
Stomatologická problematika u dítěte s mukopolysacharidózou II. typu
Stomatological Problems in Child with the II Type Mucopolysaccharidosis
Introduction:
Mucopolysaccharidoses are congenital, hereditary disorders of metabolism, caused by deficiency of vital enzyme. As a result of this the products of metabolism accumulate and settle in tissues and body organs (liver, spleen, heart, brain) and cause malfunction. Children mostly thrive well initially, clinical manifestation of illness are obvious in future development. The II type mucopolysaccharidosis, well-known as Hunter syndrome, affects predominantly boys. In the clinical picture there is typical small figure, short neck, macrocephalia, prominent forehead, flat root of nose, hard-featured, fingers with flex contraction, large abdomen and mental retardation. Stomatological report include poor labial closure and macroglossia manifested in opening of the mouth, macrocheilia, wide dental arches and dentition with spaces. Suspicion for Hunter syndrome follow from clinical examination of the patient, the diagnosis is supported by analysis of urine and definitively confirmed by molecular genetical examination. The transplantation of haematopoietic cells or more frequently the substitution of missing enzyme are used in the therapy.
Case Report:
Stomatological findings and the clinical dental management of a boy with the II type mucopolysaccharidosis are described in the case report. We take care about this patient for nine years, he was recommended in our ward by the pediatrician at the age of five years and six month. The child co-operated very bad during the examination, he had typical broad face, prominent lips, convex profile and the sign of bialveolar protrusion. In the oral cavity there were wide dental arches, carious deciduous dentition with spaces and gingivitis due to dental plaque.
Conclusion:
In this case report we wanted to point out stomatological findings of a boy with the II type mucopolysaccharidosis, rare, hereditary, incurable illness. Not only pediatrician, surgeon, psychologist and other specialists participate on the treatment of this patients, but dentist either. Interdisciplinary co-operation is very important in these cases.
Key words:
mucopolysaccharidosis – Hunter syndrome – gargoylism – multisystemic disability – enzyme replacement therapy
Authors:
L. Baborská; V. Merglová
Authors‘ workplace:
Stomatologická klinika LF UK a FN, Plzeň
Published in:
Česká stomatologie / Praktické zubní lékařství, ročník 113, 2013, 5, s. 59-64
Category:
Case Report
Overview
Úvod:
Mukopolysacharidózy jsou vrozená dědičně podmíněná onemocnění látkové výměny, která jsou způsobena chyběním konkrétního životně důležitého enzymu. Následkem toho se hromadí produkty metabolismu, jež se usazují ve tkáních četných orgánů (játra, slezina, srdce, mozek) s následnou poruchou jejich funkce. Zpočátku děti většinou dobře prospívají, klinické projevy onemocnění jsou patrné až v pozdějším vývoji.
Mukopolysacharidóza II. typu, také známá jako Hunterův syndrom, postihuje převážně chlapce. Klinickému obrazu dominuje malá postava, krátký krk, makrocefalie, prominující čelo, plochý kořen nosu, hrubé rysy obličeje, flekční kontraktura prstů, velké břicho a mentální retardace. Stomatologický nález zahrnuje chabý retní uzávěr a makroglosii projevující se otevřenými ústy, makrocheilii, široké zubní oblouky a trematózní chrup. Podezření na Hunterův syndrom vyplývá z klinického vyšetření pacienta, diagnózu podpoří analýza moči a definitivně potvrdí molekulárně genetické vyšetření. K léčbě se využívá transplantace hematopoetických kmenových buněk nebo častěji substituce chybějícího enzymu.
Vlastní pozorování:
V kazuistice uvádíme stomatologický nález u chlapce s mukopolysacharidózou II. typu a zkušenosti s jeho ošetřováním. Chlapce ošetřujeme již devět roků, na naše oddělení byl doporučen praktickým lékařem pro děti a dorost ve věku pět roků a šest měsíců. Dítě při vyšetření špatně spolupracovalo, byl nápadný široký obličej, prominující rty, konvexní profil a náznak bialveolární protruze. Intraorálně byly patrné široké zubní oblouky, kariézní dočasný chrup s trematy a plakem podmíněný zánět dásní.
Závěr:
Sdělením jsme chtěli upozornit na stomatologickou problematiku u dítěte s mukopolysacharidózou II. typu, vzácným, dědičným, nevyléčitelným onemocněním. Na léčbě těchto pacientů se podílí nejen pediatr, chirurg, psycholog a další specialisté, ale také zubní lékař. Mezioborová spolupráce je v těchto případech velmi důležitá.
