
Liečivá rastlinného pôvodu a ich využitie v terapii onkologických ochorení
Plant-derived drugs and their applicaction in the anticancer therapy
Nowadays, oncological diseases are one of the most common causes of untimely death. Current therapy based on synthetic chemotherapeuties has a number of side-effects, and a resistance to this type of treatment is very common. For this reason, new substances without these effects are constantly sought. In this regard, natural products appear to be a promising source of new active compounds. This review aims to introduce cytostatics inspired by natural substances that are in clinical trials or are already in common use.
Keywords:
Cytostatics – clinical trials – tumour – natural compounds
:
Romana Blažejová; Jan Hošek
:
Čes. slov. Farm., 2019; 68, 3-11
:
Review Articles
Onkologické ochorenia predstavujú v dnešnej dobe jednu z najčastejších príčin predčasného úmrtia. Súčasná terapia založená na syntetických chemoterapeutikách má radu vedľajšich nežiadúcich účinkov a veľmi často sa objavuje rezistencia na tento typ liečby. Z toho dôvodu sa neustále hľadajú nové látky, ktoré by tieto vlastnosti nemali. Prírodné produkty sa v tomto ohľade javia ako sľubný zdroj nových účinných zlúčenín. Toto review si kladie za cieľ predstaviť cytostatiká inšpirované prírodnými látkami, ktoré sú vo fáze klinických skúšok alebo sa už bežne používajú.
Klíčová slova:
cytostatiká – klinické štúdie – nádor – prírodné látky
Prírodné produkty ako zdroje nových liečiv
Prírodné produkty a ich deriváty sú oddávna považované za významný zdroj nových liečiv. Väčšina biologicky aktívnych prírodných produktov sú sekundárne metabolity s veľmi komplexnou štruktúrou. Predstavujú nielen finálne liečivá, ale predovšetkým poskytujú východiskové molekuly (angl. lead structures) s rozmanitými štruktúrami a často s početnými stereogénnymi centrami umožňujúcimi rôzne syntetické obmeny. Špecifické štrukturálne rysy prírodných produktov, ktoré z nich robia zaujímavé východiskové zlúčeniny a zároveň ich odlišujú od produktov syntetickej a kombinatoriálnej chémie, sú napr. početné chirálne centrá, prítomnosť aromatických kruhov a cyklických systémov, rôzny stupeň nenasýtenosti molekúl či počet a pomer heteroatómov1). V porovnaní so syntetickými zlúčeninami, ktoré často nesú funkčné skupiny obsahujúce dusík, síru a halogény, u prírodných produktov je typicky vyšší počet atómov kyslíka2).
Výskum a vývoj nových potenciálnych liečiv z prírodných produktov dosiahol vrchol v západnom farmaceutickom priemysle v sedemdesiatych a osemdesiatych rokoch 20. storočia. V nasledujúcich dvoch dekádach veľké farmaceutické spoločnosti ukončili alebo významne obmedzili svoju činnosť v oblasti liečiv prírodného pôvodu. Koehn a Carter2) popisujú obdobie zvyšujúcej sa patentovej aktivity v priebehu osemdesiatych rokov, nasledované obdobím stagnácie až mierneho poklesu v období od 1990 do 1999 a opätovné oživenie v rokoch 2000 – 2003.
Pokles záujmu farmaceutického priemyslu o výskum v oblasti prírodných produktov môžeme pripísať viacerým faktorom. Medzi ne patrí rozvoj kombinatoriálnej chémie, umožňujúci vznik veľkých súborov rôznych chemických zlúčenín, nazývaných chemické knižnice, ktoré sa využívajú pre farmakologický skríning. Ďalším faktorom je rozvoj vysokokapacitných skríningových techník (angl. high-throughput screening), umožňujúcich rýchlu identifikáciu aktívnych zlúčenín (angl. hit) proti definovaným molekulárnym cieľom, čo podnietilo mnoho firiem, aby sa presunulo z rozsiahlych knižníc prírodných produktov do tzv. syntetických chemických knižníc. Knižnice prírodných produktov totiž môžu obsahovať vzorky surových extraktov (teda zmesí desiatok až stoviek zlúčenín), čiastočne purifikovaných zmesí (5 až 10 zlúčenín) alebo purifikovaných prírodných produktov. Heterogenita vzoriek, najmä v prvom prípade, tak sťažuje proces detekcie aktívnych molekúl a pridáva na komplexnosti skríningového procesu. Po tretie je to pokrok v oblasti molekulárnej biológie, bunkovej biológie a genomiky, ktorý viedol k objaveniu množstva nových molekulárnych cieľov a skrátil dobu potrebnú na vývoj nového liečiva. V neposlednom rade je to pokles záujmu veľkých farmaceutických spoločností o vývoj účinných molekúl proti infekčným ochoreniam, čo je tradičná oblasť silne závislá na prírodných produktoch. Príčiny spomínaného trendu sú teda rovnako komerčné ako aj vedecké, najmä v prípade vývoja liečiv v oblasti infekčných ochorení. V posledných rokoch však prírodné produkty vyvolávajú opätovný záujem v dôsledku zlyhania alternatívnych metód pri objavovaní a dizajne liečiv ako zdroje východiskových štruktúr v kľúčových terapeutických oblastiach, vrátane ochorení imunitného systému, metabolických či onkologických ochorení2, 3).
V roku 2016 publikovali Newman a Cragg4) rozsiahlu aktualizovanú a rozšírenú verziu predchádzajúcich štyroch prehľadových článkov z rokov 1997, 2003, 2007 a 2012, týkajúcich sa využitia prírodných produktov ako zdrojov nových liečiv. V prípade protinádorových liečiv bol časový rámec rozšírený až na 65 rokov, od roku 1950 do decembra roku 2014. Za toto obdobie bolo schválených 175 nových protirakovinových liečiv typu malých molekúl, z ktorých 85, teda 49 % boli prírodné produkty alebo zlúčeniny z nich odvodené. V ďalších oblastiach bol význam prírodných zdrojov takisto značný, predovšetkým, ako bolo očakávané na základe predchádzajúcich štúdií, v oblasti antiinfektív, ktoré sú z veľkej časti závislé na prírodných produktoch a odvodených štruktúrach4).
Protinádorová liečivá rastlinného pôvodu
Liečivé rastliny boli k liečbe rôznych ochorení používané už od dávnych čias5). Vo svojom prehľadovom článku Hartwell5) vymenoval viac ako 3000 rastlinných druhov, ktoré boli údajne používané k terapií rakoviny, avšak v mnohých prípadoch sa jednalo o „rakovinu“ bližšie nešpecifikovanú, resp. o najrôznejšie patologické prejavy, vrátane zdurenín, abscesov, polypov, kalusov, bradavíc či tumorov. Zvyčajne boli teda popísané rôzne viditeľné kožné prejavy, ktoré každopádne môžu niekedy odpovedať kanceróznemu či prekanceróznemu stavu. V každom prípade na tvrdenia o protirakovinovom pôsobení niektorých látok sa treba pozerať s určitou mierou skepticizmu, pretože rakovina ako veľmi špecifické ochorenie bola v ľudovej a tradičnej medicíne len veľmi slabo definovaná. Na rozdiel od liečiv rastlinného pôvodu používaných napríklad k liečbe malárie či bolesti, čo sú stavy ľahšie definovateľné a v oblastiach, kde je tradičná medicína široko využívaná, aj pomerne časté6).
