
Gitelmanův syndrom s chondrokalcinózou v dospělosti - kazuistika
Gitelman syndrome with chondrocalcinosis in adult age – case report
The authors present a case of 56 years old female patient with anamnestic pain in knee joints, cramps and limbs paresthesia, with detected steady low serum levels of potassium and magnesium and metabolic alkalosis. High renal losses of both elements as well as hypocalciuria were detected. Radiographic examination gave evidence of knee joints chondrocalcinosis. Genetic examination proved the presence of homozygous point mutation in exon 10 of gene SLC12A3, which leads to change of glycine to serine at position 439 of cotransport protein NCTT. This mutation has already been repeatedly described in Gitelman syndrome. Gitelman syndrome comes under tubulopathies causing hypokalemia and is related to Bartter syndrome. Both diseases are linked to hypokalemia, renal losses of potassium, metabolic alkalosis and the activation of renin-angiotensin-aldosterone axis. In contrast to Bartter syndrome, the individuals with Gitelman syndrome have significant hypomagnesemia, hypermagnesiuria, hypocalciuria and milder clinical development with manifestation in higher age. The possible link between this syndrome and chondrocalcinosis appears interesting for a rheumatologist. In case of mentioned female patient, the therapy based on primarily massive parenteral, subsequently continual peroral substitution of potassium and magnesium and application of spironolactone and indomethacin led to total clinical symptom reduction and partial correction of laboratory values.
Key words:
Gitelman syndrome, hypokalemia, hypomagnesemia, chondrocalcinosis
Authors:
P. Horák 1; N. Jeck 2; Z. Fryšák 1; J. Zadražil 1
Authors‘ workplace:
III. interní klinika FN a LF UP Olomouc, 2Kinderklinik, Universität von Marburg
1
Published in:
Čes. Revmatol., 16, 2008, No. 1, p. 30-33.
Category:
Case Report
Overview
Autoři prezentují případ 56leté nemocné s anamnézou bolestí kolenních kloubů, křečí a parestezií končetin, u které byly zjištěny stabilně nízké sérové hladiny draslíku a hořčíku a metabolická alkalóza. Byly rovněž nalezeny vysoké ledvinné ztráty obou prvků, jakož i hypokalciurie. Radiografické vyšetření svědčilo pro chondrokalcinózu kolenních kloubů. Genetické vyšetření prokázalo přítomnost homozygotní bodové mutace v exonu 10 genu SLC12A3, která vede k záměně glycinu za serin na 439 místě kotransportního proteinu NCCT. Tato mutace byla v minulosti již opakovaně popsána u Gitelmanova syndromu. Gitelmanův syndrom patří mezi hypokalemizující tubulopatie a je příbuzný s Bartterovým syndromem. Obě choroby jsou spojeny s hypokalémií, renálními ztrátami draslíku, metabolickou alkalózou a s aktivací osy renin-angiotensin-aldosteron. Na rozdíl od Bartterova syndromu, jedinci s Gitelmanovým syndromem mají výraznou hypomagnezémii, hypermagnezurii, hypokalciurii a mírnější klinický průběh s pozdějším věkem manifestace. Pro revmatologa se jeví jako zajímavé možné spojení tohoto syndromu s chondrokalcinózou. V případě zmiňované nemocné vedla léčba spočívající ve zprvu masivní parenterální, následně kontinuální perorální substituci draslíku a hořčíku, a podání spironolaktonu a indomethacinu k vymizení klinické symptomatologie a částečné úpravě laboratorních hodnot.