Klíčová slova:
mukopolysacharidózy – Hunterův syndrom – gargoylismus – multisystémové postižení – enzymová substituční terapie
ÚVOD A CÍL
Mukopolysacharidózy jsou vzácné vrozené dědičně podmíněné vady metabolismu mukopolysacharidů, které se vyznačují poruchou aktivity lyzozomálních enzymů [1, 2, 3]. Způsobuje je nedostatek nebo absence životně důležitého enzymu potřebného pro rozklad mukopolysacharidů (glykosaminoglykanů). Výsledkem je porušení přeměny jednotlivých metabolitů v těle. Následuje hromadění a usazování meziproduktů látkové výměny ve tkáních orgánů (játra, slezina, srdce, mozek) a jejich postupná porucha funkce v důsledku toxického působení na buňky. Mukopolysacharidózy lze také definovat jako disproporční poruchy růstu s deformitami skeletu.
Děti s mukopolysacharidózou zpočátku většinou dobře prospívají, klinické projevy onemocnění jsou patrné až v pozdějším vývoji, i po řadě let. Nastává zpomalení růstu dítěte, omezení pohybu, poškození centrální nervové soustavy, srdce, gastrointestinálního a respiračního systému i dalších orgánů [1, 2, 3].
Mukopolysacharidózy se dědí autozomálně recesivně s výjimkou Hunterova syndromu, který je vázaný na X-chromozom [1, 2, 3, 4, 5, 6, 8]. Hunterův syndrom (mukopolysacharidóza II. typu) se vyskytuje po celém světě. Celkem bylo zjištěno přibližně 2000 případů. Je popsáno [10], že v USA trpí tímto onemocněním 500 pacientů, v Kanadě 30, v Irsku šest a na Novém Zélandu jeden pacient. Vyšší výskyt byl zaznamenán u židovských obyvatel žijících v Izraeli. Odhadovaná incidence mukopolysacharidózy typu II se široce liší: Izrael 1 : 34 000, Britská Kolumbie 1 : 111 000, Velká Británie 1 : 132 000, Německo a Nizozemsko 1 : 140–330 tisícům živě narozených dětí [11]. V České republice trpí v současné době touto vzácnou chorobou několik desítek dětí [12, 16]. Cílem sdělení je upozornit na složitou problematiku stomatologického ošetřování dítěte s mukopolysacharidózou II. typu.
VLASTNÍ POZOROVÁNÍ
Chlapce ve věku 14 let ošetřujeme na dětském oddělení Stomatologické kliniky FN v Plzni již devět roků. Na naše oddělení byl doporučen praktickým lékařem pro děti a dorost ve věku pěti roků a šesti měsíců po stanovení diagnózy mukopolysacharidóza II. typu.
Rodinná anamnéza je u dítěte bezvýznamná, otec, matka i dvě starší sestry jsou všichni zdrávi. Z osobní anamnézy jsme zjistili, že dítě je ze třetího těhotenství fyziologického průběhu bez komplikací. Porod proběhl v termínu, spontánně záhlavím, novorozenec měl hmotnost 3300 g a měřil 51 cm. Dítě nebylo kříšeno, poporodní adaptace byla v normě, ikterus bez nutnosti fototerapie a plně kojeno bylo tři měsíce. Od narození mělo dítě ingvinální hernie bilaterálně.
Na doporučení praktického dětského lékaře bylo pro nález kontraktur na prstech rukou, faciální dysmorfii a kostní dysplazii provedeno metabolické vyšetření dítěte. Mukopolysacharidóza byla potvrzena na enzymatické i na molekulárně genetické úrovni.
Při extraorálním vyšetření je nápadný široký obličej, prominující rty, konvexní profil a náznak bialveolární protruze (obr. 1). Dítě mělo široké zubní oblouky, kariézní dočasný chrup s trematy a plakem podmíněný zánět dásní. I při pouhém vyšetření špatně spolupracovalo.
