolides from a marine sponge. Pure Appl. Chem. 1986; 58.
70. Pettit G. R., Tan R., Gao F., Williams M. D., Doubek D. L., Boyd M. R., Schmidt J. M., Chapuis J. C., Hamel E. Isolation and structure of halistatin 1 from the eastern Indian Ocean marine sponge Phakellia carteri. J. Org. Chem. 1993; 58, 2538–2543.
71. HALAVEN SPC. http://www.ema.europa.eu/docs/cs_CZ/document_library/EPAR_-_Product_Information/human/002084/WC500105112.pdf (30. 5. 2017).
72. Cortes J., O’Shaughnessy J., Loesch D., Blum J. L., Vahdat L. T., Petrakova K., Chollet P., Manikas A., Diéras V., Delozier T., Vladimirov V., Cardoso F., Koh H., Bougnoux P., Dutcus C. E., Seegobin S., Mir D., Meneses N., Wanders J., Twelves C., EMBRACE (Eisai Metastatic Breast Cancer Study Assessing Physician’s Choice Versus E7389) investigators. Eribulin monotherapy versus treatment of physician’s choice in patients with metastatic breast cancer (EMBRACE): a phase 3 open-label randomised study. Lancet Lond. Engl. 2011; 377, 914–923.
73. Schöffski P., Chawla S., Maki R. G., Italiano A., Gelderblom H., Choy E., Grignani G., Camargo V., Bauer S., Rha S. Y., Blay J.-Y., Hohenberger P., D’Adamo D., Guo M., Chmielowski B., Le Cesne A., Demetri G. D., Patel S. R. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet Lond. Engl. 2016; 387, 1629–1637.
74. Gitlitz B. J., Tsao-Wei D. D., Groshen S., Davies A., Koczywas M., Belani C. P., Argiris A., Ramalingam S., Vokes E. E., Edelman M., Hoffman P., Ballas M. S., Liu S. V., Gandara D. R. A phase II study of halichondrin B analog eribulin mesylate (E7389) in patients with advanced non-small cell lung cancer previously treated with a taxane: a California cancer consortium trial. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2012; 7, 574–578.
75. Spira A. I., Iannotti N. O., Savin M. A., Neubauer M., Gabrail N. Y., Yanagihara R. H., Zang E. A., Cole P. E., Shuster D., Das A. A phase II study of eribulin mesylate (E7389) in patients with advanced, previously treated non-small-cell lung cancer. Clin. Lung Cancer 2012; 13, 31–38.
76. Mok T. S., Geater S. L., Iannotti N., Thongprasert S., Spira A., Smith D., Lee V., Lim W. T., Reyderman L., Wang B., Gopalakrishna P., Garzon F., Xu L., Reynolds C. Randomized phase II study of two intercalated combinations of eribulin mesylate and erlotinib in patients with previously treated advanced non-small-cell lung cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2014; 25, 1578–1584.
77. de Bono J. S., Molife L. R., Sonpavde G., Maroto J. P., Calvo E., Cartwright T. H., Loesch D. M., Feit K., Das A., Zang E. A., Wanders J., Agoulnik S., Petrylak D. P. Phase II study of eribulin mesylate (E7389) in patients with metastatic castration-resistant prostate cancer stratified by prior taxane therapy. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2012; 23, 1241–1249.
78. Hensley M. L., Kravetz S., Jia X., Iasonos A., Tew W., Pereira L., Sabbatini P., Whalen C., Aghajanian C. A., Zarwan C., Berlin S. Eribulin mesylate (halichondrin B analog E7389) in platinum-resistant and platinum-sensitive ovarian cancer: a 2-cohort, phase 2 study. Cancer 2012; 118, 2403–2410.
79. Schöffski P., Ray-Coquard I. L., Cioffi A., Bui N. B., Bauer S., Hartmann J. T., Krarup-Hansen A., Grünwald V., Sciot R., Dumez H., Blay J-Y., Le Cesne A., Wanders J., Hayward C., Marreaud S., Ouali M., Hohenberger P., European Organisation for Research and Treatment of Cancer (EORTC) Soft Tissue and Bone Sarcoma Group (STBSG). Activity of eribulin mesylate in patients with soft-tissue sarcoma: a phase 2 study in four independent histological subtypes. Lancet Oncol. 2011; 12, 1045–1052.
80. Takezaki Y., Namikawa T., Koyama T., Munekage E., Munekage M., Maeda H., Kitagawa H., Hanazaki K. Antitumor Effects of Eribulin Mesylate in Gemcitabine-resistant Pancreatic Cancer Cell Lines. Anticancer Res. 2016; 36, 6077–6082.
81. Inagaki A., Nakamura T., Wakisaka G. Studies on the mechanism of action of 1-beta-D-arabinofuranosylcytosine as an inhibitor of DNA synthesis in human leukemic leukocytes. Cancer Res. 1969; 29, 2169–2176.
82. Bergmann W., Feeney R. J. Contributions to the study of marine products. XXXII. The nucleosides of sponges. I. J. Org. Chem. 1951; 981–987.
83. CYTARABINE ACCORD SPC. http://www.sukl.cz/modules/medication/download.php?file=SPC76455.pdf&type=spc&as=cytarabine-accord-100-mg-ml-injekcni-infuzni-roztok-spc (30. 5. 2017).
84. CYTOSAR SPC. http://www.sukl.cz/modules/medication/download.php?file=SPC66037.pdf&type=spc&as=cytosar-1-g-spc (30. 5. 2017).
85. ALEXAN SPC. http://www.sukl.cz/modules/medication/download.php?file=SPC50287.pdf&type=spc&as=alexan-50-mg-ml-spc (30. 5. 2017).
86. DEPOCYTE SPC. http://www.ema.europa.eu/docs/cs_CZ/document_library/EPAR_-_Product_Information/human/000317/WC500035649.pdf (30. 5. 2017).
87. Rai K. R., Holland J. F., Glidewell O. J., Weinberg V., Brunner K., Obrecht J. P., Preisler H. D., Nawabi I. W., Prager D., Carey R. W., Cooper M. R., Haurani F., Hutchison J. L., Silver R. T., Falkson G., Wiernik P., Hoagland H. C., Bloomfield C. D., James G. W., Gottlieb A., Ramanan S. V., Blom J., Nissen N. I., Bank A., Ellison R. R., Kung F., Henry P., McIntyre O. R., Kaan S. K. Treatment of acute myelocytic leukemia: a study by cancer and leukemia group B. Blood 1981; 58, 1203–1212.
88. Yates J., Glidewell O., Wiernik P., Cooper M. R., Steinberg D., Dosik H., Levy R., Hoagland C., Henry P., Gottlieb A., Cornell C., Berenberg J., Hutchison J. L., Raich P., Nissen N., Ellison R. R., Frelick R., James G. W., Falkson G., Silver R. T., Haurani F., Green M., Henderson E., Leone L., Holland J. F. Cytosine arabinoside with daunorubicin or adriamycin for therapy of acute myelocytic leukemia: a CALGB study. Blood 1982; 60, 454–462.
89. Perry M. C., Doll D. C., Freter C. E. Chemotherapy source book. 5th ed. Philadelphia: Wolters Kluwer/Lippincott Williams & Wilkins 2012.