Napriek týmto pozorovaniam je význam liečivých rastlín vo vývoji protinádorových liečiv nespochybniteľný a hoci sa v posledných rokoch zvýšil záujem farmaceutických spoločností a vedeckých a výskumných organizácií o molekulové modelovanie, kombinatoriálnu chémiu a iné techniky syntetickej chémie, prírodné produkty a predovšetkým liečivé rastliny zostávajú naďalej významným zdrojom nových liečiv a predlohových molekúl vhodných k ďalšej optimalizácii1).
Výskum liečiv z rastlín je dnes založený hlavne na izolácii a štúdiu biologickej aktivity látok. Ďalším krokom je ich charakterizácia spojená s modifikáciami molekuly za účelom zvýšenia aktivity, potlačenia toxických vplyvov a zlepšenia farmakologického profilu a prípadná syntéza analógov7). Schopnosť potlačiť vývoj a progresiu nádorov u pacientov s rakovinou už bola pozorovaná u mnohých rastlinných druhov a identifikovaných bolo množstvo zlúčenín ako aktívnych nositeľov účinku týchto rastlinných drog. Konkrétny mechanizmus protirakovinového účinku na molekulárnej úrovni je však stále u väčšiny rastlinných produktov a odvodených látok predmetom intenzívneho výskumu. Tieto zlúčeniny vykazujú širokú a komplexnú škálu aktivít na úrovni jadrových a cytosolických faktorov nádorových buniek. Môžu selektívne eliminovať rýchlo sa deliace bunky, cielene účinkovať na abnormálne exprimované rastové faktory, pôsobiť proti oxidačnému stresu, zabraňovať novotvorbe ciev v kanceróznom tkanive či indukovať apoptózu. Môžu pôsobiť na úrovni bunkových signálnych dráh zapojených do procesov rastu a migrácie rakovinových buniek a tvorby metastáz. Ďalšou možnosťou je zabránenie malígnej transformácie preneoplastickej bunky či blokovanie metabolickej konverzie pro-karcinogénu8).
V nasledujúcej časti práce sme sa zamerali na protinádorové látky odvodené z rastlín, ktoré našli uplatnenie, alebo majú potenciál budúceho využitia v klinickej praxi.
Protinádorové liečivá rastlinného pôvodu v klinickej praxi
Prírodné produkty sú nevyčerpateľným zdrojom predlohových látok, charakteristické svojou mimoriadnou štrukturálnou diverzitou a širokou škálou farmakologických aktivít. Objavenie protirakovinových účinkov rastlinných alkaloidov vinblastínu a vinkristínu z Cataranthus roseus (L.) G. Don (Apocynaceae) a podofylotoxínu z Podophyllum peltatum L. (Berberidaceae) v päťdesiatych rokoch 20. storočia odštartovalo rozsiahly výskum potenciálnych protinádorových liečiv rastlinného pôvodu8).
Americký Národný inštitút pre výskum rakoviny (NCI) následne v roku 1960 inicioval extenzívny program zameraný na zber rastlín so zameraním na mierne podnebné pásmo. To viedlo k objaveniu množstva nových chemotypov vykazujúcich významnú škálu cytotoxických účinkov, vrátane taxánov a kamptotecínov, ale k ich zavedeniu do klinickej praxe došlo až o ďalších približne 30 rokov, v rokoch deväťdesiatych. Počas prvej fázy programu NCI, ktorá bola ukončená v roku 1982, bolo získaných viac ako 114 000 rastlinných extraktov, ktorých protinádorová aktivita bola testovaná na rôznych nádorových líniách in vitro aj na myších modeloch rakoviny in vivo. Vďaka rozvoju skríningových technológii došlo k obnoveniu programu v roku 1986 – teraz s dôrazom na rastliny, ale aj iné organizmy žijúce v subtropických a tropických regiónoch6). V období od roku 1986 do roku 2004 sa NCI podarilo získať kolekciu približne 60 000 vzoriek získaných z vyšších rastlín z rôznych regiónov po celom svete. V súčasnosti prebieha druhá, evaluačná fáza programu, kedy je rastlinný materiál podrobne charakterizovaný a starostlivo študovaný z hľadiska biologickej aktivity9).
Vinca-alkaloidy
Prvé látky, ktoré si našli miesto v klinickej praxi boli takzvané vinca-alkaloidy, vinblastín a vinkristín (obr. 1), izolované z madagaskarskej zimozelene rastliny Cataranthus roseus, ktorá bola tradične používaná rôznymi kultúrami k liečbe diabetes mellitus. Počas skúmania tejto rastliny ako zdroja potenciálnych perorálne účinných hypoglykemicky pôsobiacich látok bolo zistené, že extrakty znižovali počet bielych krviniek a spôsobovali útlm kostnej drene u potkanov. Následne sa ukázali byť účinné proti lymfocytárnej leukémii u myší. To viedlo k izolácii vinblastínu a vinkristínu ako zodpovedných aktívnych substancií6). Vinca-alkaloidy a odvodené deriváty patria medzi mitotické inhibítory schopné naviazať sa na tubulínové diméry, čím zabraňujú polymerizácii mikrotubulov a zastavujú bunku v metafáze mitózy.

Tieto látky sú primárne používané v kombinácii s inými protirakovinovými chemoterapeutikami k liečbe rôznych typov rakoviny, vrátane leukémii, lymfómov, pokročilej rakoviny semenníkov, rakoviny prsníka a pľúc či Kaposiho sarkómu7). Z novších semisyntetických analógov našli uplatnenie v klinickej praxi: vindesín k liečbe akútnej lymfoblastickej leukémie u detí, rezistentnej k iným chemoterapeutikám10), vinorelbín v kombinovanej chemoterapii karcinómu prsníka, pľúc a ovárií11) a vinflunín k liečbe karcinómu močového mechúra12).