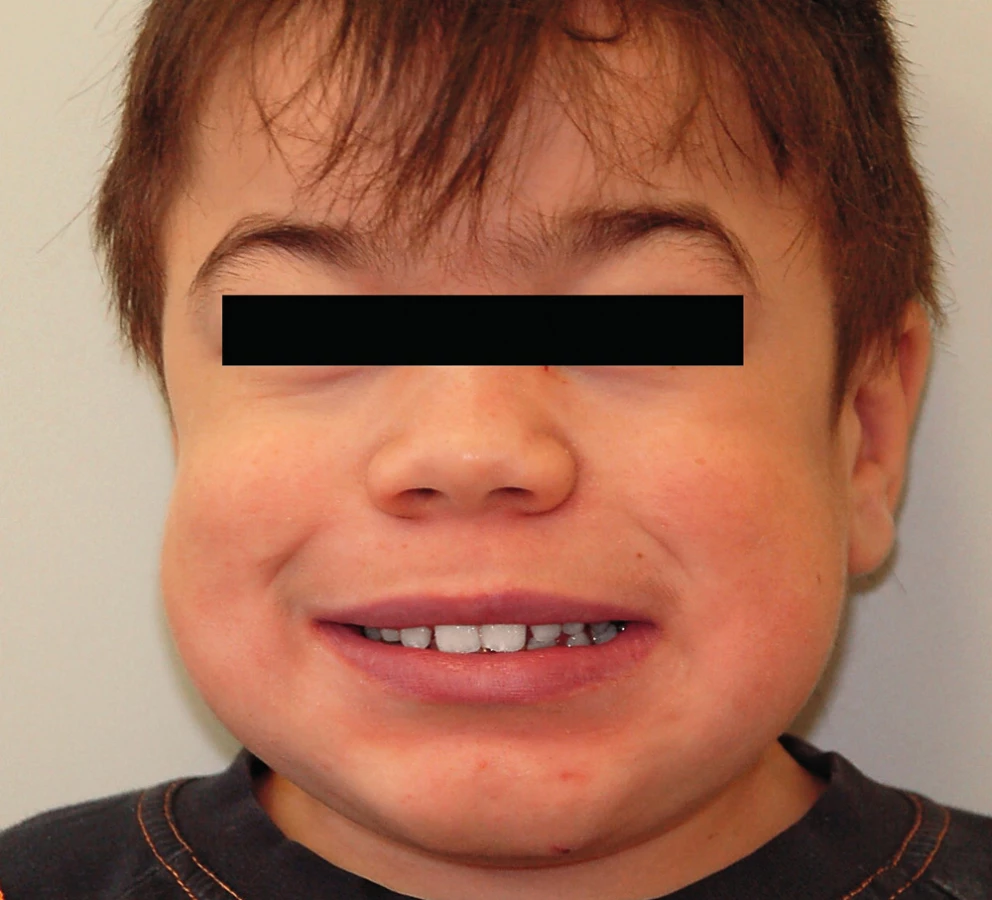
V následujícím období dítě prodělalo zánět okostice alveolárního výběžku dolní čelisti, který se léčil intraorální incizí s následnou extrakcí příčinného dočasného moláru. Ve věku šesti roků se u dítěte provedla plánovaná sanace chrupu v celkové anestezii na Stomatologické klinice dětí a dospělých 2. LF UK a FN v Praze-Motole. V nazotracheální intubaci při šetrném záklonu hlavy a v antibiotické cloně byly konzervačně ošetřeny zuby 53, 55 a extrahovány zuby 54, 64, 65, 75, 84, 85. Ve věku deset a půl roku jsme opakovali rentgenové vyšetření chlapce pro podezření na retenci zubu 36 a přítomnost folikulární cysty (obr. 2). Retence zubu 36 byla potvrzena a vzhledem k již téměř dokončenému vývoji kořenů tohoto zubu a po konzultaci s ortodontistou jsme provedli v místním znecitlivění a opět v antibiotické cloně chirurgickou extrakci.