90. AML Collaborative Group. A systematic collaborative overview of randomized trials comparing idarubicin with daunorubicin (or other anthracyclines) as induction therapy for acute myeloid leukaemia. AML Collaborative Group. Br. J. Haematol. 1998; 103, 100–109.
91. Li X., Xu S., Tan Y., Chen J. The effects of idarubicin versus other anthracyclines for induction therapy of patients with newly diagnosed leukaemia. Cochrane Database Syst. Rev. 2015; CD010432.
92. Holowiecki J., Grosicki S., Robak T., Kyrcz-Krzemien S., Giebel S., Hellmann A., Skotnicki A., Jedrzejczak W. W., Konopka L., Kuliczkowski K., Zdziarska B., Dmoszynska A., Marianska B., Pluta A., Zawilska K., Komarnicki M., Kloczko J., Sulek K., Haus O., Stella-Holowiecka B., Baran W., Jakubas B., Paluszewska M., Wierzbowska A., Kielbinski M., Jagoda K., Polish Adult Leukemia Group (PALG). Addition of cladribine to daunorubicin and cytarabine increases complete remission rate after a single course of induction treatment in acute myeloid leukemia. Multicenter, phase III study. Leukemia 2004; 18, 989–997.
93. Holowiecki J., Grosicki S., Giebel S., Robak T., Kyrcz-Krzemien S., Kuliczkowski K., Skotnicki A. B., Hellmann A., Sulek K., Dmoszynska A., Kloczko J., Jedrzejczak W. W., Zdziarska B., Warzocha K., Zawilska K., Komarnicki M., Kielbinski M., Piatkowska-Jakubas B., Wierzbowska A., Wach M., Haus O. Cladribine, but not fludarabine, added to daunorubicin and cytarabine during induction prolongs survival of patients with acute myeloid leukemia: a multicenter, randomized phase III study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012; 30, 2441–2448.
94. Wiedower E., Jamy O., Martin M. G. Induction of Acute Myeloid Leukemia with Idarubicin, Cytarabine and Cladribine. Anticancer Res. 2015; 35, 6287–6290.
95. Fridle C., Medinger M., Wilk M. C., Seipel K., Passweg J., Manz M. G., Pabst T. Cladribine, cytarabine and idarubicin (CLA-Ida) salvage chemotherapy in relapsed acute myeloid leukemia (AML). Leuk. Lymphoma 2016; 1–8.
96. Caballero M. D., Rubio V., Rifon J., Heras I., García-Sanz R., Vázquez L., Vidriales B., del Cañizo M. C., Corral M., Gonzalez M., León A., Jean-Paul E., Rocha E., Moraleda J. M., San Miguel J. F. BEAM chemotherapy followed by autologous stem cell support in lymphoma patients: analysis of efficacy, toxicity and prognostic factors. Bone Marrow Transplant. 1997; 20, 451–458.
97. Sharma A., Kayal S., Iqbal S., Malik P. S., Raina V. Comparison of BEAM vs. LEAM regimen in autologous transplant for lymphoma at AIIMS. SpringerPlus 2013; 2, 489.
98. Kim J.-W., Lee H. J., Yi H. G., Kim B.-S., Bang S.-M., Kim J. S., Kim I., Yoon S.-S., Lee J. S., Kim C. S., Park S., Kim B. K. Mitoxantrone, etoposide, cytarabine, and melphalan (NEAM) followed by autologous stem cell transplantation for patients with chemosensitive aggressive non-Hodgkin lymphoma. Am. J. Hematol. 2012; 87, 479–483.
99. Probíhající studie s cytarabinem v EU. https://www.clinicaltrialsregister.eu/ctr-search/search?query=Cytarabine+&status=ongoing (30. 5. 2017).
100. Probíhající studie s cytarabinem v USA. https://www.clinicaltrials.gov/ct2/results?term=cytarabine&recr=Open&no_unk=Y&show_flds=Y (30. 5. 2017).
101. Sagar S., Kaur M., Minneman K. P. Antiviral lead compounds from marine sponges. Mar. Drugs 2010; 8, 2619–2638.
102. Privat de Garilhe M., De Rudder J. Effect of 2 arabinose nucleosides on the multiplication of herpes virus and vaccine in cell culture. Comptes Rendus Hebd. Seances Acad. Sci. 1964; 259, 2725–2728.
103. Cozzarelli N. R. The mechanism of action of inhibitors of DNA synthesis. Annu. Rev. Biochem. 1977; 46, 641–668.
104. Iwatsubo K., Bravo C., Uechi M., Baljinnyam E., Nakamura T., Umemura M., Lai L., Gao S., Yan L., Zhao X., Park M., Qiu H., Okumura S., Iwatsubo M., Vatner D. E., Vatner S. F., Ishikawa Y. Prevention of heart failure in mice by an antiviral agent that inhibits type 5 cardiac adenylyl cyclase. Am. J. Physiol. Heart Circ. Physiol. 2012; 302, H2622–2628.
105. Vatner S. F., Pachon R. E., Vatner D. E. Inhibition of adenylyl cyclase type 5 increases longevity and healthful aging through oxidative stress protection. Oxid. Med. Cell. Longev. 2015; 2015, 250310.
106. Bravo C. A., Vatner D. E., Pachon R., Zhang J., Vatner S. F. A Food and Drug Administration-Approved Antiviral Agent that Inhibits Adenylyl Cyclase Type 5 Protects the Ischemic Heart Even When Administered after Reperfusion. J. Pharmacol. Exp. Ther. 2016; 357, 331–336.
107. Nakamura T., Fujita T., Kishimura M., Suita K., Hidaka Y., Cai W., Umemura M., Yokoyama U., Uechi M., Ishikawa Y. Vidarabine, an Anti-Herpes Virus Agent, Protects Against the Development of Heart Failure With Relatively Mild Side-Effects on Cardiac Function in a Canine Model of Pacing-Induced Dilated Cardiomyopathy. Circ. J. Off. J. Jpn. Circ. Soc. 2016; 80, 2496–2505.
108. Leal J. F. M., Martínez-Díez M., García-Hernández V., Moneo V., Domingo A., Bueren-Calabuig J. A., Negri A., Gago F., Guillén-Navarro M. J., Avilés P., Cuevas C., García-Fernández L. F., Galmarini C. M. PM01183, a new DNA minor groove covalent binder with potent in vitro and in vivo anti-tumour activity. Br. J. Pharmacol. 2010; 161, 1099–1110.
109. Santamaría Nuñez G., Robles C. M. G., Giraudon C., Martínez-Leal J. F., Compe E., Coin F., Aviles P., Galmarini C. M., Egly J.-M. Lurbinectedin specifically triggers the degradation of phosphorylated rna polymerase ii and the formation of DNA breaks in cancer cells. Mol. Cancer Ther. 2016; 15, 2399–2412.
110. Céspedes M. V., Guillén M. J., López-Casas P. P., Sarno F., Gallardo A., Álamo P., Cuevas C., Hidalgo M., Galmarini C. M., Allavena P., Avilés P., Mangues R. Lurbinectedin induces depletion of tumor-associated macrophages (TAM), an essential component of its in vivo synergism with gemcitabine. Dis. Model. Mech. 2016.