Deriváty epipodofylotoxínu
Rôzne druhy rodu Podophyllum (Podophyllaceae), P. peltatum L. (bežne známy ako noholist americký), P. emodii Wallich z Indického subkontinentu, majú dlhú históriu použitia v medicíne, vrátane liečby rakoviny kože či kožných bradavíc. Hlavná aktívna zložka – podofylotoxín (PTOX) (obr. 2) – bol prvýkrát
izolovaný v roku 1880, ale jeho štruktúra bola objasnená až okolo roku 1950. Izolované boli aj ďalšie veľmi podobné látky typu lignanov a mnohé z nich boli podrobené klinickému testovaniu, avšak bezúspešne, či už z dôvodu nízkej účinnosti alebo neprijateľnej toxicity6). Hlavným problémom u podofylotoxínov je výrazná gastrointestinálna toxicita7). Samotný podofylotoxín sa z dôvodu vysokej toxicity v súčasnosti používa iba ako lokálne antivirotikum13). Tri klinicky účinné látky – etopozid (VM 26) (obr. 2), etopofos (etopozid fosfát) a tenipozid (VP 16-213) – sú semisyntetické deriváty prírodného epipodofylotoxínu (izomér podofylotoxínu), glykozidy so zníženou toxicitou, ktoré sa v súčasnosti používajú predovšetkým k liečbe lymfómov a rakoviny semenníkov a bronchov7). Úspech dosiahnutý s týmito liečivami, no zároveň aj vedľajšie účinky spojené s ich používaním stimulovali vývoj nových látok s PTOX-skeletom s lepším farmakologickým profilom a širším terapeutickým uplatnením. Dodnes bolo nasyntetizovaných množstvo derivátov, z ktorých sa až do štádia klinických hodnotení dostali napríklad TOP-53, GL-331 (obr. 2), NK-611, NPF, azatoxín či taflupozid14). Mnohé z týchto látok preukázali významnejšiu cytotoxickú aktivitu než etopozid, avšak doteraz žiadna neuspela v klinických skúškach15).

Taxány
O niečo neskôr sa do arzenálu protirakovinových chemoterapeutík odvodených z rastlín zaradili taxány. Paclitaxel (Taxol®) (obr. 3) pôvodne izolovaný z kôry Taxus brevifolia Nutt. – tisu tichomorského (Taxaceae) v rámci programu zberu rastlín pre NCI pod záštitou amerického ministerstva poľnohospodárstva (U.S. Department of Agriculture, USDA). Mnohé domorodé americké kmene používali rôzne časti Taxus brevifolia a iných druhov rodu Taxus (napr. T. canadensis Marshall, T. baccata L.) k liečbe rôznych nekanceróznych stavov, zatiaľ čo listy T. baccata sú tradične používané v indickom medicínskom systéme Ayurvéda, pričom bolo hlásené aj jedno použitie k liečbe určitého typu „rakoviny“ 5). Paclitaxel (obr. 3) spolu s ďalšími prekurzormi (baccatínmi) sa vyskytuje v listoch viacerých druhov rodu Taxus a možnosť semisyntetickej konverzie relatívne hojných baccatínov na paclitaxel, rovnako ako aktívne analógy paclitaxelu, napr. docetaxel (Taxotere®), poskytuje významný obnoviteľný prírodný zdroj tejto dôležitej triedy liečiv6). Taxány stabilizujú mikrotubuly a inhibujú ich depolymerizáciu späť na tubulín. Tým vznikajú nefunkčné proteínové štruktúry, čo vedie k zastaveniu bunkového cyklu v G2/M fáze7). Paclitaxel sa používa k terapii rakoviny prsníka, vaječníkov, nemalobunkového karcinómu pľúc (NSCLC) a zdá sa byť účinný aj proti Kaposiho sarkómu, zatiaľ čo docetaxel je primárne používaný k liečbe rakoviny prsníka a NSCLC6). Pozitívne sa prejavilo i použitie docetaxelu pri liečbe ďalších typov nádorov, napr. pokročilého štádia rakoviny žalúdka, rakoviny hrubého čreva, alebo rakoviny močových ciest7). Paclitaxel ďalej priťahuje pozornosť aj pre svoje potenciálne využitie k liečbe roztrúsenej sklerózy, psoriázy a reumatoidnej artritídy6).

Cabazitaxel (Jevtana®) (obr. 3) je semisyntetický taxanový derivát vyvinutý Sanofi-Aventis a schválený americkou FDA k liečbe hormónalne-refraktérneho karcinómu prostaty v júni 2010. Vo fáze II klinických skúšok sa momentálne nachádza ortataxel, ktorý preukázal aktivitu aj u nádorov rezistentných k paclitaxelu a docetaxelu. Ďalšie taxány sú v súčasnosti v štádiu preklinického vývoja ako potenciálne protirakovinové liečivá16).
Kamptotecíny
Ďalšou dôležitou triedou v zozname protinádorových liečiv odvodených z rastlín, ktoré si našli miesto v klinickej praxi, sú deriváty kamptotecínu, pôvodne izolovaného z kôry čínskeho okrasného stromu Camptotheca acuminata Decne (Nyssaceae). Jeho významným zdrojom je však aj indický strom Nothapodytes foetida (Wight) Sleumer (Icacinaceae). Kamptotecín patrí do skupiny chinolínových alkaloidov a ovplyvňuje aktivitu topoizomerázy-I (Topo-I), ktorá štiepi, odtáča a znovu liguje DNA. Po naviazaní kamptotecínu na Topo-I dôjde k štiepeniu, avšak nie k ligácii DNA, čo spôsobuje vznik jednoduchých zlomov DNA vlákna7). Do klinických skúšok vedených americkým NCI sa kamptotecín (vo forme sodnej soli) dostal v sedemdesiatych rokoch 20. storočia ako inhibítor DNA topoizomerázy I, avšak čoskoro bol vyradený v dôsledku závažnej urotoxicity. Intenzívny výskum však nakoniec viedol k vývoju efektívnejších derivátov, topotekanu (Hycamtin®) a irinotekanu (CPT-11, Camptosar®) (obr. 4)6).

Topotekan sa používa k liečbe ovariálneho a malobunkového pľúcneho karcinómu6), ďalej tiež v kombinácií s cisplatinou k liečbe recidivujúceho karcinómu krčka maternice17), irinotekan je používaný k liečbe kolorektálneho karcinómu, kde je považovaný za jedno z najdôležitejších liečiv6). Medzi klinicky používané deriváty kamptotecínu patrí ďalej belotekan, ktorý bol schválený k liečbe malobunkového pľúcneho karcinómu a ovariálneho karcinómu a nachádza sa v III. fázy klinických štúdií18). Americká spoločnosť BioNumerik vyvinula syntetický derivát kamptotecínu, Karenitecín (BNP1350), ktorý v roku 2007 vstúpil do tretej fázy klinického skúšania v liečbe pokročilého ovariálneho karcinómu19).
V štádiu klinického hodnotenia je niekoľko ďalších derivátov z rodiny kamptotecínov, napr. semisyntetické deriváty kamptotecínu, gimatekan a diflomotekan či homokamptotecínový analóg elomotekan v liečbe pokročilých solídnych nádorov20).