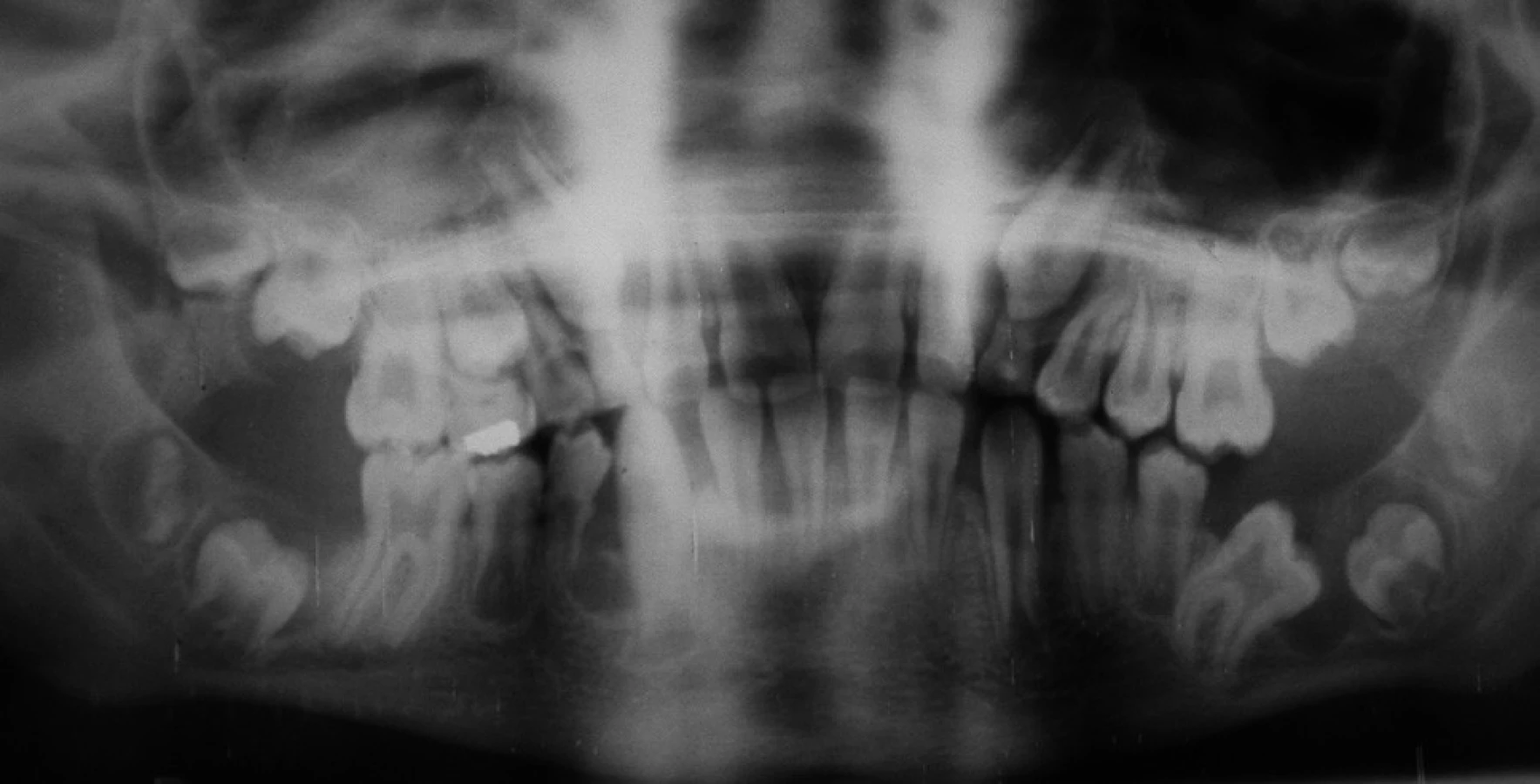
U našeho pacienta jsme dále řešili malformaci zubu 44, hluboký úpon frenula horního rtu, mělké dolní vestibulum a přítomnost atypických slizničních řas v dolním vestibulu. Zub 44 měl anomálii tvaru klinické korunky podmíněnou rozsáhlými hypoplastickými změnami v krčkové krajině a krátký radix (obr. 3). Zub 44 jsme extrahovali opět v antibiotické cloně a místní anestezii. Ve věku 13 let jsme za stejných podmínek (antibiotická clona, místní anestezie) provedli frenulektomii a prohloubili dolní vestibulum. Na ortopantomogramu ve věku 14 let je patrná retence zubů 37, 47 a změny na hlavicích obou temporomandibulárních kloubů (obr. 4). V současné době chlapec dochází na pravidelná preventivní vyšetření.