111. Vidal A., Muñoz C., Guillén M-J., Moretó J., Puertas S., Martínez-Iniesta M., Figueras A., Padullés L., García-Rodriguez F. J., Berdiel-Acer M., Pujana M. A., Salazar R., Gil-Martin M., Martí L., Ponce J., Molleví D. G., Capella G., Condom E., Viñals F., Huertas D., Cuevas C., Esteller M., Avilés P., Villanueva A. Lurbinectedin (PM01183), a new DNA minor groove binder, inhibits growth of orthotopic primary graft of cisplatin-resistant epithelial ovarian cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012; 18, 5399–5411.
112. Elez M. E., Tabernero J., Geary D., Macarulla T., Kang S. P., Kahatt C., Pita A. S-M., Teruel C. F., Siguero M., Cullell-Young M., Szyldergemajn S., Ratain M. J. First-in-human phase I study of Lurbinectedin (PM01183) in patients with advanced solid tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014; 20, 2205–2214.
113. CORAIL. Clinical Trial of Lurbinectedin (PM01183) in Platinum Resistant Ovarian Cancer Patients (CORAIL). https://clinicaltrials.gov/ct2/show/NCT02421588 (30. 5. 2017).
114. NCT02448537. A Phase II Multi-Strata Study of PM01183 as a Single Agent or in Combination With Conventional Chemotherapy in Metastatic and/or Unresectable Sarcomas. https://clinicaltrials.gov/ct2/show/NCT02448537 (30. 5. 2017).
115. NCT01525589. A Phase II Clinical Trial of PM01183 in BRCA 1/2-Associated or Unselected Metastatic Breast Cancer. https://clinicaltrials.gov/ct2/show/NCT01525589 (30. 5. 2017).
116. Lago J., Rodríguez L. P., Blanco L., Vieites J. M., Cabado A. G. Tetrodotoxin, an Extremely Potent Marine Neurotoxin: Distribution, Toxicity, Origin and Therapeutical Uses. Mar. Drugs 2015; 13, 6384–6406.
117. Narahashi T. Tetrodotoxin – A brief history. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2008; 84, 147–154.
118. Mosher H. S., Fuhrman F. A., Buchwald H. D., Fischer H. G. Tarichatoxin-tetrodotoxin: a potent neurotoxin. Science 1964; 144, 1100–1110.
119. Kim Y. H., Brown G. B., Mosher F. A. Tetrodotoxin: Occurrence in atelopid frogs of Costa Rica. Science 1975; 189, 151–152.
120. Sheumack D. D., Howden M. E., Spence I., Quinn R. J. Maculotoxin: a neurotoxin from the venom glands of the octopus Hapalochlaena maculosa identified as tetrodotoxin. Science 1978; 199, 188–189.
121. Noguchi T., Jeon J. K., Arakawa O., Sugita H., Deguchi Y., Shida Y., Hashimoto K. Occurrence of tetrodotoxin and anhydrotetrodotoxin in Vibrio sp. isolated from the intestines of a xanthid crab, Atergatis floridus. J. Biochem. (Tokyo) 1986; 99, 311–314.
122. Chau R., Kalaitzis J. A., Neilan B. A. On the origins and biosynthesis of tetrodotoxin. Aquat. Toxicol. Amst. Neth. 2011; 104, 61–72.
123. Yu V. C.-H., Yu P. H.-F., Ho K.-C., Lee F. W.-F. Isolation and Identification of a New Tetrodotoxin-Producing Bacterial Species, Raoultella terrigena, from Hong Kong Marine Puffer Fish Takifugu niphobles. Mar. Drugs 2011; 9, 2384–2396.
124. Chau J., Ciufolini M. A. The Chemical Synthesis of Tetrodoxin: An Ongoing Quest. Mar. Drugs 2011; 9, 2046–2074.
125. Shi J., Liu T.-T., Wang X., Epstein D. H., Zhao L.-Y., Zhang X.-L., Lu L. Tetrodotoxin reduces cue-induced drug craving and anxiety in abstinent heroin addicts. Pharmacol. Biochem. Behav. 2009; 92, 603–607.
126. Song H., Li J., Lu C.-L., Kang L., Xie L., Zhang Y.-Y., Zhou X.-B., Zhong S. Tetrodotoxin alleviates acute heroin withdrawal syndrome: a multicentre, randomized, double-blind, placebo-controlled study. Clin. Exp. Pharmacol. Physiol. 2011; 38, 510–514.
127. Kostyuk P. G., Veselovsky N. S., Tsyndrenko A. Y. Ionic currents in the somatic membrane of rat dorsal root ganglion neurons-I. Sodium currents. Neuroscience 1981; 6, 2423–2430.
128. Luiz A. P., Wood J. N. Sodium Channels in Pain and Cancer: New Therapeutic Opportunities. Adv. Pharmacol. San Diego Calif 2016; 75, 153–178.
129. McEntire D. M., Kirkpatrick D. R., Dueck N. P., Kerfeld M. J., Smith T. A., Nelson T. J., Reisbig M. D., Agrawal D. K. Pain transduction: a pharmacologic perspective. Expert Rev. Clin. Pharmacol. 2016; 9, 1069–1080.
130. Akopian A. N., Souslova V., England S., Okuse K., Ogata N., Ure J., Smith A., Kerr B. J., McMahon S. B., Boyce S., Hill R., Stanfa L. C., Dickenson A. H., Wood J. N. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat. Neurosci. 1999; 2, 541–548.
131. Ogata N., Ohishi Y. Molecular diversity of structure and function of the voltage-gated Na+ channels. Jpn. J. Pharmacol. 2002; 88, 365–377.
132. Strassman A. M., Raymond S. A. Electrophysiological evidence for tetrodotoxin-resistant sodium channels in slowly conducting dural sensory fibers. J. Neurophysiol. 1999; 81, 413–424.
133. Pinto V., Derkach V. A., Safronov B. V. Role of TTX-sensitive and TTX-resistant sodium channels in Adelta - and C-fiber conduction and synaptic transmission. J. Neurophysiol. 2008; 99, 617–628.
134. Blair N. T., Bean B. P. Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J. Neurosci. Off. J. Soc. Neurosci. 2002; 22, 10277–10290.
135. Zimmermann K., Leffler A., Babes A., Cendan C. M., Carr R. W., Kobayashi J., Nau C., Wood J. N., Reeh P. W. Sensory neuron sodium channel Nav1.8 is essential for pain at low temperatures. Nature 2007; 447, 855–858.
136. TEC-006. Safety & Efficacy Study of Subcutaneous Tetrodotoxin for Moderate to Severe Inadequately Controlled Cancer-related Pain (TEC-006). https://clinicaltrials.gov/ct2/show/NCT00725114 (30. 5. 2017).
137. TTX-CINP-201. The Purpose of This Study is to Determine if Tetrodotoxin (TTX) is Effective in the Treatment of Pain Resulting From Chemotherapy Treatment (TTX-CINP-201). https://clinicaltrials.gov/ct2/show/NCT01655823 (30. 5. 2017).
138. Ott P. A., Hamid O., Pavlick A. C., Kluger H., Kim K. B., Boasberg P. D., Simantov R., Crowley E., Green J. A., Hawthorne T., Davis T. A., Sznol M., Hwu P. Phase I/II Study of the Antibody-Drug Conjugate Glembatumumab Vedotin in Patients With Advanced Melanoma. J. Clin. Oncol. 2014; 32, 3659–3666.