Nezaradená protinádorová liečivá rastlinného pôvodu v klinickej praxi
Medzi ďalšie klinicky používané látky odvodené z rastlín patrí homoharringtonín (HHT), cytotoxický alkaloid pôvodne izolovaný z čínskeho stromu Cephalotaxus harringtonia (Cephalotaxaceae)6). HHT je inhibítor proteosyntézy s jedinečným mechanizmom účinku, ktorý spočíva v zabránení procesu elongácie peptidového reťazca. Racemická zmes harringtonínu a homoharringtonínu bola po prvýkrát s úspechom použitá v Číne k liečbe akútnej myeoloidnej leukémie (AML) a chronickej myeloidnej leukémie (CML)21). Purifikovaný HHT vykazoval účinnosť proti rôznym typom leukémie, vrátane rezistentných k štandardnej liečbe, a viedol ku kompletnej hematologickej remisii (normalizácii krvného obrazu) u pacientov s neskorými chronickými štádiami CML6). Klinický vývoj HHT načas pozastavil objav inhibítoru tyrozínkinázy (TKI), imatinib mesylátu (Gleevec®). U veľkého množstva pacientov s CML sa však vyvinie rezistencia k TKI, často prostredníctvom mutácií v kinázovej doméne BCR-ABL122). K obnoveniu záujmu o HHT v terapií CML tak viedli pozitívne výsledky u pacientov, u ktorých liečba imatinibom zlyhala. Omacetaxín mepesukcinát (Synribo®), semisyntetická forma HHT s výbornou biodostupnosťou pri subkutánnom podaní bol v októbri roku 2010 schválený FDA a získal status orphan drug („liečivo-sirota”) k liečbe pacientov s chronickou myeloidnou leukémiou s rezistenciou k TKI21).
Ako ďalší príklad môže slúžiť ingenol mebutát (PEP005, Picato®), derivát diterpénu ingenolu, extrahovaného zo živice Euphorbia peplus L. (Euphorbiaceae). V roku 2012 bol ingenol mebutát (obr. 5) schválený úradmi FDA aj EMA vo forme gélu k topickej liečbe aktinickej keratózy, považovanej za včasný in situ skvamocelulárny karcinóm23). Ingenol mebutát predstavuje sľubnú terapeutickú možnosť aj pre superficiálny bazocelulárny karcinóm. Štúdie poukazujú na dvojitý mechanizmus účinku ingenol mebutátu, a to indukciou epidermálnej nekrózy a výraznej zápalovej reakcie, charakterizovanej infiltráciou imunokompetentných buniek24).
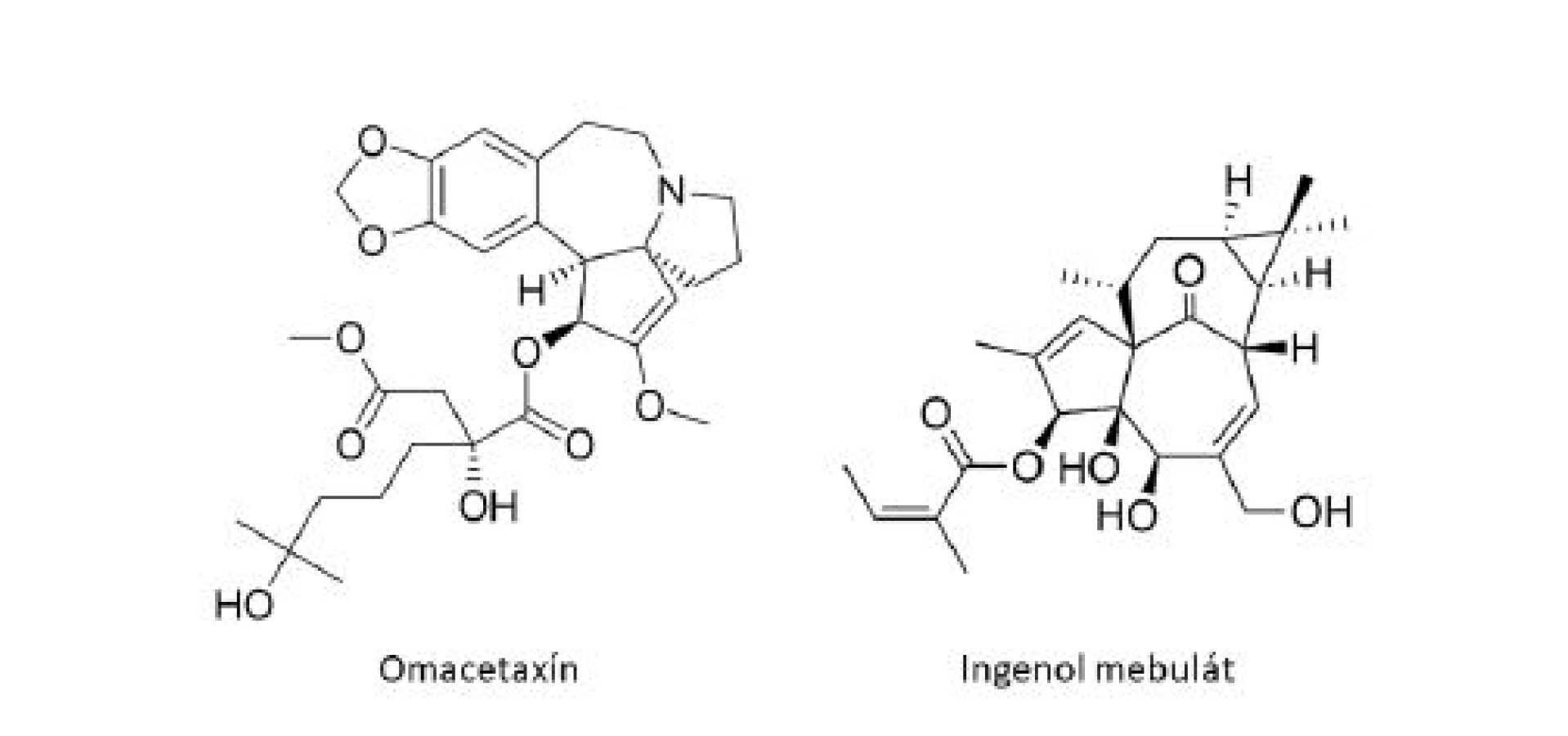
Príklady protinádorových liečiv odvodených z rastlín v štádiu klinického vývoja
Combretastatíny sa podarilo izolovať z juhoafrickej vŕby Combretum caffrum (Eckl. & Zeyh.) Kuntze (Combretaceae), v sedemdesiatych rokoch 20. storočia vďaka spolupráci USDA s juhoafrickým výskumným ústavom Botanical Research Institute of South Africa. Rastliny rodov Combretum a Terminalia, ktoré oba patria do čeľade Combretaceae, sú používané v africkej i indickej tradičnej medicíne k liečeniu rôznych ochorení, napr. hepatitídy či malárie6). Combretastatíny predstavujú rodinu stilbénových derivátov s esenciálnym trimetoxyarylovým postranným reťazcom, ktoré svojimi antiangiogénnymi účinkami zabraňujú vaskularizácii nádoru, a tým spôsobujú jeho nekrózu. Hlavným predstaviteľom tejto skupiny vaskulárne namierených liečiv je vo vode rozpustný analóg, combretastatín-A4-fosfát (CA4P alebo fosbretabulín), ktorý sa nachádza vo fáze klinických štúdií25). Pozitívne výsledky boli reportované pri použití CA4P napríklad v liečbe nemalobunkového pľúcneho karcinómu26) či pokročilého ovariálneho karcinómu27). Combretastatín-A1-difosfát (OXi4503 alebo CA1P) je protivaskulárne liečivo s duálnym mechanizmom účinku, ktoré je v súčasnosti vo fáze I/II klinického skúšania v liečbe akútnej myeloidnej leukémie (AML) a myelodysplastického syndrómu28).
