
Wegenerova granulomatóza s trombotickou trombopenickou purpurou – neurologické projevy
Wegener’s granulomatosis with thrombotic thrombocytopenic purpura – neurologic manifestations
Wegener’s granulomatosis (WG) is a systemic vasculitis of medium-sized and small-sized vessels. Most of all, the kidneys and respiratory tract, peripheral nerves, less frequently CNS are affected with either vasculitis or granulomas. Other organs may be affected, as well. Thrombotic thrombocytopenic purpura (TTP) is a disease caused by deficiency of von Willebrand factor’s protease resulting in hyperadhesiveness of thrombocytes with various clinical consequences. The case report of a patient with coincidental course of WG and TTP is described. The disease was recurrent, diffusing and potentiating the clinical manifestations, which took on dramatic picture. The case draws attention to the possibility of combination of two rare conditions and the need of thorough examination of clinical manifestations and laboratory findings, which do not fit in the image of one disease.
Key words:
Wegener’s granulomatosis, thrombotic thrombocytopenic purpura, neurologic manifestations
Authors:
J. Vítová
Authors‘ workplace:
Interní oddělení Nemocnice Č. Budějovice, a. s.
Published in:
Čes. Revmatol., 16, 2008, No. 2, p. 92-96.
Category:
Case Report
Overview
Wegenerova granulomatóza (WG) je systémová vaskulitida středních a malých cév. Postiženy jsou především ledviny a dýchací ústrojí, periferní nervy, méně často CNS, ať už vaskulitidou či granulomy. Mohou být ale zasaženy i jiné orgány. Trombotická trombopenická purpura (TTP) je onemocnění způsobené deficitem proteázy von Willebrandova faktoru jejímž výsledkem je hyperadhezivita trombocytů s různými klinickými důsledky. Je popsána kazuistika pacienta se současným průběhem WG i TTP. Onemocnění bylo recidivující s prolínáním a potencováním klinických projevů které nabíraly dramatický obraz. Případ upozorňuje na možnost kombinace dvou vzácných stavů a potřebu pečlivého vyšetření klinických projevů a laboratorních nálezů nezapadajících do obrazu jednoho onemocnění.
Klíčová slova:
Wegenerova granulomatóza, trombotická trombopenická purpura, neurologické projevy
ÚVOD
Wegenerova granulomatóza (WG) byla poprvé popsána r. 1930. Je charakterizována nekrotizujícími granulomatózními ložisky respiračního traktu, glomerulonefritidou a vaskulitidou, ale i lokalizovaným postižením očí, CNS, centrálního a více periferního nervstva, srdce, kůže, GIT, močového ústrojí a případně dalších orgánů (1).
WG byla zaznamenána u osob ve věku od 3 měsíců až do 75 let, zvýšená incidence je však ve 4. a 5. dekádě věku.
Přes jisté pokroky v diagnostice je etiologie WG stále nejasná. Jistá je patogenetická účast protilátkové aktivity, tj. přítomnost c-ANCA protilátek namířených proti serinové proteináze 3 (PR3) (2). Pravděpodobně se ale v patogenezi účastní i buněčná imunita vzhledem ke granulomatóznímu charakteru zánětu (3) a možná i exprese fractalkinu CX3CL1 a jeho receptoru CX3CR1 (4). Geneticky se zvýšeně u pacientů s WG nachází antigen HLA-DR2 a HLA-B8, HLA-B8 (5). Svou roli jistě zastává i infekce (6).
Prognóza WG je závažná svou podstatou i komplikacemi a mortalita i přes imunosupresivní léčbu a účinnou likvidaci komplikací je stále vysoká. Významným prognostickým faktorem je především postižení ledvin a postižení plic zvláště v prvním roce po stanovení diagnózy.
Trombotická trombopenická purpura (TTP) je charakterizována hyperadhezivitou trombocytů na podkladě kumulace von Willebrandova faktoru (vWF) v důsledku deficitu proteázy vWF. Deficit vWF je výjimečně vrozený, častěji je získaný na autoimunitním podkladě (jiná autoimunita) nebo vlivem infekce, léků a toxinů poškozujících endotel, či v přítomnosti nádoru. Klinika onemocnění je charakterizována mechanickou hemolýzou, nefropatií, postižením CNS především centra řeči či trombopenií, neuropatií, hepatopatií. Může vést k selhání ledvin či ireverzibilnímu postižení CNS (7).