139. Weterman M. A., Ajubi N., van Dinter I. M., Degen W. G., van Muijen G. N., Ruitter D. J., Bloemers H. P. nmb, a novel gene, is expressed in low-metastatic human melanoma cell lines and xenografts. Int. J. Cancer 1995; 60, 73–81.
140. Tse K. F., Jeffers M., Pollack V. A., McCabe D. A., Shadish M. L., Khramtsov N. V., Hackett C. S., Shenoy S. G., Kuang B., Boldog F. L., MacDougall J. R., Rastelli L., Herrmann J., Gallo M., Gazit-Bornstein G., Senter P. D., Meyer D. L., Lichenstein H. S., LaRochelle W. J. CR011, a fully human monoclonal antibody-auristatin E conjugate, for the treatment of melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006; 12, 1373–1382.
141. Kuan C.-T., Wakiya K., Dowell J. M., Herndon J. E., Reardon D. A., Graner M. W., Riggins G. J., Wikstrand C. J., Bigner D. D. Glycoprotein nonmetastatic melanoma protein B, a potential molecular therapeutic target in patients with glioblastoma multiforme. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006; 12, 1970–1982.
142. Rose A. A. N., Grosset A-A., Dong Z., Russo C., Macdonald P. A., Bertos N. R., St-Pierre Y., Simantov R., Hallett M., Park M., Gaboury L., Siegel P. M. Glycoprotein nonmetastatic B is an independent prognostic indicator of recurrence and a novel therapeutic target in breast cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010; 16, 2147–2156.
143. Roth M., Barris D. M., Piperdi S., Kuo V., Everts S., Geller D., Houghton P., Kolb E. A., Hawthorne T., Gill J., Gorlick R. Targeting Glycoprotein NMB With Antibody-Drug Conjugate, Glembatumumab Vedotin, for the Treatment of Osteosarcoma. Pediatr. Blood Cancer 2016; 63, 32–38.
144. Oyewumi M. O., Manickavasagam D., Novak K., Wehrung D., Paulic N., Moussa F. M., Sondag G. R., Safadi F. F. Osteoactivin (GPNMB) ectodomain protein promotes growth and invasive behavior of human lung cancer cells. Oncotarget 2016; 7, 13932–13944.
145. Bendell J., Saleh M., Rose A. A. N., Siegel P. M., Hart L., Sirpal S., Jones S., Green J., Crowley E., Simantov R., Keler T., Davis T., Vahdat L. Phase I/II study of the antibody-drug conjugate glembatumumab vedotin in patients with locally advanced or metastatic breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014; 32, 3619–3625.
146. Yardley D. A., Weaver R., Melisko M. E., Saleh M. N., Arena F. P., Forero A., Cigler T., Stopeck A., Citrin D., Oliff I., Bechhold R., Loutfi R., Garcia A. A., Cruickshank S., Crowley E., Green J., Hawthorne T., Yellin M. J., Davis T. A., Vahdat L. T. EMERGE: A randomized phase ii study of the antibody-drug conjugate glembatumumab vedotin in advanced glycoprotein nmb-expressing breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015; 33, 1609–1619.
147. NCT01997333. Study of Glembatumumab Vedotin (CDX-011) in Patients With Metastatic, gpNMB Over-Expressing, Triple Negative Breast Cancer (METRIC). https://clinicaltrials.gov/ct2/show/NCT01997333 (30. 5. 2017).
148. Kitagaki J., Shi G., Miyauchi S., Murakami S., Yang Y. Cyclic depsipeptides as potential cancer therapeutics. Anticancer. Drugs 2015; 26, 259–271.
149. Rinehart K. L., Kishore V., Bible K. C., Sakai R., Sullins D. W., Li K. M. Didemnins and tunichlorin: novel natural products from the marine tunicate Trididemnum solidum. J. Nat. Prod. 1988; 51, 1–21.
150. Crampton S. L., Adams E. G., Kuentzel S. L., Li L. H., Badiner G., Bhuyan B. K. Biochemical and cellular effects of didemnins A and B. Cancer Res. 1984; 44, 1796–1801.
151. Jiang T. L., Liu R. H., Salmon S. E. Antitumor activity of didemnin B in the human tumor stem cell assay. Cancer Chemother. Pharmacol. 1983; 11, 1–4.
152. Motzer R., Scher H., Bajorin D., Sternberg C., Bosl G. J. Phase II trial of Didemnin B in patients with advanced renal cell carcinoma. Invest. New Drugs 1990; 8, 391–392.
153. Shin D. M., Holoye P. Y., Murphy W. K., Forman A., Papasozomenos S. C., Hong W. K., Raber M. Phase I/II clinical trial of didemnin B in non-small-cell lung cancer: neuromuscular toxicity is dose-limiting. Cancer Chemother. Pharmacol. 1991; 29, 145–149.
154. Cain J. M., Liu P. Y., Alberts D. E., Gallion H. H., Laufman L., O’Sullivan J., Weiss G., Bickers J. N. Phase II trial of didemnin-B in advanced epithelial ovarian cancer. A Southwest Oncology Group study. Invest. New Drugs 1992; 10, 23–24.
155. Jacobs A. J., Blessing J. A., Munoz A. A phase II trial of didemnin B (NSC No. 325319) in advanced and recurrent cervical carcinoma: a Gynecologic Oncology Group study. Gynecol. Oncol. 1992; 44, 268–270.
156. Sakai R., Rinehart K. L., Kishore V., Kundu B., Faircloth G., Gloer J. B., Carney J. R., Namikoshi M., Sun F., Hughes R. G., García Grávalos D., de Quesada T. G., Wilson G. R., Heid R. M. Structure-activity relationships of the didemnins. J. Med. Chem. 1996; 39, 2819–2834.
157. Broggini M., Marchini S. V., Galliera E., Borsotti P., Taraboletti G., Erba E., Sironi M., Jimeno J., Faircloth G. T., Giavazzi R., D’Incalci M. Aplidine, a new anticancer agent of marine origin, inhibits vascular endothelial growth factor (VEGF) secretion and blocks VEGF-VEGFR-1 (flt-1) autocrine loop in human leukemia cells MOLT-4. Leukemia 2003; 17, 52–59.
158. Cuadrado A., Garcia-Fernandez L. F., Gonzalez L., Suarez Y., Losada A., Alcaide V., Martinez T., Fernandez-Sousa J. M., Sanchez-Puelles J. M., Munoz A. Aplidin induces apoptosis in human cancer cells via glutathione depletion and sustained activation of the epidermal growth factor receptor, Src, JNK, and p38 MAPK. J. Biol. Chem. 2003; 278, 241–250.
159. González-Santiago L., Suárez Y., Zarich N., Muñoz-Alonso M. J., Cuadrado A., Martínez T., Goya L., Iradi A., Sáez-Tormo G., Maier J. V., Moorthy A., Cato A. C. B., Rojas J. M., Muñoz A. Aplidin induces JNK-dependent apoptosis in human breast cancer cells via alteration of glutathione homeostasis, Rac1 GTPase activation, and MKP-1 phosphatase downregulation. Cell Death Differ. 2006; 13, 1968–1981.