Ďalším klinicky študovaným rastlinným derivátom so sľubným protinádorovým potenciálom je β-lapachón, 1,2-naftochinón izolovaný z kôry stromu Tabebuia avellanedae Lorentz ex Griseb. (Bignoniaceae). Cytotoxický účinok β-lapachónu súvisí s NAD(P)H-chinónoxidoreduktázou-1, flavoproteínom nadmerne exprimovaným (až dvadsaťnásobne) v bunkách rakoviny vaječníkov, hrubého čreva, pľúc, rakoviny prostaty či prsníkov. Prostredníctvom NQO1 dochádza ku konverzií β-lapachónu medzi hydrochinónom, semichinónom a chinónovou formou, následkom ktorej je masívna tvorba reaktívnych foriem kyslíka poškodzujúcich DNA29). To vedie k hyperaktivácií PARP-1, deplécii NAD+/ATP a nakoniec k smrti bunky30). V súčasnosti je β-lapachón (ARQ761) predmetom klinického skúšania v liečbe pokročilých solídnych nádorov29).
Berberín, prírodný izochinolínový alkaloid izolovaný z koreňov a kôry medicínsky dôležitých rastlín, ako sú Berberis vulgaris L., B. aquifolium Pursh, B. aristata DC. (Berberidaceae) či Tinospora cordifolia (Willd.) Miers (Menispermaceae), bol od nepamäti používaný ako potenciálne liečivo v čínskej medicíne7). Nedávne štúdie ukázali, že berberín vykazuje protinádorovú aktivitu nielen in vitro, ale aj v in vivo experimentoch u rôznych typov rakoviny a prostredníctvom viacerých mechanizmov. Berberín inhibuje proliferáciu a reprodukciu určitých tumorigénnych mikroorganizmov a vírusov ako Helicobacter pylori či vírus hepatitídy typu B. Ďalej reguluje transkripciu niektorých onkogénov ako aj expresiu iných génov spojených s kancerogenézou, popísaná bola interakcia s DNA aj RNA. Okrem toho je berberín enzýmový inhibítor so širokým spektrom pôsobenia, vrátane N-acetyltransferázy, COX-2, topoizomeráz a iných. Tieto mechanizmy spoločne s reguláciou tvorby ROS, reguláciou transmembránového potenciálu mitochondrií a nukleárneho faktora kappa B (NF-κB) podmieňujú jeho antiproliferatívne a proapoptické účinky31). V doterajších štúdiách bola preukázaná schopnosť berberínu potlačiť rast nádoru aj tvorbu metastáz32, 33). Navyše sa zdá byť výhodné aj jeho použitie v kombinácií so štandardnou liečbou34) či rádioterapiou pre zvýšenie senzitivity nádorových buniek35). Výborné výsledky dlhoročného výskumu vlastností berberínu in vitro
aj in vivo jasne ukazujú jeho potenciál v chemoterapií nádorov.
Flavopiridol (FP, známy aj pod názvom alvocidib) je semisyntetická látka, ktorej flavonoidná štruktúra je odvodená od chromonového alkaloidu rohitukínu7). Rohitukín bol izolovaný ako účinná zložka zodpovedná za protizápalovú a imunomodulačnú aktivitu Dysoxylum binectariferum Hook. f. (Meliaceae), ktorý je fylogeneticky príbuzný rastline známej a používanej v Ayurvéde k liečbe reumatoidnej artritídy – D. malabaricum Bedd.6). Flavopiridol je inhibítor cyklín-dependentných kináz (CDK) so silnou schopnosťou blokovať bunkový cyklus v G1-fáze a indukovať apoptózu nádorových buniek7). Okrem svojej schopnosti eliminovať proliferujúce bunky, FP je účinný aj proti dormantným rakovinovým bunkám36). Flavopiridol sa stal prvým chemickým inhibítorom CDK zaradeným do klinických štúdii, i keď dnes už nie je jediným37). Klinické skúšanie v súčasnej dobe naďalej pokračuje a nakoľko sa in vitro prejavil synergický účinok s niektorými konvenčnými cytotoxickými liečivami, zahájené bolo aj klinické hodnotenie kombinácií flavopiridolu s docetaxelom, cisplatinou či s cytarabínom a mitoxantrónom (FLAM)38). V roku 2014 americká FDA (Správa potravín a liečiv) udelila lieku Alvocidib proti akútnej myeloidnej leukémii spoločnosti Tolero Pharmaceuticals status orphan drug.
Cyklín-dependentné kinázy predstavujú rodinu proteínových kináz, ktoré majú hlavné slovo v riadení bunkového cyklu. Vďaka ich častým poruchám u nádorových buniek sa stali sľubným cieľom protirakovinových liečiv. Prakticky zároveň s objavom prirodzených proteínových inhibítorov regulujúcich aktivitu cyklín-dependentných kináz (CKI) v bunkách sa začalo hľadanie inhibítorov chemických. Na počiatku tejto éry bol objavený purínový derivát olomoucín, rýchlo nasledovaný ďalšími inhibítormi. Obmenami jeho štruktúry boli získané účinnejšie zlúčeniny ako roskovitín (seliciclib), ktorý sa v súčasnej dobe nachádza vo fáze II klinického skúšania u pacientov s pokročilými solídnymi nádormi. Ďalší vývoj tejto série zlúčenín viedol k syntéze ešte potentnejších purvalanolov, ktoré sú zatiaľ v preklinickej fáze6).
Epidemiologické štúdie odhalili inverznú koreláciu medzi diétou bohatou na sóju a rizikom rakoviny u ázijskej populácie39). Preventívna úloha genisteínu a ďalších sójových izoflavónov bola demonštrovaná aj na viacerých animálnych modeloch rakoviny40, 41). Na základne pozitívnych výsledkov štúdií in vitro boli genisteín a daidzeín zaradené do klinických hodnotení pre chemoterapeutické a chemopreventívne účely u rôznych typov rakoviny42). Genisteín (4′,5,7-trihydroxyisoflavón) je antineoplastický fytoestrogén, ktorý bol prvýkrát izolovaný z Genista tinctoria L. (Leguminosae) v roku 1899 a neskôr objavený ako jeden z hlavných sekundárnych metabolitov rodu Trifolium a Glycine max L. (Leguminosae). Jeho chemická štruktúra je príbuzná estradiolu, z čoho vyplýva jeho schopnosť väzby na estrogénové receptory.