Klíčová slova:
Gitelmanův syndrom, hypokalémie, hypomagnezémie, chondrokalcinóza
Popis případu
Padesátišestiletá nemocná byla přijata na III. interní kliniku FN Olomouc pro pětiletou anamnézu bolestí kolenních kloubů, křečí a parestézií horních i dolních končetin a celkové slabosti. Z anamnézy vyplynulo, že ataky křečí se objevují několikrát během dne, bolesti kolen jsou někdy velmi intenzivní a znemožňují úplnou flexi kloubů. Někdy má pocit oteklých kolenních kloubů. Připouštěla rovněž přítomnost křečí v minulosti v dolních končetinách, nastupující snad kolem čtyřiceti let věku, kterým však nevěnovala větší pozornost. Posledních pět let se však potíže natolik zhoršily, že se rozhodla svěřit se lékaři. Sourozence nemá, její matka zemřela ve věku 78 let na cévní mozkovou příhodu, otec ve věku 72 let na pneumonii. Má dvě děti, které podobné potíže nemají a jejichž laboratorní nálezy nejsou známy. Klimakterium u ní nastoupilo ve věku 51 let, hormonální substituce jí nebyla předepsána. Stěžovala si na postupně se zhoršující zrak. Při vyšetření u oftalmologa, který stanovil příčinu horšícího se zraku jako počínající glaukom, byla v laboratorních testech zjištěna poměrně výrazná hypokalémie nejasného původu. V té době neužívala žádné léky. Poté co byly zjištěny nízké hodnoty draslíku (2,55 mmol/l) a hořčíku (0,55 mmol/l), byla zahájena orální substituce tabletami kalium chloratum (1500 mg denně) a magnesium lacticum (1000 mg denně). I přes uvedenou terapii nedošlo k normalizaci laboratorních hodnot ani k ústupu či zmírnění potíží nemocné. Tři týdny před plánovanou hospitalizací jí doporučil odesílající lékař substituci vysadit.
Úvodní fyzikální vyšetření prokázalo zvýšenou křečovou pohotovost na horních i dolních končetinách, byl pozitivní Chvostkův i Trousseaův příznak. Nález na kolenních kloubech vykazoval přítomnost drásotů a vrzotů bez příznaků svědčících pro přítomnost volné tekutiny v kloubní dutině. Jiná zjevná patologie prokázaná nebyla. Krevní tlak byl v normě. Pozoruhodné však byly laboratorní testy. Byla nalezena konstantně nízká hladina kalia pohybující se mezi 2,6–3,05 mmol/l. Dalším výrazným abnormálním nálezem byla výrazná hypomagnezémie a metabolická alkalóza (viz tab. 1). Sérové hladiny urey, kreatininu, sodíku, chloridů, kyseliny močové, glykémie, alanin aminotransferázy, aspartát aminotransferázy, gamma-glutamyltransferázy, laktát dehydrogenázy, alkalické fosfatázy, cholesterolu a triglyceridů stejně jako parametry krevního obrazu byly v mezích laboratorních norem. Při elektrokardiografickém vyšetření nebyly patrny EKG známky hypokalémie či ischémie. Biochemické vyšetření moči a močového sedimentu nevykazovalo významnou patologii. Bylo provedeno vyšetření odpadu moči za 24 hodin, které prokázalo vysoké ztráty draslíku a magnézia. Dalším nálezem byla značná hypokalciurie. Hladiny kortizolu byly v mezích normy, byla však zjištěna elevace reninu a aldosteronu nad horní hranici fyziologické normy dospělých. Ultrazvukové a ani CT vyšetření neukázalo patologické změny v oblasti ledvin či nadledvin. Koloskopické vyšetření indikované k vyloučení přítomnosti vilonodulárního adenomu tlustého střeva vedoucího k ztrátám draslíku bylo negativní. Radiografické vyšetření odhalilo přítomnost rentgenových známek chondrokalcinózy na obou kolenních kloubech (obr. 1). Jelikož klinicky nebyla přítomna volná tekutina v kolenních kloubech, nebyla indikována artrocentéza a nález tak nemohl být verifikován vyšetřením v polarizovaném mikroskopu. Na základě uvedených nálezů byla stanovena pracovní diagnóza hypokalemické, minerály ztrácející tubulopatie Bartterova typu. Klinický obraz doplněný výraznou hypomagnezémií svědčil pro možnost přítomnosti Gitelmanovy varianty Bartterova syndromu (tzv. Gitelmanův syndrom). Vzorek krve byl odeslán k provedení genetického testování do laboratoře v Marburgu (Kinderklinik, Universität von Marburg). Toto vyšetření prokázalo přítomnost homozygotní bodové mutace v exonu 10 genu SLC12A3. Tato bodová mutace vede k záměně glycinu za serin na 439 místě kotransportního proteinu. Záměna za serin v jinak velmi konzervovaném regionu G439 v rámci rodiny transportérů kationů a chloridu byla již v minulosti popsána u nemocných s Gitelmanovým syndromem (1).