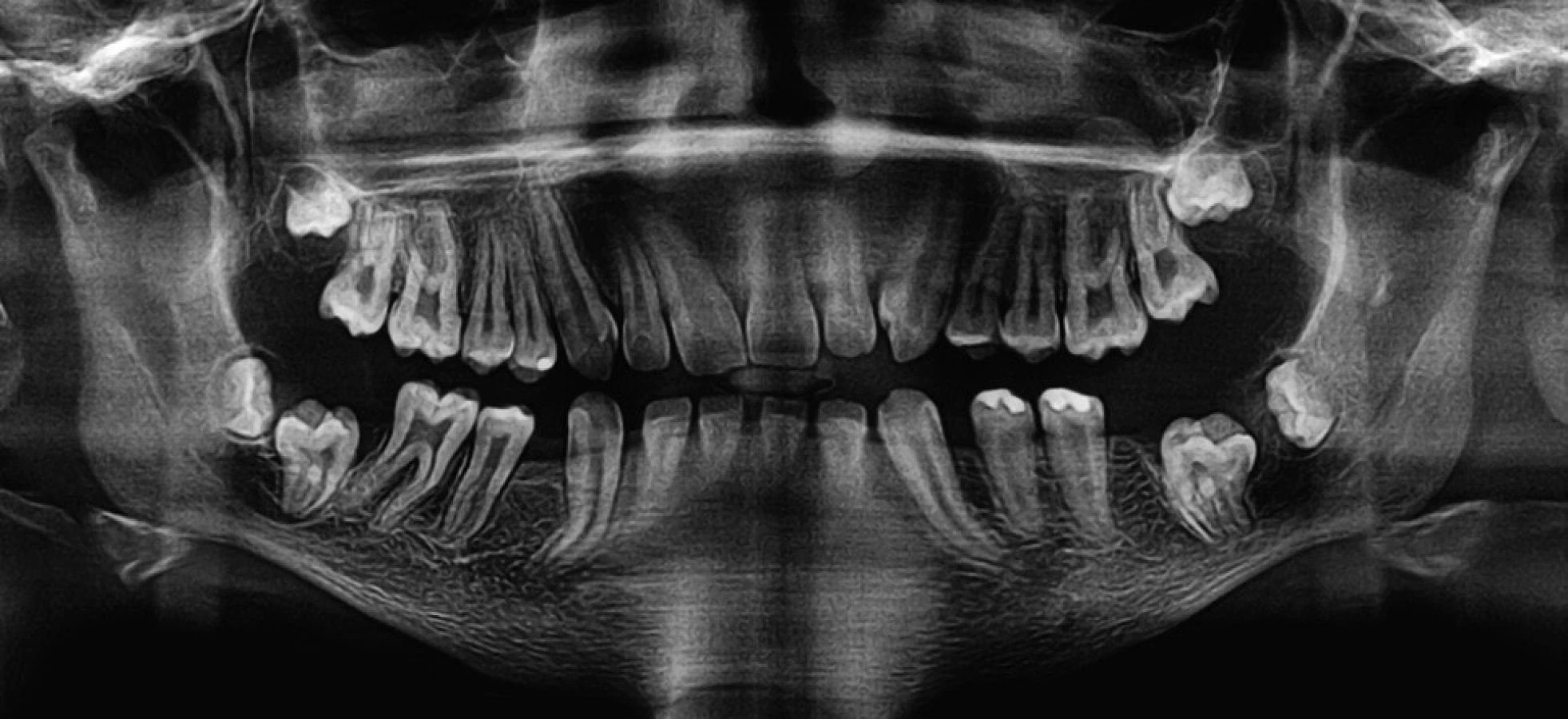
Léčení základního pacientova onemocnění se zahájilo ve věku deseti let a spočívá v substituční terapii aplikací preparátu Elaprase (výrobce Shire Human Genetic Therapies AB, Švédsko). U chlapce byly provedeny operace oboustranného syndromu karpálního tunelu a kolenního kloubu. Nosí rovněž naslouchadla. Nedílnou součástí léčení je také sociální a symptomatická terapie.
DISKUSE
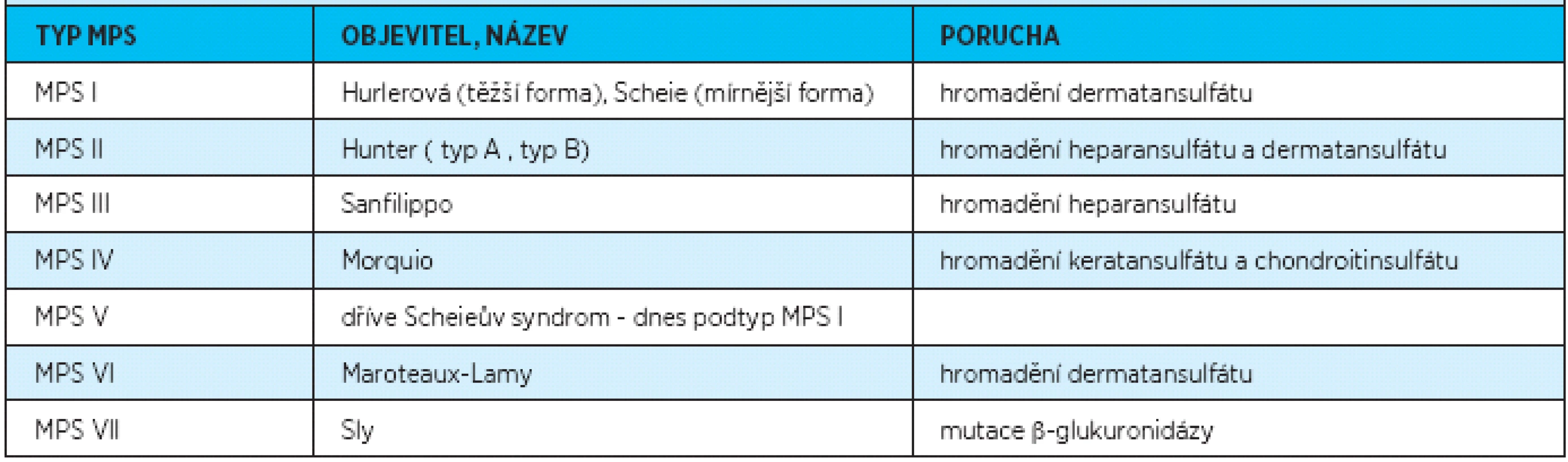
Mukopolysacharidóza je v dřívější literatuře [1, 2, 6] také známá jako Hunterův-Hurlerové-Pfaundlerův syndrom, gargoylismus, Ellisův Sheldonův syndrom, dysostosis multiplex, lipochondrodystrofie nebo dysostotická idiocie. Rozlišujeme sedm typů mukopolysacharidóz podle jejich objevitelů [1, 2, 3, 4, 5, 8] (tab. 1).
Mukopolysacharidóza II. typu, neboli Hunterův syndrom, byla poprvé diagnostikována roku 1917 skotsko-kanadským lékařem Charlesem Hunterem (1873–1955). Onemocnění je gonozomálně recesivně dědičné, vázané na X–chromozom, a proto postihuje téměř výhradně muže. U žen je výskyt Hunterova syndromu vzácný. Mukopolysacharidóza II. typu je závažné progresivní smrtelné onemocnění [2, 5]. Primárním problémem je nedostatečná funkce nebo úplné chybění enzymu iduronát-2-sulfatázy. V tkáňových buňkách se hromadí heparansulfát a dermatansulfát za vzniku multisystémového postižení včetně mentální retardace [1, 2].
Diagnostika Hunterova syndromu je možná již prenatálně. Měří se aktivita enzymu iduronát-2-sulfatázy v amniocytech plodové vody nebo ve tkáni choriových klků [1]. Postnatální diagnostika je v první fázi založena na klinickém vyšetření. V klinickém obraze onemocnění mohou být již přibližně od prvního roku věku typické somatické změny, ve většině případů je však klinický obraz typický až kolem třetího roku života, kdy se také ve větší míře projevují poruchy chování a