160. Bravo S. B., García-Rendueles M. E. R., Seoane R., Dosil V., Cameselle-Teijeiro J., López-Lázaro L., Zalvide J., Barreiro F., Pombo C. M., Alvarez C. V. Plitidepsin has a cytostatic effect in human undifferentiated (anaplastic) thyroid carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005; 11, 7664–7673.
161. Biscardi M., Caporale R., Balestri F., Gavazzi S., Jimeno J., Grossi A. VEGF inhibition and cytotoxic effect of aplidin in leukemia cell lines and cells from acute myeloid leukemia. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2005; 16, 1667–1674.
162. Taraboletti G., Poli M., Dossi R., Manenti L., Borsotti P., Faircloth G. T., Broggini M., D’Incalci M., Ribatti D., Giavazzi R. Antiangiogenic activity of aplidine, a new agent of marine origin. Br. J. Cancer 2004; 90, 2418–2424.
163. Plummer R., Lorigan P., Brown E., Zaucha R., Moiseyenko V., Demidov L., Soriano V., Chmielowska E., Andrés R., Kudryavtseva G., Kahatt C., Szyldergemajn S., Extremera S., de Miguel B., Cullell-Young M., Calvert H. Phase I-II study of plitidepsin and dacarbazine as first-line therapy for advanced melanoma. Br. J. Cancer 2013; 109, 1451–1459.
164. Mateos M. V., Cibeira M. T., Richardson P. G., Prosper F., Oriol A., de la Rubia J., Lahuerta J. J., García-Sanz R., Extremera S., Szyldergemajn S., Corrado C., Singer H., Mitsiades C. S., Anderson K. C., Bladé J., San Miguel J. Phase II clinical and pharmacokinetic study of plitidepsin 3-hour infusion every two weeks alone or with dexamethasone in relapsed and refractory multiple myeloma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010; 16, 3260–3269.
165. Schöffski P., Guillem V., Garcia M., Rivera F., Tabernero J., Cullell M., Lopez-Martin J. A., Pollard P., Dumez H., del Muro X. G., Paz-Ares L. Phase II randomized study of Plitidepsin (Aplidin), alone or in association with L-carnitine, in patients with unresectable advanced renal cell carcinoma. Mar. Drugs 2009; 7, 57–70.
166. Pardanani A., Tefferi A., Guglielmelli P., Bogani C., Bartalucci N., Rodríguez J., Extremera S., Pérez I., Alfaro V., Vannucchi A. M. Evaluation of plitidepsin in patients with primary myelofibrosis and post polycythemia vera/essential thrombocythemia myelofibrosis: results of preclinical studies and a phase II clinical trial. Blood Cancer J. 2015; 5, e286.
167. Dumez H., Gallardo E., Culine S., Galceran J. C., Schöffski P., Droz J. P., Extremera S., Szyldergemajn S., Fléchon A. Phase II study of biweekly plitidepsin as second-line therapy for advanced or metastatic transitional cell carcinoma of the urothelium. Mar. Drugs 2009; 7, 451–463.
168. Eisen T., Thatcher N., Leyvraz S., Miller W. H., Couture F., Lorigan P., Lüthi F., Small D., Tanovic A., O’Brien M. Phase II study of weekly plitidepsin as second-line therapy for small cell lung cancer. Lung Cancer Amst. Neth. 2009; 64, 60–65.
169. Hamann M. T., Otto C. S., Scheuer P. J., Dunbar D. C. Kahalalides: Bioactive Peptides from a Marine Mollusk Elysia rufescens and Its Algal Diet Bryopsis sp.(1). J. Org. Chem. 1996; 61, 6594–6600.
170. Suárez Y., González L., Cuadrado A., Berciano M., Lafarga M., Muñoz A. Kahalalide F, a new marine-derived compound, induces oncosis in human prostate and breast cancer cells. Mol. Cancer Ther. 2003; 2, 863–872.
171. Janmaat M. L., Rodriguez J. A., Jimeno J., Kruyt F. A. E., Giaccone G. Kahalalide F induces necrosis-like cell death that involves depletion of ErbB3 and inhibition of Akt signaling. Mol. Pharmacol. 2005; 68, 502–510.
172. Martín-Algarra S., Espinosa E., Rubió J., López López J. J., Manzano J. L., Carrión L. A., Plazaola A., Tanovic A., Paz-Ares L. Phase II study of weekly Kahalalide F in patients with advanced malignant melanoma. Eur. J. Cancer Oxf. Engl. 2009; 1990 45, 732–735.
173. Serova M., de Gramont A., Bieche I., Riveiro M. E., Galmarini C. M., Aracil M., Jimeno J., Faivre S., Raymond E. Predictive Factors of Sensitivity to Elisidepsin, a Novel Kahalalide F-Derived Marine Compound. Mar. Drugs 2013; 11, 944–959.
174. Petty R., Anthoney A., Metges J-P., Alsina M., Gonçalves A., Brown J., Montagut C., Gunzer K., Laus G., Iglesias Dios J. L., Miguel-Lillo B., Bohan P., Salazar R. Phase Ib/II study of elisidepsin in metastatic or advanced gastroesophageal cancer (IMAGE trial). Cancer Chemother. Pharmacol. 2016; 77, 819–827.
175. Oku N., Gustafson K. R., Cartner L. K., Wilson J. A., Shigematsu N., Hess S., Pannell L. K., Boyd M. R., McMahon J. B. Neamphamide A, a new HIV-inhibitory depsipeptide from the Papua New Guinea marine sponge Neamphius huxleyi. J. Nat. Prod. 2004; 67, 1407–1411.
176. Yamano Y., Arai M., Kobayashi M. Neamphamide B, new cyclic depsipeptide, as an anti-dormant mycobacterial substance from a Japanese marine sponge of Neamphius sp. Bioorg. Med. Chem. Lett. 2012; 22, 4877–4881.
177. Moore K. S., Wehrli S., Roder H., Rogers M., Forrest J. N., McCrimmon D., Zasloff M. Squalamine: an aminosterol antibiotic from the shark. Proc. Natl. Acad. Sci. U. S. A. 1993; 90, 1354–1358.
178. Sills A. K., Williams J. I., Tyler B. M., Epstein D. S., Sipos E. P., Davis J. D., McLane M. P., Pitchford S., Cheshire K., Gannon F. H., Kinney W. A., Chao T. L., Donowitz M., Laterra J., Zasloff M., Brem H. Squalamine inhibits angiogenesis and solid tumor growth in vivo and perturbs embryonic vasculature. Cancer Res. 1998; 58, 2784–2792.
179. Bhargava P., Marshall J. L., Dahut W., Rizvi N., Trocky N., Williams J. I., Hait H., Song S., Holroyd K. J., Hawkins M. J. A phase I and pharmacokinetic study of squalamine, a novel antiangiogenic agent, in patients with advanced cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2001; 7, 3912–3919.
180. Herbst R. S., Hammond L. A., Carbone D. P., Tran H. T., Holroyd K. J., Desai A., Williams J. I., Bekele B. N., Hait H., Allgood V., Solomon S., Schiller J. H. A phase I/IIA trial of continuous five-day infusion of squalamine lactate (MSI-1256F) plus carboplatin and paclitaxel in patients with advanced non-small cell lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003; 9, 4108–4115.
181. NCT01769183. Squalamine for the Treatment in Proliferative Diabetic Retinopathy. https://clinicaltrials.gov/ct2/show/NCT01769183 (30. 5. 2017).