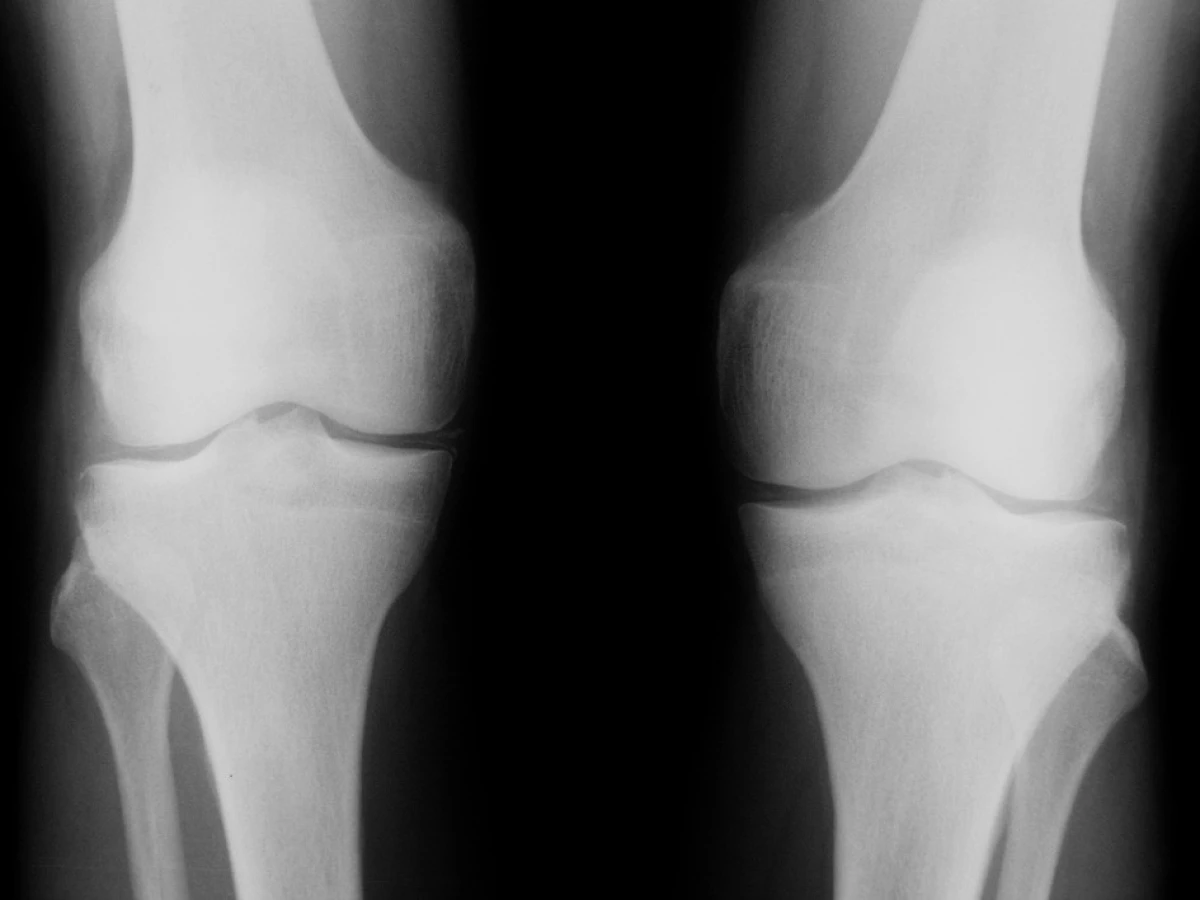
Byla zahájena zprvu parenterální substituce chloridu draselného (3000 mg denně ve dvou infuzích) a magnézia sulfátu (4000 mg denně), terapie byla potencována přidáním spironolaktonu. Po 5 dnech se přešlo na substituci per os (kalium chloratum 3x500 mg, magnezium lacticum s pyridoxinem 3x1000 mg denně). Na základě literárních údajů byla rovněž zahájena terapie indomethacinem v dávce 100 mg denně. V době propuštění z nemocnice byly hodnoty draslíku 3,15 mmol/l hladiny magnézia 0,72 mmol/l. Při kontrolách za 1, 3 a 6 měsíců po propuštění se nemocná cítila dobře a křeče popisované v úvodu ustoupily. Bolesti kolenních kloubů nebyly za dobu od propuštění nápadnější. Laboratorní testy byly nejdříve stále abnormální s hladinou draslíku 3,2 mmol/l a magnézia 0,61 mmol/l. Při návštěvě po 6 měsících byly dokonce hladiny draslíku i magnézia v mezích laboratorní normy.
Diskuse
Hypokalémie je častým laboratorním nálezem spojeným často s klinickými příznaky. Diferenciální diagnostika příčin tohoto nálezu je nesmírně široká, vysvětlení je však většinou celkem prozaické. Bartterův syndrom je vzácnou příčinou hypokalémie. Jedná se o stav, který se manifestuje již v raném dětství pomalým