KAZUISTIKA
Dosud zdravý 49letý lesní technik s nevýznamnou rodinnou a osobní anamnézou měl v dubnu 2004 provedenu resekci horního laloku levé plíce pro suspektní tumor. Histologicky nebyl prokázán nádor, bylo však popsáno ložisko nekrotické tkáně s epiteloidně-granulomatózní reakcí bez příměsi Langhansových buněk. Byly zachyceny částečně ztrombotizované cévy bez zánětlivé reakce. Nebyla zastižena mykotická či bakteriální infekce.
Počátkem června 2004 byla při chirurgické kontrole zjištěna léze brachiálního plexu vlevo, která byla hodnocena jako pooperační. Již tehdy si pacient stěžoval na únavu a pocení, ale základní laboratoř včetně ANCA protilátek byla negativní.
Dne 22. 6. 2004 byl pacient poprvé vyšetřen revmatologem pro vysokou FW, oligoartritidu, kožní vaskulitidu, subfebrilie, neproduktivní kašel a subikterus sklér. V laboratoři byla lehká eosinofilie, renální insuficience s kreatininem 192 μmol/l, vysoké zánětlivé markery s CRP 158 mg/l. Současně byl přítomen nefritický syndrom s erytrocyturií 566,7 a s leukocyturií 25 bez přítomnosti válců v Hamburgerově sedimentu. V imunologickém rozboru byly CIK 60 a cANCA protilátky silně pozitivní, antikardiolipinové protilátky negativní, kvantitativní stanovení imunoglobulinů v normě, stejně jako ANA, anti-ENA a složky komplementu. Byla provedena biopsie ledvin s nálezem těžké fokálně segmentální glomerulonefritidy nekrotizující s přechodem na Bowmanovo pouzdro, kde byl místy zachycen zánět granulomatózního charakteru. Na RTG hrudníku (obr. 1) bylo motýlovité zastření obou středních a pravého dolního pole.