182. NCT01678963. Efficacy and Safety of Squalamine Lactate Eye Drops in Subjects With Neovascular (Wet) Age-related Macular Degeneration (AMD). https://clinicaltrials.gov/ct2/show/NCT01678963 (30. 5. 2017).
183. Kem W. R. A study of the occurrence of anabaseine in Paranemertes and other nemertines. Toxicon Off. J. Int. Soc. Toxinology 1971; 9, 23–32.
184. Wheeler J. W., Olubajo O., Storm C. B., Duffield R. M. Anabaseine: venom alkaloid of aphaenogaster ants. Science 1981; 211, 1051–1052.
185. Kem W., Soti F., Wildeboer K., LeFrancois S., MacDougall K., Wei D.-Q., Chou K.-C., Arias H. R. The Nemertine Toxin Anabaseine and Its Derivative DMXBA (GTS-21): Chemical and Pharmacological Properties. Mar. Drugs 2006; 4, 255–273.
186. Berg D. K., Conroy W. G. Nicotinic alpha 7 receptors: synaptic options and downstream signaling in neurons. J. Neurobiol. 2002; 53, 512–523.
187. Wonnacott S., Barik J., Dickinson J., Jones I. W. Nicotinic receptors modulate transmitter cross talk in the CNS: nicotinic modulation of transmitters. J. Mol. Neurosci. MN 2006; 30, 137–140.
188. Hajós M., Rogers B. N. Targeting alpha7 nicotinic acetylcholine receptors in the treatment of schizophrenia. Curr. Pharm. Des. 2010; 16, 538–554.
189. Deutsch S. I., Schwartz B. L., Schooler N. R., Brown C. H., Rosse R. B., Rosse S. M. Targeting alpha-7 nicotinic neurotransmission in schizophrenia: a novel agonist strategy. Schizophr. Res. 2013; 148, 138–144.
190. Beinat C., Banister S. D., Herrera M., Law V., Kassiou M. The therapeutic potential of α7 nicotinic acetylcholine receptor (α7 nAChR) agonists for the treatment of the cognitive deficits associated with schizophrenia. CNS Drugs 2015; 29, 529–542.
191. Hashimoto K. Targeting of α7 Nicotinic Acetylcholine Receptors in the Treatment of Schizophrenia and the Use of Auditory Sensory Gating as a Translational Biomarker. Curr. Pharm. Des. 2015; 21, 3797–3806.
192. Kem W. R. The brain alpha7 nicotinic receptor may be an important therapeutic target for the treatment of Alzheimer’s disease: studies with DMXBA (GTS-21). Behav. Brain Res. 2000; 113, 169–181.
193. Vallés A. S., Borroni M. V., Barrantes F. J. Targeting brain α7 nicotinic acetylcholine receptors in Alzheimer’s disease: rationale and current status. CNS Drugs 2014; 28, 975–987.
194. Wessler I., Kirkpatrick C. J. Acetylcholine beyond neurons: the non-neuronal cholinergic system in humans. Br. J. Pharmacol. 2008; 154, 1558–1571.
195. Reardon C. Neuro-immune interactions in the cholinergic anti-inflammatory reflex. Immunol. Lett. 2016; 178, 92–96.
196. Yue Y., Liu R., Cheng W., Hu Y., Li J., Pan X., Peng J., Zhang P. GTS-21 attenuates lipopolysaccharide-induced inflammatory cytokine production in vitro by modulating the Akt and NF-κB signaling pathway through the α7 nicotinic acetylcholine receptor. Int. Immunopharmacol. 2015; 29, 504–512.
197. Giebelen I. A. J., van Westerloo D. J., LaRosa G. J., de Vos A. F., van der Poll T. Stimulation of alpha 7 cholinergic receptors inhibits lipopolysaccharide-induced neutrophil recruitment by a tumor necrosis factor alpha-independent mechanism. Shock Augusta Ga 2007; 27, 443–447.
198. Kox M., Pompe J. C., Gordinou de Gouberville M. C., van der Hoeven J. G., Hoedemaekers C. W., Pickkers P. Effects of the α7 nicotinic acetylcholine receptor agonist GTS-21 on the innate immune response in humans. Shock Augusta Ga 2011; 36, 5–11.
199. Kihara T., Shimohama S., Sawada H., Kimura J., Kume T., Kochiyama H., Maeda T., Akaike A. Nicotinic receptor stimulation protects neurons against beta-amyloid toxicity. Ann. Neurol. 1997; 42, 159–163.
200. Jin Y., Tsuchiya A., Kanno T., Nishizaki T. Amyloid-β peptide increases cell surface localization of α7 ACh receptor to protect neurons from amyloid β-induced damage. Biochem. Biophys. Res. Commun. 2015; 468, 157–160.
201. Lombardo S., Maskos U. Role of the nicotinic acetylcholine receptor in Alzheimer’s disease pathology and treatment. Neuropharmacology 2015; 96, 255–262.
202. Kitagawa H., Takenouchi T., Azuma R., Wesnes K. A., Kramer W. G., Clody D. E., Burnett A. L. Safety, pharmacokinetics, and effects on cognitive function of multiple doses of GTS-21 in healthy, male volunteers. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2003; 28, 542–551.
203. Olincy A., Harris J. G., Johnson L. L., Pender V., Kongs S., Allensworth D., Ellis J., Zerbe G. O., Leonard S., Stevens K. E., Stevens J. O., Martin L., Adler L. E., Soti F., Kem W. R., Freedman R. Proof-of-concept trial of an alpha7 nicotinic agonist in schizophrenia. Arch. Gen. Psychiatry 2006; 63, 630–638.
204. Freedman R., Olincy A., Buchanan R. W., Harris J. G., Gold J. M., Johnson L., Allensworth D., Guzman-Bonilla A., Clement B., Ball M. P., Kutnick J., Pender V., Martin L. F., Stevens K. E., Wagner B. D., Zerbe G. O., Soti F., Kem W. R. Initial phase 2 trial of a nicotinic agonist in schizophrenia. Am. J. Psychiatry 2008; 165, 1040–1047.
205. NCT00414622. GTS21-201 for Alzheimer Disease:GTS-21 Administered Daily for 28 Days to Participants With Probable Alzheimer’s Disease.https://clinicaltrials.gov/ct2/show/NCT00414622 (30. 5. 2017).
206. NCT00419445. Safety and Efficacy of GTS21 in Adults With Attention-deficit Hyperactivity Disorder. https://clinicaltrials.gov/ct2/show/NCT00419445 (30. 5. 2017).
207. Pettit G. R., Herald C. L., Doubek D. L., Herald D. L., Arnold E., Clardy J. Isolation and structure of bryostatin 1. J. Am. Chem. Soc. 1982; 104, 6846–6848.
208. Davidson S. K., Allen S. W., Lim G. E., Anderson C. M., Haygood M. G. Evidence for the biosynthesis of bryostatins by the bacterial symbiont ‘Candidatus Endobugula sertula’ of the bryozoan Bugula neritina. Appl. Environ. Microbiol. 2001; 67, 4531–4537.
209. Sudek S., Lopanik N. B., Waggoner L. E., Hildebrand M., Anderson C., Liu H., Patel A., Sherman D. H., Haygood M. G. Identification of the putative bryostatin polyketide synthase gene cluster from ‘Candidatus Endobugula sertula’, the uncultivated microbial symbiont of the marine bryozoan Bugula neritina. J. Nat. Prod. 2007; 70, 67–74.