růstem, polyurií, polydipsií a hypotenzí. Zpravidla je diagnostikován před šestým rokem věku. Gitelmanův syndrom je charakterizován mírnějším klinickým průběhem a pozdějším věkem klinické manifestace a je ve většině případů diagnostikován během školního věku. Někdy je také nalezen u asymptomatických dospělých, kteří mají jinak nevysvětlitelnou hypokalémii a hypomagnezémii a kteří mohou anamnesticky zmiňovat přítomnost období spojených s výraznou slabostí, tetanií, abdominální bolestí, zvracením či zvýšenou teplotou(2). Růstová retardace a polyurie nejsou typicky v rámci Gitelmanova syndromu přítomny. U některých nemocných se vyskytuje chondrokalcinóza, jejíž přítomnost souvisí zřejmě s dlouhodobou hypomagnezémií. Tento syndrom byl poprvé popsán H. Gitelmanem jako autosomální recesivní porucha (3). Byl popsán jako samostatná jednotka příbuzná s Bartterovým syndromem. Obě choroby jsou spojeny s hypokalémií, renálními ztrátami draslíku, metabolickou alkalózou a s aktivací osy renin-angiotensin-aldosteron. Na rozdíl od Bartterova syndromu, jedinci s Gitelmanovým syndromem mají výraznou hypomagnezémii, hypermagnezurii, hypokalciurii, a jak již bylo zmíněno výše, mírnější klinický průběh a pozdější věk manifestace.