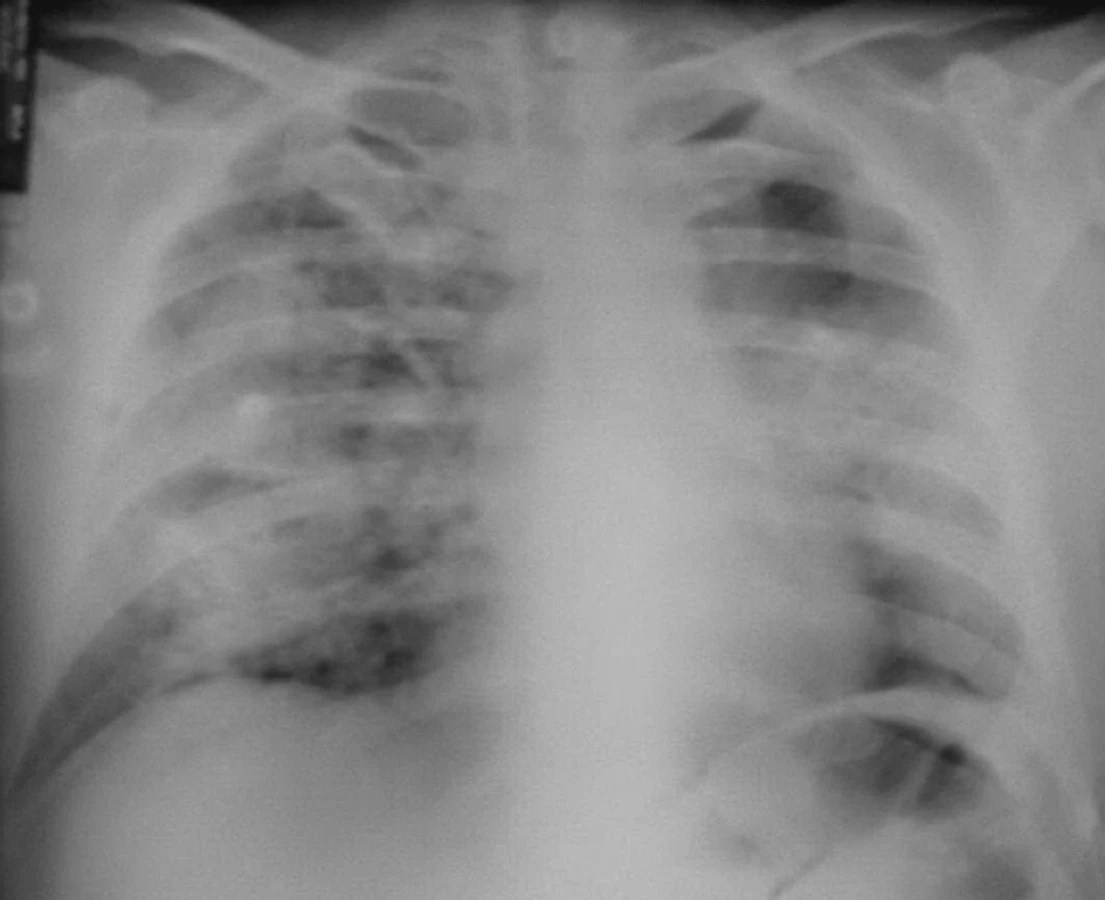
Byla tedy stanovena diagnóza Wegenerovy granulomatózy a zahájena obvyklá léčba, tj. cyklofosfamid a glukokortikoidy za krytí antibiotiky a antimykotiky. Přesto ale došlo velmi rychle k progresi renálního selhání, rychlé anemizaci a trombocytopenii. Dále se objevila bilirubinemie 44/16, která zcela nezapadala do obrazu WG. Vyšetřeními bylo vysloveno podezření na TTP, které bylo testem ADAMST 13 ověřeno. Léčba byla tedy doplněna o plazmaferézy s podáním čerstvé plazmy. Přesto ale vyvstala nutnost přechodné akutní hemodialyzační péče. I přes komplikace, jako byla rozsáhlá ulcerace jazyka s mykotickou a stafylokokovou infekcí (aureus) a recidivující generalizovaný exantém, spojený s dušností, který nebyl zcela objasněn, se stav postupně upravil a stabilizoval. Pacient byl propuštěn po tříměsíční hospitalizační léčbě s negativním RTG hrudníku (obr. 2) s kreatininem kolem 200 μmol/l bez nutnosti hemodialýzy a s normálním krevním obrazem i normálním haptoglobinem a hodnotou LDH. Při dimisi dostával Imuran 100 mg za den, Medrol 24 mg denně s další detrakcí, Biseptol 480 mg, Vessel due F a Helicid 20mg denně.
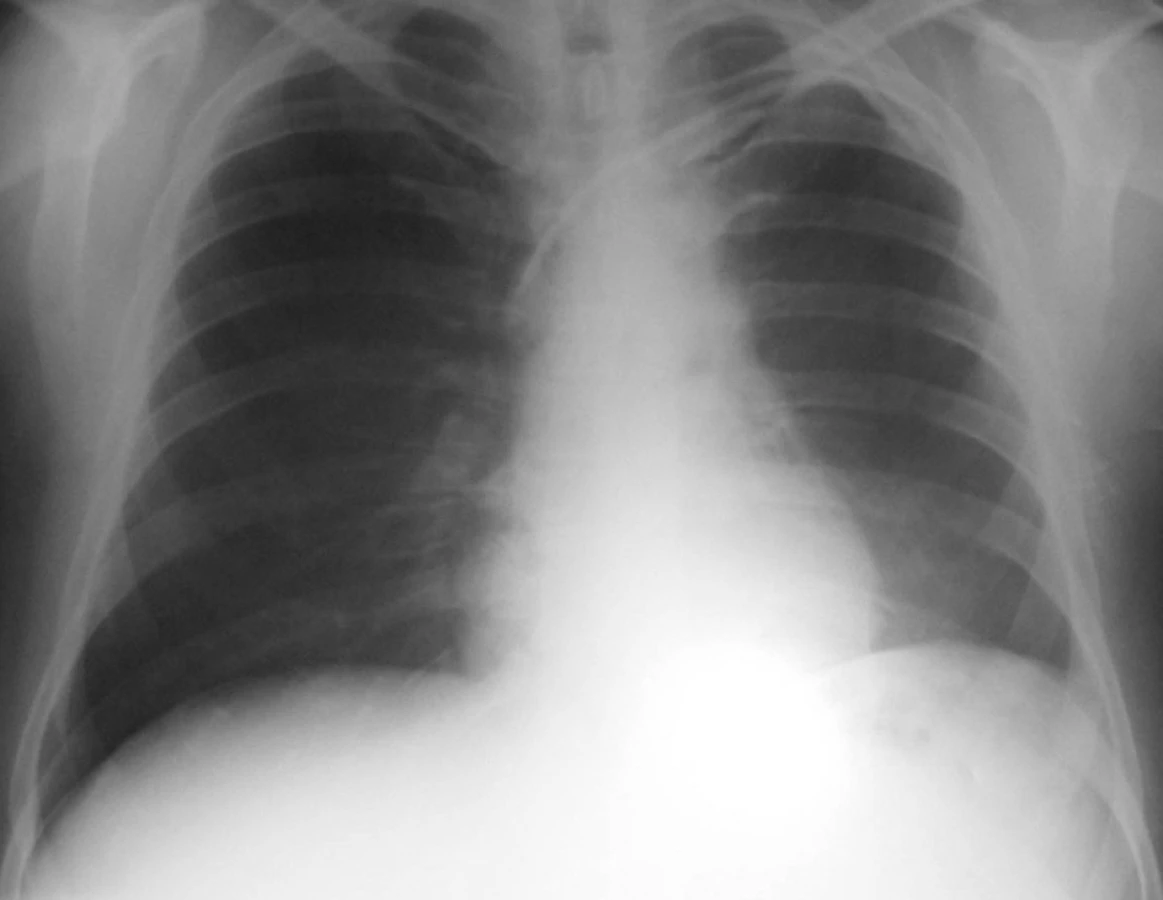
Od září 2004 do prosince 2004 byl pacient stále stabilizován s kreatininem 150–200 μmol/l s normální hodnotou trombocytů bez hemodialýzy a plazmaferéz.
Před Vánocemi roku 2004 se po velké fyzické námaze objevila náhlá aktivace WG a současně i TTP. Došlo již k nezvratnému selhání ledvin s nutností trvalé hemodialýzy a plazmaferéz.