210. Kollár P., Rajchard J., Balounová Z., Pazourek J. Marine natural products: bryostatins in preclinical and clinical studies. Pharm. Biol. 2014; 52, 237–242.
211. Russo P., Kisialiou A., Lamonaca P., Moroni R., Prinzi G., Fini M. New Drugs from Marine Organisms in Alzheimer’s Disease. Mar. Drugs 2015; 14, 5.
212. Stone R. M., Sariban E., Pettit G. R., Kufe D. W. Bryostatin 1 activates protein kinase C and induces monocytic differentiation of HL-60 cells. Blood 1988; 72, 208–213.
213. Nelson T. J., Alkon D. L. Neuroprotective versus tumorigenic protein kinase C activators. Trends Biochem. Sci. 2009; 34, 136–145.
214. Black J. D. Protein kinase C-mediated regulation of the cell cycle. Front. Biosci. J. Virtual Libr. 2000; 5, D406–423.
215. Zhang X., Zhang R., Zhao H., Cai H., Gush K. A., Kerr R. G., Pettit G. R., Kraft A. S. Preclinical pharmacology of the natural product anticancer agent bryostatin 1, an activator of protein kinase C. Cancer Res. 1996; 56, 802–808.
216. Asiedu C., Biggs J., Lilly M., Kraft A. S. Inhibition of leukemic cell growth by the protein kinase C activator bryostatin 1 correlates with the dephosphorylation of cyclin-dependent kinase 2. Cancer Res. 1995; 55, 3716–3720.
217. Philip P. A., Rea D., Thavasu P., Carmichael J., Stuart N. S., Rockett H., Talbot D. C., Ganesan T., Pettit G. R., Balkwill F. Phase I study of bryostatin 1: assessment of interleukin 6 and tumor necrosis factor alpha induction in vivo. The Cancer Research Campaign Phase I Committee. J. Natl. Cancer Inst. 1993; 85, 1812–1818.
218. Mohr H., Pettit G. R., Plessing-Menze A. Co-induction of lymphokine synthesis by the antineoplastic bryostatins. Immunobiology 1987; 175, 420–430.
219. Alkon D. L., Sun M-K., Nelson T. J. PKC signaling deficits: a mechanistic hypothesis for the origins of Alzheimer’s disease. Trends Pharmacol. Sci. 2007; 28, 51–60.
220. Sun M-K., Alkon D. L. The ‘memory kinases’: roles of PKC isoforms in signal processing and memory formation. Prog. Mol. Biol. Transl. Sci. 2014; 122, 31–59.
221. Pandey G. N., Dwivedi Y. Focus on protein kinase A and protein kinase C, critical components of signal transduction system, in mood disorders and suicide. Int. J. Neuropsychopharmacol. 2005; 8, 1–4.
222. Shelton R. C. The molecular neurobiology of depression. Psychiatr. Clin. North Am. 2007; 30, 1–11.
223. Sun M-K., Alkon D. L. Dual effects of bryostatin-1 on spatial memory and depression. Eur. J. Pharmacol. 2005; 512, 43–51.
224. NCT02431468. A Study Assessing Bryostatin in the Treatment of Moderately Severe to Severe Alzheimer’s Disease. https://clinicaltrials.gov/ct2/show/NCT02431468 (30. 5. 2017).
225. Mehla R., Bivalkar-Mehla S., Zhang R., Handy I., Albrecht H., Giri S., Nagarkatti P., Nagarkatti M., Chauhan A. Bryostatin modulates latent HIV-1 infection via PKC and AMPK signaling but inhibits acute infection in a receptor independent manner. PloS One 2010; 5, e11160.
226. Díaz L., Martínez-Bonet M., Sánchez J., Fernández-Pineda A., Jiménez J. L., Muñoz E., Moreno S., Álvarez S., Muñoz-Fernández M. Á. Bryostatin activates HIV-1 latent expression in human astrocytes through a PKC and NF-ĸB-dependent mechanism. Sci. Rep. 2015; 5, 12442.
227. Boto W. M., Brown L., Chrest J., Adler W. H. Distinct modulatory effects of bryostatin 1 and staurosporine on the biosynthesis and expression of the HIV receptor protein (CD4) by T cells. Cell Regul. 1991; 2, 95–103.
228. Gutiérrez C., Serrano-Villar S., Madrid-Elena N., Pérez-Elías M. J., Martín M. E., Barbas C., Ruipérez J., Muñoz E., Muñoz-Fernández M. A., Castor T., Moreno S. Bryostatin-1 for latent virus reactivation in HIV-infected patients on antiretroviral therapy. AIDS Lond. Engl. 2016; 30, 1385–1392.
229. Ajani J. A., Jiang Y., Faust J., Chang B. B., Ho L., Yao J. C., Rousey S., Dakhil S., Cherny R. C., Craig C., Bleyer A. A multi-center phase II study of sequential paclitaxel and bryostatin-1 (NSC 339555) in patients with untreated, advanced gastric or gastroesophageal junction adenocarcinoma. Invest. New Drugs 2006; 24, 353–357.
230. Barr P. M., Lazarus H. M., Cooper B. W., Schluchter M. D., Panneerselvam A., Jacobberger J. W., Hsu J. W., Janakiraman N., Simic A., Dowlati A., Remick S. C. Phase II study of bryostatin 1 and vincristine for aggressive non-Hodgkin lymphoma relapsing after an autologous stem cell transplant. Am. J. Hematol. 2009; 84, 484–487.
231. Lam A. P., Sparano J. A., Vinciguerra V., Ocean A. J., Christos P., Hochster H., Camacho F., Goel S., Mani S., Kaubisch A. Phase II study of paclitaxel plus the protein kinase C inhibitor bryostatin-1 in advanced pancreatic carcinoma. Am. J. Clin. Oncol. 2010; 33, 121–124.
232. Nezhat F., Wadler S., Muggia F., Mandeli J., Goldberg G., Rahaman J., Runowicz C., Murgo A. J., Gardner G. J. Phase II trial of the combination of bryostatin-1 and cisplatin in advanced or recurrent carcinoma of the cervix: a New York Gynecologic Oncology Group study. Gynecol. Oncol. 2004; 93, 144–148.
233. DeChristopher B. A., Fan A. C., Felsher D. W., Wender P. A. ‘Picolog’, a synthetically-available bryostatin analog, inhibits growth of MYC-induced lymphoma in vivo. Oncotarget 2012; 3, 58–66.
234. Tan Z., Turner R. C., Leon R. L., Li X., Hongpaisan J., Zheng W., Logsdon A. F., Naser Z. J., Alkon D. L., Rosen C. L., Huber J. D. Bryostatin improves survival and reduces ischemic brain injury in aged rats after acute ischemic stroke. Stroke 2013; 44, 3490–3497.
235. Tan Z., Lucke-Wold B. P., Logsdon A. F., Turner R. C., Tan C., Li X., Hongpaison J., Alkon D. L., Simpkins J. W., Rosen C. L., Huber J. D. Bryostatin extends tPA time window to 6 h following middle cerebral artery occlusion in aged female rats. Eur. J. Pharmacol. 2015; 764, 404–412.