duševního vývoje [1]. Dalšími diagnostickými metodami jsou radiologické vyšetření, analýza moči testující vylučování glykosaminoglykanů nebo specifické enzymatické vyšetření leukocytů [1]. Konečná diagnóza je však stanovena na základě měření aktivity enzymu iduronát-2-sulfatázy v séru, bílých krvinkách nebo fibroblastech při biopsii kůže [1]. Při radiologické diagnostice se vy-užívá zadopřední a bočná projekce lebky k vizualizaci sella turcica, snímek krční páteře a dále zadopřední a bočná projekce odontoidu, hrudní páteře, pánve, hlavice femuru a acetabula, stehenní a holenní kosti. Doplňujícími diagnostickými metodami jsou oftalmologické vyšetření, echokardiogram a elektrokardiogram, vyšetření dýchacích funkcí a sluchu. V histologickém vyšetření periferních granulocytů nebo buněk kostní dřeně lze odhalit typické Olšeho-Reillyho granulace [1, 2, 5, 9].
Obecně lze rozlišit dva typy Hunterova syndromu. Závažnější formou onemocnění s častějším výskytem je typ A, který lze diagnostikovat již mezi druhým. a čtvrtým rokem života. Typický je různý stupeň mentální retardace. Mírnější formou s pozdějšími příznaky je typ B, který bývá zjištěn většinou kolem desátého roku života a intelekt bývá zachován [1, 2, 6].
Charakteristickými znaky Hunterova syndromu jsou deformity skeletu, mentální retardace, hepa-tosplenomegalie, hluchota a postižení srdečních chlopní. Dále je to malá postava (120–140 cm), velká hlava, krátký krk, prominující čelo, hrubé rysy obličeje, široký nos, flekční kontraktura prstů, velké břicho, kýly, ztuhlost kloubů a syndrom karpálního tunelu. Následkem změn kostní struktury je nejen malý růst, ale také hypoplazie odontoidu. Zesílená pokožka je predispozicí pro vznik poruch termoregulace. Typické je také zpomalení psychomotorického vývoje dítěte a rapidní ztráta duševních a motorických schopností [1, 2, 6, 7].