V březnu 2005 byl krátce hospitalizován pro trombózu AV brachiocefalické fistule vpravo, která byla řešená trombektomií. Jinak byl ale klinicky i laboratorně stále stabilizován při hemodialýze 3x týdně a plazmaferézách 1x měsíčně.
Dne 1. 11. 2005 po nekomplikované hemodialýze doma kolaboval, objevila se přechodná porucha řeči. Následující den se stav poněkud vylepšil, takže lékaře nevyhledal. Dne 3. 11. 2005 se opět celkově zhoršil a neudržel se na nohou. Byl převezen na naše pracoviště. Při přijetí byl při vědomí, TK 170/90, konstatována dysfazie, frustní pravostranná hemiparéza, agrafie, dyslexie, dyskalkulie a sporná okohybná centrální porucha.
Diferenciálně diagnosticky byly zvažovány následující stavy: krvácení, vaskulitida při WG, trombopatie při TTP, či kombinace několika příčin. CT mozku vyloučilo krvácení. Magnetická rezonance mozku (obr. 3 a,b) odhalila pokročilou atrofii mozkovou a rozsáhlé změny v mozkové tkáni různého charakteru. Hypersignální izolovaná splývavá ložiska v hloubce bílé hmoty zvláště vpravo temporoparietálně odpovídala vaskulárně ischemickým změnám staršího data. Dále bylo patrné edematózní prosáknutí kortikálních a subkortikálních oblastí s převahou parietálně vlevo. Tyto změny byly hodnoceny jako typické pro postižení mozkové tkáně při vaskulitidě. Laboratorně se tento stav prezentoval trombocytopenií 26 giga/l, vysokou hodnotou LDH 48,1 μkat/l, normálním CRP, negativním nálezem c-ANCA protilátek a CIK 4 arb.j., negativními antikardiolipinovými protilátkami a normálními hodnotami při vyšetření hyperkoagulačních stavů.