Několik podobných klinických jednotek, které jsou charakterizovány jako normotenzní hypokalemické soli ztrácející tubulopatie jsou někdy označovány jako “Bartteręs like“ syndromy (4). Tyto mohou být rozděleny do tří základních klinických fenonotypů: infantilní varianta Bartterova syndromu (IBS), klasický Bartterův syndrom a Gitelmanův syndrom. V současné době výzkum renálních transportérů, kotransportérů a kanálů transepiteliálního transportu iontů identifikoval mutace několika genů zodpovědných za rozvoj těchto syndromů a přispěl zásadním způsobem k fenotypické a genotypické klasifikaci těchto poruch. SLC12A1 gen kóduje natrium-draslík-chlorid kotransportér NKCC2, gen KCNJ1 kóduje draselný kanál zevní ledvinné dřeně ROMK, gen CLCNKB kóduje bazolaterální chloridový kanál C1C-Kb a konečně gen SLC12A3 kóduje thiazid senzitivní natrium-chloridový kotransportér NCCT distálního stočeného tubulu. Tyto mutace se liší svojí klinickou manifestací jak demonstrovala například práce Petersové (5).
Defekty NKCC2 a ROMK jsou často charakterizovány klinickou triádou příznaků, které se projevují v perinatálním období či raném dětství: polyhydramnion, hypo - či izostenurie a nefrokalcinóza. Život ohrožující ztráty vody a elektrolytů se objevují v bezprostředním postnatálním období a tyto mutace jsou detekovány v případě IBS. Naopak věk nástupu klinických a laboratorních symptomů nemocných s mutacemi C1C-Kb vykazuje větší variabilitu a všeobecně pozdější věk počátku choroby. Některé z nich mají klasický Bartterův fenotyp v raném dětství, nicméně později nastupující symptomy mohou připomínat spíše Gitelmanův syndrom (6). V rámci IBS syndromu byla identifikována samostatná skupina nemocných s senzoricko-neurální hluchotou (BSND). Nedávno byl identifikován gen kódující ß-podjednotku C1C chloridového kanálu. Byl pojmenován barttin a bylo popsáno jeho několik mutací spojených se syndromem BSND (7). V případě některých nemocných s hypokalcémií s deficientní sekrecí parathormonu, kteří mají známky Bartterova syndromu, byla nalezena aktivační mutace genu pro CASR (calcium sensing receptor). Aktivace CASR inhibuje aktivitu ROMK, který bývá mutovaný rovněž u některých pacientů s IBS syndromem, jak popsáno výše. Tento nález nabízí také nový pohled na rozvoj vlastního Bartterova syndrom (8).
Nemocná referovaná v této práci měla bodovou mutaci SLC12A3 genu (16q13), který kóduje NCCT. Bodová mutace exonu 10 nalezená u našeho pacienta vede k záměně glycinu za serin na 439 pozici NCCT. Oblast 439 je evolučně jednou z velmi striktně konzervovaných oblastí, její sekvence je stejná u primátů jako u paryb (1). Tento defekt byl popsán u několika nemocných s Gitelmanovým syndromem v Evropě (přehled viz Reissinger A. et al., 2002, 9). Pacienti s touto genetickou variantou Gitelmanova syndromu trpí únavou, zvýšenou neuromuskulární dráždivostí, kloubními bolestmi, vykazují hypokalémii, hypomagnezémii, hypokalciurii, vyšší plazmatickou reninovou aktivitu a hyperaldosteronismus. Popsaná genetická varianta patří mezi téměř devadesát mutací NCCT popsaných v souvislosti s Gitelmanovým syndromem (10). Gitelmanův syndrom se manifestuje později v průběhu dětského věku než klasický Bartterův syndrom. Nicméně první manifestace v pozdní dospělosti jsou velmi vzácné. Dle našich informací, není popisovaný případ nejstarším nemocným s nově diagnostikovaným Gitelmanovým syndromem, ale případy po padesátém roce věku jsou stále velmi raritní zejména s přesně stanovenou genetickou diagnózou. Můžeme spekulovat o vlivu menopauzy na rozvoj klinické manifestace u popisované nemocné. Lze rovněž předpokládat, že řada případů této poruchy zůstane nediagnostikovaná. Popisovaný případ rovněž poukazuje na možnosti diagnostiky, které poskytuje klinikovi molekulární klasifikace založená na detekci molekulárních defektů.