Stomatologický nález u mukopolysacharidóz zahrnuje otevřená ústa, makroglosii, makrocheilii, dlouhý horní ret, široké zubní oblouky, trematózní chrup, časté malokluze, hypertrofický alveolární výběžek, retence zubů a omezenou pohyblivost temporomandibulárních kloubů [1, 2, 5, 6]. Kromě poruch otevírání úst jsme u našeho pacienta zjistili všechny uvedené symptomy a dále ještě atypické úpony retních uzdiček, slizničních řas a změny na kloubních hlavicích temporomandibulárních kloubů. U mukopolysacharidózy typu IV byly popsány [12, 13, 14, 15] poruchy vývoje zubní skloviny ve smyslu hypoplazie spojené s větším rizikem vzniku zubního kazu [12, 13, 14, 15]. U námi ošetřovaného dítěte jsme se setkali se závažnou hypoplazií tvrdých zubních tkání pouze u dolního premoláru, která se pravděpodobně vyvinula na lokálním podkladě. Stomatologické ošetřování dětí s mukopolysacharidózou je obtížné nejen pro špatnou spolupráci způsobenou mentální retardací. Při ošetřování těchto dětí v zubní ordinaci nebo v celkové anestezii je možný pouze mírný záklon hlavy, neboť vzhledem k instabilitě atlantookcipitálního skloubení hrozí fraktura či subluxace dentis epistrophei a prolomení krčních obratlů [1, 2, 6].
V diferenciální diagnostice Hunterova syndromu je nutno pomýšlet na ostatní typy mukopolysacharidóz, kretenismus, hypothyreoidismus a myxedém. Abnormality kostí mohou napodobovat rachitidu. V úvahu přicházejí také jiná metabolická onemocnění, jako je mannosidóza nebo Niemannův-Pickův syndrom. Diferenciální diagnostika je složitá, protože kojenci s morbus Hunter jsou obvykle bez typických klinických příznaků, takže diagnostika onemocnění je možná až později, v souvislosti s postupujícím vývojem fenotypu. Naopak kojenci s fenotypem morbus Hunter, přítomným obvykle brzy po narození, trpí s největší pravděpodobností mukolipidózou typu II (tzv. I-cell disease) [1, 2, 7].
Kauzální léčba v terapii Hunterova syndromu v současné době neexistuje. Léčba pacientů je soustředěna do specializovaných center, kde je možný multidisciplinární přístup. Chirurgická terapie zahrnuje výkony v oblasti břicha, uší, nosu a krku. Dále jsou v léčbě často využívány ortopedické a neurochirurgické zákroky. Neméně důležitou a v některých případech a fázích onemocnění jedinou léčbou jsou paliativní postupy. Taková léčba má za cíl snížit dopady zhoršení tělesných funkcí a zlepšit kvalitu života pacientů. Nedílnou součástí péče o tyto nemocné je psychiatrické vyšetření, sledování a léčba.
Jednou z léčebných možností je transplantace hematopoetických kmenových buněk. Další variantou v léčbě Hunterova syndromu je enzymová substituční terapie, která je však omezena neschopností enzymu překročit hematoencefalickou bariéru, je-li podána intravenózně [1, 2]. Nejčastěji se využívá idursulfáza, kopie lidského enzymu iduronát-2-sulfatázy. Substitucí enzymu je dosaženo zlepšení nebo kontroly příznaků onemocnění. Enzymová substituční terapie nevstupuje do centrální nervové soustavy, a nemá proto žádný vliv na kognitivní funkce [1].
Hunterův syndrom provázejí závažné komplikace. Vyskytují se především při těžké formě mukopolysacharidózy II. typu. Typické je poškození srdečních chlopní, neurologické komplikace, zahušťování tracheální stěny vedoucí k obstrukčnímu onemocnění dýchacích cest. Další komplikací je postupující hepatosplenomegalie, vyklenutí břišní stěny a větší prominence kýl. Dále jsou v různém stupni postiženy klouby, zejména kyčle, kolena, zápěstí a lokty. Při častém syndromu karpálního tunelu je postižena funkce ruky [2].
Dlouhodobá prognóza pacientů s Hunterovým syndromem je rozdílná podle typu onemocnění.