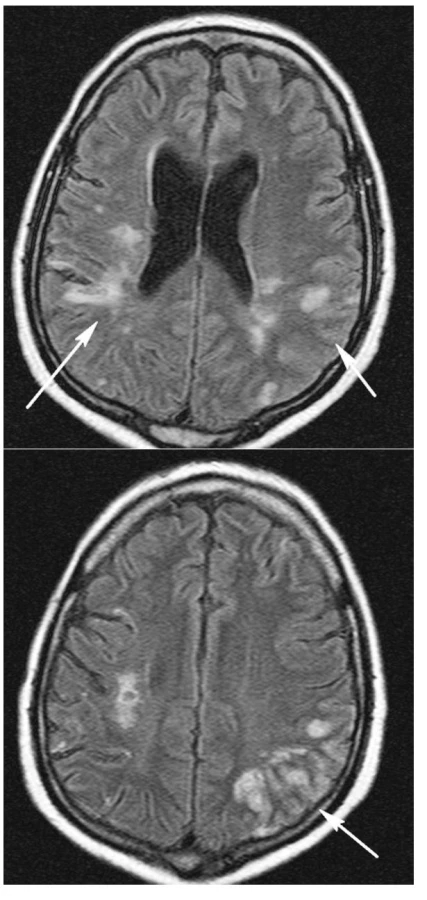
Stav pacienta byl komplikován uzávěrem stávající AV fistule pravé horní končetiny s recidivou trombózy i po její chirurgické revizi. Dále došlo k akutní trombóze ve v. subclavia a v. jugularis vlevo s masivním otokem levé horní končetiny. Současně se ale objevilo krvácení ze všech vpichů a z původní i nové AV fistule a centrálního žilního katétru ve v. jugularis vpravo. Další komplikací léčby byla pak i osteoporóza s akutní kompresivní frakturou L3.
Při komplexní léčbě včetně intenzivní rehabilitace došlo během jednoho roku k úpravě hemiparézy ad integrum. Dosud přetrvává postižení kognitivních funkcí, nicméně dyslexie, dyskalkulie a dysgrafie jsou značně zlepšeny, pacient je schopen péče o sebe sama i běžných životních i právních úkonů. Na kontrolním MR mozku provedeném v prosinci 2006 (obr. 4 a,b) je nápadná regrese postižení mozkové tkáně kortikálně a subkortikálně odpovídající původním změnám při vaskulitidě s demarkací gliových jizev. Ostatní dříve popisované jevy jsou stacionární, odpovídají předpokládaným starším vaskulárně ischemickým změnám.
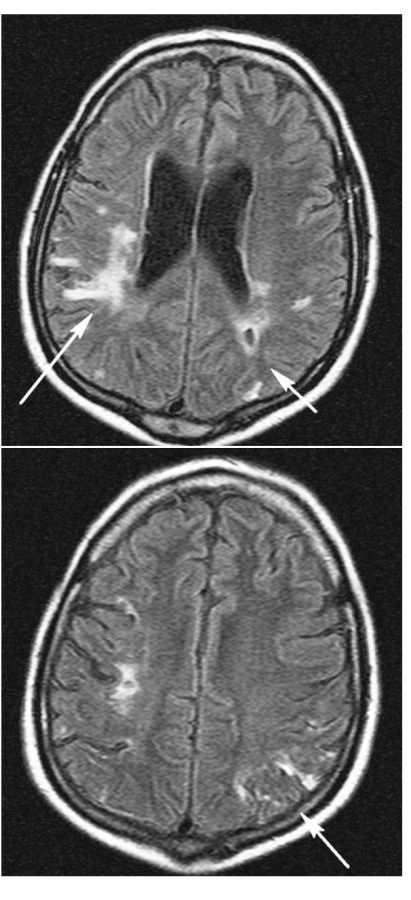
Poslední zaznamenaná terapie zahrnuje: Medrol 4 mg/den, Imuran 25 mg/den, Furorese 125 mg/den, Rocaltrol 0,5 mg/den, Ca carbonicum 1500 mg/den, Biseptol 480 mg /den Helicid 20 mg/den, Atram 12,5 mg/den, hemodialýzy 3x týdně, plazmaferézy každé 4 týdny s případnou úpravou frekvence dle stavu.
DISKUSE
K diskusi se nabízí řada otázek
Předně je to otázka možnosti včas diagnostikovat WG (8–11).V našem případě na počátku onemocnění bylo pouze histologicky podezřelé ložisko v levé plíci. V této fázi onemocnění byla veškerá laboratoř včetně c-ANCA protilátek v normě, resp. negativní. V době objevení se léze brachiálního plexu nejsou c-ANCA protilátky k dispozici, vyšetření provedl chirurg a lézi plexu vztahoval k resekci plíce. Protilátky c-ANCA se již jasně objevují v době manifestace renální insuficience a difuzního plicního procesu. Naopak jsme je nezachytili při klinické manifestaci neurologického postižení.