Léčba Gitelmanova syndromu je založena na substituci draslíku a magnézia a na kalium šetřících diureticích spironolaktonu, amiloridu či triamterenu, někteří autoři popisují rovněž dobrý efekt indomethacinu založený zřejmě na ovlivnění prostaglandinů renálních tubulů. Gitelmanův syndrom je většinou méně závažným onemocněním než klasický Bartterův či IBS syndrom a růstová prognóza dětí je normální. Často je nemožné dosáhnout normálních sérových hladin minerálů a je smysluplnější soustředit se na zlepšení klinických příznaků. Popsaný případ poukazuje na skutečnost, že Gitelmanův syndrom by měl být zvažován i v případě dospělých jedinců v diferenciální diagnostice etiologicky nejasné hypokalémie a hypomagnezémie. Z pohledu revmatologa je zajímavá možnost přítomnosti chondrokalcinózy, jejíž vznik zřejmě souvisí s dlouhodobě nízkou hladinou hořčíku. Uvedená kazuistika ukazuje důležitou roli molekulární biologie jako cenného nástroje přesné diagnostiky ve vybraných situacích.
Doc. MUDr. Pavel Horák, CSc.
III. interní klinika, FN a LF UP Olomouc
I. P. Pavlova 6
772 00 Olomouc
e-mail: horak@pfnol.cz
Sources
1. Mastroianni N, Bettinelli A, Colussi G, et al. Novel molecular variants of the Na-Cl cotransporter gene are responsible for Gitelman syndrome Am J Hum Gen 1996; 59 : 1019-1026.
2. Ismail HM, Jagadeesch T. Gitelman’s syndrome. J Royal Soc Med 2002; 92 : 299–300.
3. Gitelman HJ, Graham JB, Welt LG. A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physic 1966; 79 : 221–235.
4. Kamel KS, Oh MS, Halperin ML. Bartter’s, Gitelman’s, and Gordon’s. Syndromes. Nephron 2002; 92, Suppl. 1 : 18–27.
5. Peters M, Jeck N, Reinalter S, et al. Clinical presentation of genetically defined patients with hypokalemic salt-losing tubulopathies. Am J Med 2002; 112 : 183–190.
6. Warnock DG. Renal genetic disorders related to K(+) and Mg(+). Ann Rev Physiol 2002; 64 : 845–876.
7. Brennan TMH, Landau D, Shalev H, et al. Linkage of infantile Bartter syndrome with sensorineural deafness to chromosome 1p. Am J Hum Gen 1998; 62 : 355–361.
8. Watanabe S, Fukumoto S, Chang H, et al. Association between activating mutations of calcium-sensing receptor and Bartter’s syndrome. Lancet 2002; 360 : 692–694.
9. Reissinger A, Ludwig M, Utsch B, et al. Novel NCCT gene mutations as a cause of Gitelman’s syndrome and a systemic review of mutant and polymorphic NCCT alleles, Kidney Blood Press Res 2002, 25 : 354–362.
10. Maki N, Komatsuda A, Wakui H, et al. Four novel mutations in the thiazide-sensitive Na-Cl co-transporter gene in Japanese patients with Gitelman’s syndrome. Nephrol Dial Transplant 2004; 19 : 1761–1766.
Labels
Dermatology & STDs Paediatric rheumatology RheumatologyArticle was published in
Czech Rheumatology

2008 Issue 1
Most read in this issue
- Protilátky proti složkám komplementového systému a systémový lupus erythematodes
- Idiopatická retroperitoneální fibróza: Méně častá příčina bolestí dolní části zad. Použití tamoxifenu v terapii onemocnění
- Vliv pohybové terapie na pohyblivost páteře a subjektivní vnímání bolesti u jedinců s ankylozující spondylitidou
- Cytokiny BAFF (B-cell activating factor) a APRIL (a proliferation-inducing ligand) a jejich role u autoimunitních onemocnění