Typické je progresivní viscerální postižení, duševní a motorické poruchy, dítě jakoby se vracelo ve vývoji zpět. Intelekt se postupně snižuje, pacient je inkontinentní, objevují se poruchy hybnosti, porucha a ztráta polykacího reflexu, v pozdějším stadiu také poruchy dýchání. Konečným stadiem je spastická kvadruplegie s poruchami polykání. Smrt nastává nejčastěji následkem pneumonie ve druhé životní dekádě.
U pacientů s těžkou formou Hunterova syndromu se v literatuře [1, 2, 5, 6] uvádí délka života 10 až 15 let. V případě mírnější formy se pacienti mohou dožít 50 až 70 let. Motto Společnosti pro mukopolysacharidózu, sdružující rodiče postižených dětí: „není naděje, zbývá láska,“ je výstižné.
Vzhledem ke sporadickému výskytu tohoto onemocnění a nevelké skupině pacientů jsou zkušenosti s ošetřováním takto postižených dětí relativně malé. Možná právě proto bychom se jim měli pečlivě věnovat, důsledně dbát na dodržování dietních návyků, na řádnou hygienu ústní dutiny a včasnou sanaci kazů. Rodiče nemocných často vzhledem k velkému množství lékařských vyšetření a zákroků nutných k diagnostice nebo léčbě tohoto těžkého smrtelného onemocnění zapomínají, že zdravé zuby i celá dutina ústní jsou pro jejich dítě neméně důležité.
ZÁVĚR
Tímto sdělením jsme chtěli upozornit na stomatologickou problematiku u dítěte s mukopolysacharidózou II. typu, vzácným dědičným nevyléčitelným onemocněním. Na léčbě těchto pacientů se podílí nejen pediatr, chirurg, psycholog a další specialisté, ale také zubní lékař, který by měl postiženým dětem ve spolupráci s rodiči zajistit co nejlepší úroveň orálního zdraví. Mezioborová spolupráce je v těchto případech velmi důležitá.
MDDr. Lucie Baborská
Stomatologická klinika LF UK a FN
Alej Svobody 80
304 60 Plzeň
e-mail: baborskal@seznam.cz
Sources
1. Fernandes, J., Saudubray, J.-M., Walter, J. H.: Diagnostika a léčba dědičných metabolických poruch, 4. vyd. Praha, Triton, 2008.
2. Gorlin, R. J., Cohen, M. M., Jr., Hennekam, R. C. M.: Syndromes of the head and neck. Oxford, 2001, s. 119–139.
3. Hoffmann, G. F., Nyhan, W. L., Zshocke, J., Kahler, S .G., Mayetapek, E.: Dědičné metabolické poruchy, 1. vyd. Praha, Grada, 2005.
4. Hyánek, J.: Dědičné metabolické poruchy, 1. vyd. Praha, Avicenum, 1990.
5. Jones, K. L.: Smiths recognizable patterns of human malformation, sixth ed. Philadelphia, Pennsylvania, 2006, s. 518–544.
6. Kožich, V., Zeman, J.: Dědičné metabolické poruchy v pediatrii. Postgrad. Med., roč. 12, 2010, č. 7, s. 793–799.
7. Leiber, W., Olbrich, G.: Wörterbuch der klinischen Syndrome, 2. vyd. München, Urban und Schwarzenberg, 1959.
8. Muntan, A. C.: Pediatrie, 1. české vyd. Praha, Grada, 2009, s. 136–138.
9. Zitelli, B. J., Davis, H. W.: Atlas of pediatric physical diagnosis, 4. vyd., Missouri, 2002, s. 684. Dostupné online na URL adrese http://www.zitelliatlas.com/.
Internetové zdroje
10. http://en.wikipedia.org/wiki/Hunter_syndrome
11. http://emedicine.medscape.com/article/944723-overview#a0199
12. http://svp-vzacnaonemocneni.cz/portal/?tag=mps
13. http://emedicine.medscape.com/article/947254-clinical
14. http://en.wikipedia.org/wiki/Morquio_syndrome
15. http://www.patient.co.uk/doctor/Morquio‘s-Syndrome.htm
16. http://www.mukopoly.cz/mps/mps.html
Labels
Maxillofacial surgery Orthodontics Dental medicineArticle was published in
Czech Dental Journal

2013 Issue 5
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Ektodermání dysplazie
- Funkční potraviny a funkční komponenty potravy v prevenci zubního kazu
- Dentální implantologie při léčbě následků parézy nervus facialis v dětském věku
- Stomatologická problematika u dítěte s mukopolysacharidózou II. typu