236. Sun M.-K., Hongpaisan J., Alkon D. L. Postischemic PKC activation rescues retrograde and anterograde long-term memory. Proc. Natl. Acad. Sci. U. S. A. 2009; 106, 14676–14680.
237. Nicholson B., Lloyd G. K., Miller B. R., Palladino M. A., Kiso Y., Hayashi Y., Neuteboom S. T. C. NPI-2358 is a tubulin-depolymerizing agent: in-vitro evidence for activity as a tumor vascular-disrupting agent. Anticancer. Drugs 2006; 17, 25–31.
238. Fairchild C. R., Johnston K. J., Peterson R. W., Cornell L. A., Bifario M., Raventos-Suarez C., et al. Halimide, a novel cytotoxic marine natural product, destabilizes microtubules and demonstrates in vivo antitumor activity. Proc Am Cancer Res 1998; 165.
239. Kanoh K., Kohno S., Asari T., Harada T., Katada J. Phenylahistin: a new mammalian cell cycle inhibitor produced by Aspergillus ustus. Bioorg Med Chem Lett 1997; 2847–2852.
240. Mita M. M., Spear M. A., Yee L. K., Mita A. C., Heath E. I., Papadopoulos K. P., Federico K. C., Reich S. D., Romero O., Malburg L., Pilat M., Lloyd G. K., Neuteboom S. T. C., Cropp G., Ashton E., LoRusso P. M. Phase 1 first-in-human trial of the vascular disrupting agent plinabulin(NPI-2358) in patients with solid tumors or lymphomas. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010; 16, 5892–5899.
241. Millward M., Mainwaring P., Mita A., Federico K., Lloyd G. K., Reddinger N., Nawrocki S., Mita M., Spear M. A. Phase 1 study of the novel vascular disrupting agent plinabulin (NPI-2358) and docetaxel. Invest. New Drugs 2012; 30, 1065–1073.
242. Feling R. H., Buchanan G. O., Mincer T. J., Kauffman C. A., Jensen P. R., Fenical W. Salinosporamide A: a highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus salinospora. Angew. Chem. Int. Ed Engl. 2003; 42, 355–357.
243. King R. W., Deshaies R. J., Peters J. M., Kirschner M. W. How proteolysis drives the cell cycle. Science 1996; 274, 1652–1659.
244. Levin N., Spencer A., Harrison S. J., Chauhan D., Burrows F. J., Anderson K. C., Reich S. D., Richardson P. G., Trikha M. Marizomib irreversibly inhibits proteasome to overcome compensatory hyperactivation in multiple myeloma and solid tumour patients. Br. J. Haematol. 2016; 174, 711–720.
245. Chauhan D., Catley L., Li G., Podar K., Hideshima T., Velankar M., Mitsiades C., Mitsiades N., Yasui H., Letai A., Ovaa H., Berkers C., Nicholson B., Chao T.-H., Neuteboom S. T. C., Richardson P., Palladino M. A., Anderson K. C. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell 2005; 8, 407–419.
246. Ahn K. S., Sethi G., Chao T.-H., Neuteboom S. T. C., Chaturvedi M. M., Palladino M. A., Younes A., Aggarwal B. B. Salinosporamide A (NPI-0052) potentiates apoptosis, suppresses osteoclastogenesis, and inhibits invasion through down-modulation of NF-kappaB regulated gene products. Blood 2007; 110, 2286–2295.
247. Bassères D. S., Baldwin A. S. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene 2006; 25, 6817–6830.
248. Hideshima T., Ikeda H., Chauhan D., Okawa Y., Raje N., Podar K., Mitsiades C., Munshi N. C., Richardson P. G., Carrasco R. D., Anderson K. C. Bortezomib induces canonical nuclear factor-kappaB activation in multiple myeloma cells. Blood 2009; 114, 1046–1052.
249. Cusack J. C., Liu R., Xia L., Chao T-H., Pien C., Niu W., Palombella V. J., Neuteboom S. T. C., Palladino M. A. NPI-0052 enhances tumoricidal response to conventional cancer therapy in a colon cancer model. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006; 12, 6758–6764.
250. Ruiz S., Krupnik Y., Keating M., Chandra J., Palladino M., McConkey D. The proteasome inhibitor NPI-0052 is a more effective inducer of apoptosis than bortezomib in lymphocytes from patients with chronic lymphocytic leukemia. Mol. Cancer Ther. 2006; 5, 1836–1843.
251. Miller C. P., Ban K., Dujka M. E., McConkey D. J., Munsell M., Palladino M., Chandra J. NPI-0052, a novel proteasome inhibitor, induces caspase-8 and ROS-dependent apoptosis alone and in combination with HDAC inhibitors in leukemia cells. Blood 2007; 110, 267–277.
252. Millward M., Price T., Townsend A., Sweeney C., Spencer A., Sukumaran S., Longenecker A., Lee L., Lay A., Sharma G., Gemmill R. M., Drabkin H. A., Lloyd G. K., Neuteboom S. T. C., McConkey D. J., Palladino M. A., Spear M. A. Phase 1 clinical trial of the novel proteasome inhibitor marizomib with the histone deacetylase inhibitor vorinostat in patients with melanoma, pancreatic and lung cancer based on in vitro assessments of the combination. Invest. New Drugs 2012; 30, 2303–2317.
253. Richardson P. G., Zimmerman T. M., Hofmeister C. C., Talpaz M., Chanan-Khan A. A., Kaufman J. L., Laubach J. P., Chauhan D., Jakubowiak A. J., Reich S., Trikha M., Anderson K. C. Phase 1 study of marizomib in relapsed or relapsed and refractory multiple myeloma: NPI-0052-101 Part 1. Blood 2016; 127, 2693–2700.
254. Harrison S. J., Mainwaring P., Price T., Millward M. J., Padrik P., Underhill C. R., Cannell P. K., Reich S. D., Trikha M., Spencer A. Phase I Clinical Trial of Marizomib (NPI-0052) in Patients with Advanced Malignancies Including Multiple Myeloma: Study NPI-0052-102 Final Results. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016; 22, 4559–4566.
255. NCT00461045. Phase 1/2 Clinical Trial of NPI-0052 in Patients With Relapsed or Relapsed/Refractory Multiple Myeloma. https://clinicaltrials.gov/ct2/show/NCT00461045 (30. 5. 2017).
256. NCT02330562. Stage 1: Marizomib + Bevacizumab in WHO Gr IV GBM; Stage 2: Marizomib Alone. https://clinicaltrials.gov/ct2/show/NCT02330562 (30. 5. 2017).
257. NCT02903069. Study of Marizomib With Temozolomide and Radiotherapy in Patients With Newly Diagnosed Brain Cancer. https://clinicaltrials.gov/ct2/show/NCT02903069 (30. 5. 2017).
Labels
Pharmacy Clinical pharmacologyArticle was published in
Czech and Slovak Pharmacy

2017 Issue 5
Most read in this issue
- Byliny podporujúce tvorbu materského mlieka
- Idiopatická trombocytopénia refraktérna na terapiu cyklosporínom A v klinickej praxi
- Léčivé látky z mořských organismů v klinických studiích a praxi
- Vývoj složení kombinovaného intramamárního přípravku na bázi citrátu stříbra pro veterinární použití