Genisteín je inhibítor tyrozín kináz a topoizomerázy-II, ovplyvňuje bunkový cyklus a apoptózu, inhibuje angiogenézu a rast metastáz. V súčasnosti sa nachádza vo fáze II/III klinických hodnotení k liečbe rôznych nádorov43).
Phenoxodiol (syntetický derivát daidzeínu) selektívne inhibuje sfingozín-1-fosfát (S-1-P), čo má za následok zvýšenie senzitivity rakovinových buniek k chemoterapií44). S-1-P je bioaktívny sfingolipid, ktorý sa významne podieľa na progresií rakoviny, je zapojený v procesoch bunkovej proliferácie a transformácie, neovaskularizácie, apoptózy, bunkovej migrácie a vzniku metastáz45). V literatúre sa však diskutuje aj o ďalších mechanizmoch protirakovinového účinku phenoxodiolu, vrátane blokovania bunkového cyklu v G1-fáze, indukcie bunkovej smrti prostredníctvom inhibície antiapoptických proteínov46). Americká agentúra FDA udelila phenoxodiolu v januári 2009 „fast track“ – status umožňujúci jeho rýchlejší vývoj a uvedenie na trh47). Phenoxodiol bol testovaný vo fáze III k zvýšeniu chemosenzitivity u pacientiek s ovariálnym karcinómom rezistentným k liečivám na báze platiny, kde sa však jeho účinnosť nepreukázala48). Testovaná je aj jeho účinnosť proti rakovine prostaty a krčka maternice9).
Gossypol, prírodný fenol zo semien bavlníku (rod Gossypium, Malvaceae), vykazuje významné protirakovinové účinky (najmä jeho (–)-enantiomér) a úspešne prešiel druhou fázou klinického skúšania k liečbe rakoviny prostaty49). Sľubné výsledky preukázal aj pri použití v liečbe nádorov pľúc či mozgu44). Výsledky in vitro štúdií poukázali na možný mechanizmus protinádorového účinku prostredníctvom regulácie expresie proteínov z rodiny Bcl-2 a aktivácie kaspáz50).
Kurkumín je prírodný fenolický antioxidant izolovaný z podzemných častí stonky kurkumy dlhej (Curcuma longa L., Zingiberaceae). V preklinických in vitro a in vivo štúdiach bola preukázaná celá škála biologických účinkov kurkumínu, vrátane antioxidačného, protizápalového a protirakovinového účinku proti viacerým typom nádorov, napr. nádoru prostaty, kolorekta, pečene, prsníka i ovárií prostredníctvom modulácie rôznych signálnych dráh9, 51). Na základe týchto výsledkov bola testovaná protirakovinová aktivita kurkumínu v klinických štúdiách. Hlavnou prekážkou jeho klinickej aplikácie je nízka biologická dostupnosť, ktorú sa vedci snažia prekonať napríklad formuláciou liekových foriem na báze nanočastíc či lipozómov. V súčasnosti sa kurkumín nachádza vo fáze I/II klinického testovania k liečbe viacerých typov nádorov samostatne i v kombinácií s inými chemoterapeutikami51).
Mnoho derivátov odvodených od prírodných produktov bolo podrobených klinickému skúšaniu, ktoré však bolo ukončené z dôvodu nedostatočnej účinnosti či neprijateľnej toxicity. Objavujú sa však nové technológie, vďaka ktorým sa dokonca i zlúčeniny vylúčené v predchádzajúcom období z klinických štúdií pre vysokú toxicitu dostávajú opäť do pozornosti vedcov. Možnosť technologicky pripojiť rôzne molekuly k vhodným „nosičom“ ich umožňuje priviesť špecificky priamo k nádorom. Príkladom môžu byť monoklonálne protilátky špecificky namierené na epitopy skúmaného nádoru. Takéto efektívne značenie vysoko cytotoxických prírodných látok je výhodné hlavne z hľadiska neškodnosti pre zdravé bunky organizmu, čím sa rieši problém toxicity a problém ich nízkej rozpustnosti vo vodných roztokoch7).
Prípad maytansínu ilustruje, ako nové technológie môžu oživiť záujem o tieto „staré“ látky. Maytansín bol vyradený z klinického hodnotenia ešte v osemdesiatych rokoch 20. storočia. Jeho derivát DM1 konjugovaný s monoklonálnou protilátkou namierenou proti bunkám malobunkového karcinómu pľúc (SCLC) (huN901-DM1) je v súčasnosti v štádiu vývoja k liečbe SCLC. Ďalší konjugát DM1 s J591, monoklonálnou protilátkou namierenou proti špecifickému membránovému antigénu buniek karcinómu prostaty (PSMA) dokonca dosiahol fázy I/II klinického hodnotenia proti rakovine prostaty6). Za zmienku tiež stojí, že vývoj vysoko účinných liečiv ako paclitaxel (Taxol®) či derivaty kamptotecínu, topotekan a irinotekan vyžadoval 20 – 30 rokov intenzívneho výskumu, trpezlivosti a značný objem zdrojov, kým bola preukázaná ich klinická účinnosť. Podobným príkladom látky, ktorá bola „znovuobjavená“ vďaka technologickému pokroku, je bruceantín, ktorý bol pôvodne izolovaný zo stromu Brucea antidysenterica J.F. Mill. (Simaroubaceae), ktorý je ako liečivá rastlina tradične používaný v Etiópii. Ako sa stáva pomerne často, aktivita bruceantínu bola pozorovaná u animálnych modelov rôznych typov tumorov, ale v klinickom hodnotení nebola preukázaná objektívna účinnosť, a vývoj tak bol pozastavený. Nedávne pozorovania však poukázali na významnú aktivitu proti panelu bunkových línii ľudskej leukémie, lymfómu a myelómu – rovnako ako jeho protirakovinové účinky u animálnych modelov s ranými i neskoršími štádiami rovnakých typov rakoviny. Táto aktivita bola asociovaná so zníženou expresiou kľúčového onkoproteínu c-MYC. Tieto dáta sú silnou evidenciou podporujúcou vývoj bruceantínu ako potenciálneho liečiva hematologických malignít6).
Záver
Rozvoj moderných izolačných a analytických techník umožňuje získať veľmi čisté prírodné produkty a vyhodnotiť ich biologickú aktivitu. To spolu so skutočnosťou, že pacienti sa stále častejšie obracajú k alternatívnej prírodnej terapii nádorových ochorení z dôvodu zlyhania štandardnej liečby, prispieva k prudkému rozvoju vývoja nových cytostatík na báze prírodných látok. Trendom poslednej doby je potom chemoterapia kombinovaná s prírodným cytostatikom a presné cielenie na nádorové tkanivo. Rovnako sa rozvíja využívanie komplexných nano - a mikronosičov týchto látok s riadeným uvoľňovaním.
Stret záujmov: žiadny.
Rukopis vznikol na základe textu diplomovej práce Mgr. R. Blažejovej s názvom „Indukcia apoptózy pomocou komplexov prechodných kovov“, ktorú obhájila v roku 2017 na Farmaceutickej fakulte VFU Brno.
Poďakovanie
Práca bola podporovaná MŠMT ČR, projekt číslo LO1305.
Mgr. Romana Blažejová
Ústav molekulární biologie a farmaceutické biotechnologie
Farmaceutická fakulta, Veterinární a farmaceutická univerzita Brno
RNDr. Jan Hošek, Ph.D. (*)
Oddělení biologicky aktivních komplexů a molekulových magnetů
Regionální centrum pokročilých technologií a materiálů
Přírodovědecká fakulta, Univerzita Palackého
Šlechtitelů 27, 783 71 Olomouc
e-mail: jan.hosek@upol.cz
Sources
1. Balunas M. J., Kinghorn A. D. Drug discovery from medicinal plants. Life Sci. 2005; 78, 431–441.
2. Koehn F. E., Carter G. T. The evolving role of natural products in drug discovery. Nat. Rev. Drug Discov. 2005; 4, 206–220.
3. Lahlou M. The success of natural products in drug Discovery. Pharmacol. Pharm. 2013; 4, 17–31.
4. Newman D. J., Cragg G. M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016; 79, 629–661.
5. Hartwell J. L. Plants used against cancer: A survey (Bioactive Plants, Vol. 2). Michigan: Quarterman Publications Inc. 1984.
6. Cragg G. M., Newman D. J. Plants as a source of anti-cancer agents. J. Ethnopharmacol. 2005; 100, 72–79.
7. Cibikova P., Sturdikova M., Maruna M. Natural compounds of plant origin and their application in therapy of oncological diseases. Chem. Listy 2010; 104, 12–20.
8. Singh S., Sharma B., Kanwar S. S., Kumar A. Lead phytochemicals for anticancer drug development. Front. Plant. Sci. 2016; 7, 1667.
9. Pan L., Chai H. Y., Kinghorn A. D. The continuing search for antitumor agents from higher plants. Phytochem. Lett. 2010; 3, 1–8.
10. Drugs.com. ELDISINE POWDER FOR SOLUTION FOR INJECTION 5.0 MG. https://www.drugs.com/uk/eldisine-powder-for-solution-for-injection-5-0-mg-leaflet.html (15.01.2019).
11. Gregory R. K., Smith I. E. Vinorelbine – a clinical review. Brit. J. Cancer 2000; 82, 1907–1913.
12. Gerullis H., Wawroschek F., Kohne C. H., Ecke T. H. Vinflunine in the treatment of advanced urothelial cancer: clinical evidence and experience. Ther. Adv. Urol. 2017; 9, 28–35.
13. Hearn B. R., Shaw S. J., Myles D. C. Microtubule targeting agents. In: Taylor J. B., Triggle D. J. (eds.) Comprehensive Medicinal Chemistry II. Amsterdam: Elsevier Science 2007.
14. Yousefzadi M., Sharifi M., Behmanesh M., Moyano E., Bonfill M., Cusido R. M., Palazon J. Podophyllotoxin: Current approaches to its biotechnological production and future challenges. Eng. Life Sci. 2010; 10, 281–292.
15. Liu Y. Q., Tian J., Qian K., Zhao X. B., Morris-Natschke S. L., Yang L., Nan X., Tian X., Lee K.-H. Recent progress on C-4-modified podophyllotoxin analogs as potent antitumor agents. Med. Res. Rev. 2015; 35, 1–62.
16. Ojima I., Lichtenthal B., Lee S., Wang C. W., Wang X. Taxane anticancer agents: a patent perspective. Expert Opin. Ther. Pat. 2016; 26, 1–20.
17. Coronel J., Cetina L., Candelaria M., Gonzalez-Fierro A., Arias D., Cantu D., Dueñas-González A. Weekly topotecan as second - or third-line treatment in patients with recurrent or metastatic cervical cancer. Med. Oncol. 2009; 26, 210–214.
18. Chan B. A., Coward J. I. G. Chemotherapy advances in small-cell lung cancer. J. Thorac. Dis. 2013; 5, S565–S578.
19. BioNumerik Pharmaceuticals. Karenitecin versus topotecan in patients with advanced epithelial ovarian cancer. https://clinicaltrials.gov/ct2/show/NCT00477282 (15. 01. 2019).
20. Rajan R., Varghese S. C., Kurup R., Gopalakrishnan R., Venkataraman R., Satheeshkumar K., Baby S. HPTLC-based quantification of camptothecin in Ophiorrhiza species of the southern Western Ghats in India. Cogent. Chemistry 2016; 2, 1275408.
21. Lu S. Q., Wang J. M. Homoharringtonine and omacetaxine for myeloid hematological malignancies. J. Hematol. Oncol. 2014; 7, 2.
22. Nazha A., Kantarjian H., Cortes J., Quintas-Cardama A. Omacetaxine mepesuccinate (synribo) – newly launched in chronic myeloid leukemia. Expert Opin. Pharmacol. 2013; 14, 1977–1986.
23. Khazir J., Mir B. A., Pilcher L., Riley D. L. Role of plants in anticancer drug discovery. Phytochem. Lett. 2014; 7, 173–181.
24. Alchin D. R. Ingenol mebutate: a succinct review of a succinct therapy. Dermatol. Ther. (Heidelb.) 2014; 4, 157–164.
25. Case Comprehensive Cancer Center. Combretastatin A4 phosphate in treating patients with advanced anaplastic thyroid cancer. https://clinicaltrials.gov/ct2/show/NCT00060242?term=combretastatin-A4-phosphate&rank=5 (15. 01. 2019).
26. Garon E. B., Neidhart J. D., Gabrail N. Y., de Oliveira M. R., Balkissoon J., Kabbinavar F. A randomized Phase II trial of the tumor vascular disrupting agent CACA4P (fosbretabulin tromethamine) with carboplatin, paclitaxel, and bevacizumab in advanced nonsquamous non-small-cell lung cancer. Oncotargets Ther. 2016; 9, 7275–7283.
27. Chase D. M., Chaplin D. J., Monk B. J. The development and use of vascular targeted therapy in ovarian cancer. Gynecol. Oncol. 2017; 145, 393–406.
28. Madlambayan G. J., Meacham A. M., Hosaka K., Mir S., Jorgensen M., Scott E. W., Siemann W., Cogle C. R. Leukemia regression by vascular disruption and antiangiogenic therapy. Blood 2010; 116, 1539–1547.
29. Li J. Z., Ke Y. B., Misra H. P., Trush M. A., Li Y. R., Zhu H., Jia Z. Mechanistic studies of cancer cell mitochondria - and NQO1-mediated redox activation of beta-lapachone, a potentially novel anticancer agent. Toxicol. Appl. Pharmacol. 2014; 281, 285–293.
30. Bey E. A., Bentle M. S., Reinicke K. E., Dong Y., Yang C. R., Girard L., Minna J. D., Bornmann W. G., Gao J., Boothman D. A. An NQO1-and PARP-1-mediated cell death pathway induced in non-small-cell lung cancer cells by beta-lapachone. PNAS 2007; 104, 11832–11837.
31. Sun Y. Y., Xun K. L., Wang Y. T., Chen X. P. A systematic review of the anticancer properties of berberine, a natural product from Chinese herbs. Anti-Cancer Drug 2009; 20, 757–769.
32. Liu C. H., Tang W. C., Sia P., Huang C. C., Yang P. M., Wu M. H., Lai I. L., Lee K. H. berberine inhibits the metastatic ability of prostate cancer cells by suppressing epithelial-to-mesenchymal transition (EMT) – Associated Genes with Predictive and Prognostic Relevance. Int. J. Med. Sci. 2015; 12, 63–71.
33. Chu S. C., Yu C. C., Hsu L. S., Chen K. S., Su M. Y., Chen P. N. Berberine reverses epithelial-to-mesenchymal transition and inhibits metastasis and tumor –induced angiogenesis in human cervical cancer cells. Mol. Pharmacol. 2014; 86, 609–623.
34. Bao J. L., Huang B. R., Zou L. D., Chen S. H., Zhang C., Zhang Y. L., Chen M., Wan J.-B., Su H., Wang Y., He C. Hormetic effect of berberine attenuates the anticancer activity of chemotherapeutic agents. PLOS One 2015; 10, e0139298.
35. Liu Q., Jiang H. Y., Liu Z. J., Wang Y., Zhao M. N., Hao C. Y., Feng S., Guo H., Xu B., Yang Q., Gong Y., Shao C. Berberine radiosensitizes human esophageal cancer cells by downregulating homologous recombination repair protein RAD51. PLOS One. 2011; 6, e23427.
36. Najmi S., Korah R., Chandra R., Abdellatif M., Wieder R. Flavopiridol blocks integrin-mediated survival in dormant breast cancer cells. Clin. Cancer Res. 2005; 11, 2038–2046.
37. Kelland L. R. Flavopiridol, the first cyclin-dependent kinase inhibitor to enter the clinic: current status. Expert. Opin. Inv. Drug 2000; 9, 2903–2911.
38. Wang L. M., Ren D. M. Flavopiridol, the first cyclin-dependent kinase inhibitor: recent advances in combination chemotherapy. Mini-Rev. Med. Chem. 2010; 10, 1058–1070.
39. Hirose K., Imaeda N., Tokudome Y., Goto C., Wakai K., Matsuo K., Ito H., Toyama T., Iwata H., Tokudome S., Tajima K. Soybean products and reduction of breast cancer risk: a case control study in Japan. Brit. J. Cancer 2005; 93, 15–22.
40. Perabo F. G. E., Von Low E. C., Ellinger J., Von Rucker A., Muller S. C., Bastian P. J. Soy isoflavone genistein in prevention and treatment of prostate cancer. Prostate Cancer Prostatic Dis. 2008; 11, 6–12.
41. Sarkar F. H., Li Y. W. The role of isoflavones in cancer chemoprevention. Front. Biosci.-Landmrk. 2004; 9, 2714–2724.
42. Schnekenburger M., Diederich M. Nutritional epigenetic regulators in the field of cancer: new avenues for chemopreventive approaches. In: Gray S. G. (ed.) Epigenetic Cancer Therapy. London: Academic Press 2015.
43. Spagnuolo C., Russo G. L., Orhan I. E., Habtemariam S., Daglia M., Sureda A., Nabavi S. F., Devi K. P., Loizzo M. R., Tundis R., Nabavi S. M. Genistein and cancer: current status, challenges, and future directions. Adv. Nutr. 2015; 6, 408–419.
44. Mishra B. B., Tiwari V. K. Natural products: An evolving role in future drug discovery. Eur. J. Med. Chem. 2011; 46, 4769–807.
45. Pyne N. J., Tonelli F., Lim K. G., Long J. S., Edwards J., Pyne S. Sphingosine 1-phosphate signalling in cancer. Biochem. Soc. Trans. 2012; 40, 94–100.
46. Gamble J. R., Xia P., Hahn C. N., Drew J. J., Drogemuller C. J., Brown D., Vadas M. A. Phenoxodiol, an experimental anticancer drug, shows potent antiangiogenic properties in addition to its antitumour effects. Int. J. Cancer 2006; 118, 2412–2420.
47. Georgaki S., Skopeliti M., Tsiatas M., Nicolaou K. A., Ioannou K., Husband A., Bamias A., Dimopoulos M. A., Constantinou A. I., Tsitsilonis O. E. Phenoxodiol, an anticancer isoflavene, induces immunomodulatory effects in vitro and in vivo. J. Cell. Mol. Med. 2009; 13, 3929–3938.
48. Fotopoulou C., Vergote I., Mainwaring P., Bidzinski M., Vermorken J. B., Ghamande S. A., Harnett P., Del Prete S. A., Green J. A., Spaczynski M., Blagden S., Gore M., Ledermann J., Kaye S., Gabra H. Weekly AUC2 carboplatin in acquired platinum resistant ovarian cancer with or without oral phenoxodiol, a sensitizer of platinum cytotoxicity: the phase Ill OVATURE multicenter randomized study. Ann. Oncol. 2014; 25, 160–165.
49. Xiong J., Li J. S., Yang Q., Wang J., Su T. F., Zhou S. Gossypol has anti-cancer effects by dual targeting MDM2 and VEGF in human breast cancer. Breast Cancer Res. 2017; 19, 27.
50. Meng Y., Tang W. H., Dai Y., Wu X. Q., Liu M. L., Ji Q., Ji M., Pienta K., Lawrence T., Xu L. Natural BH3 mimetic (–)-gossypol chemosensitizes human prostate cancer via Bcl-xL inhibition accompanied by increase of Puma and Noxa. Mol. Cancer Ther. 2008; 7, 2192–2202.
51. Bimonte S., Barbieri A., Leongito M., Piccirillo M., Giudice A., Pivonello C., de Angelis C., Granata V., Palaia R., Izzo F. Curcumin anticancer studies in pancreatic cancer. Nutrients 2016; 8, E433
Labels
Pharmacy Clinical pharmacologyArticle was published in
Czech and Slovak Pharmacy

2019 Issue 1
Most read in this issue
- Plant-derived drugs and their applicaction in the anticancer therapy
- Microneedles as a perspective for transdermal therapeutic systems
- Film wound dressing containing dexpanthenol – preparation and evaluation
- Theory and practice of pharmacopoeial control of quality of drugs and excipients VIII. End-point indication and other conditions for the titration of primary aromatic amines in the European Pharmacopoeia (Ph. Eur.)