Zde může být nejspíše důvodů několik. Jednak jde o pacienta s pravidelnou plazmaferézou, proto se nabízí otázka, zda opravdu hodnotíme pacientovu plazmu, resp. sérum. Nevíme na čem všem je závislý poločas imunoglobulinů a patrně ani nevíme za jak dlouho se vytvoří autoprotilátky, o které se při monitoraci léčby těchto pacientů opíráme. Navíc při lokalizovaném a mozkovém postižení a v počátcích choroby (WG) nemusí být c-ANCA detekovatelné. Zdá se tedy, že nelze opírat diagnózu WG pouze o c-ANCA protilátky (2).
Diskutabilní je i otázka užití c-ANCA a vhodnějších PR 3 protilátek k monitoraci aktivity choroby (12). Z našich zkušeností je zde spojitost s klinickou aktivitou renální a respirační, která se ovšem jeví poněkud rychlejší než laboratorní nárůst autoprotilátkové aktivity.
V případě našeho pacienta je dále situace složitější o současně probíhající TTP, která je jistě sekundární záležitostí u autoimunitního onemocnění. I když bylo na tuto možnost diferenciálně diagnosticky pomýšleno a byla včas odhalena, nelze určit skutečný podíl TTP na klinickém stavu, laboratorních hodnotách, ale i projevech odhalených zobrazovacími metodami, jako je např. MR (13). V této souvislosti by pak bylo vhodnější změny CNS odhalené MR vyšetřením hodnotit spíše jako vaskulopatické než pouze vaskulitické.Názory na léčbu TTP jsou také nejednoznačné. Byly zaznamenány relapsy i po opakovaných plazmaferézách i po splenektomii (7) či redukci kortikoidů. Diskutuje se i aplikace rituximabu.
Další diskutabilní otázkou je léčba WG (6, 8, 14–16). Klasická léčba cyklofosfamidem a kortikoidy je jistě na místě u aktivní generalizované formy WG. Naopak nejednotný je názor na léčbu lokalizované formy (17). Zde se odborná veřejnost spíše přiklání k azathioprinu spolu s kortikoidy (18). Literárně se uvádí ale i zkušenosti s MTX (19). Nověji pak se objevují četnější zkušenosti s použitím rituximabu.
Kazuistikou chci připomenout klinickou rozmanitost WG, která si svými diagnostickými úskalími stále více vynucuje nutnost mezioborové spolupráce.
Poděkování si zaslouží MUDr. Jiří Kubále, Radiologické oddělení Nemocnice Č. Budějovice a.s za přípravu a poskytnutí MR dokumentace.
MUDr. Jiřina Vítová
Interní oddělení
Nemocnice Č. Budějovice a.s.
B. Němcové 54
370 87 České Budějovice
e-mail: vitji@centrum.cz
Sources
1. Oimomi M, Suehiro I, Mizuno N, Baba S, Okada S, Kanazawa Y. Wegener’s granulomatosis with intracerebral granuloma and mammary manifestation. Report of a case. Arch Intern Med. 1980 Jun; 140 (6): 853–854.
2. Shuang Ye, Cheng de Yang. How could we make a diagnosis of Wegener’s granulomatosis? Clin Rheumatol 2007; 26 : 784–786.
3. Bartůňková J, Tesař V, Šedivá A. Diagnosis and pathogenetic role of antineutrophil cytoplasmic autoantibodies. Clin Immunol 2003; 106 : 73–82.
4. Bjerkeli V, Damäs JK, Fevang B, Holter JC, Aukrust P, Froland SS. Increased e expression of fractalkine (CX3CL1) and its receptor, CX3CR1 in Wegener’s granulomatosis – possible role in vascular inflammation. Rheumatology (Oxford) 2007 Sep; 46 (9); 1422–1427.
5. Elkon K, Sutherland DC, Rees A, et al. HLA-A antigens of patients with Wegener’s granulomatosis. Tissue Antigens 1978; 11 : 129.
6. Ronco P,Verroust P, Mignon F, Kourilsky O, Vanhille P, Meyrier A, et al. Immunopathological studies of polyarteritis nodosa and Wegener’s granulomatosis: a report of 43 patients with 51 renal biopsies. Q J Med.1983 Spring; 52 (206): 212–223
7. Dervenoulas J, Tsirigotis P, Bollas G, Pappa V, Xiros N, Economopoulos T, et al. Thrombotic thrombocytopenic purpura/hemolytic uremic syndrome (TTP/HUS): treatment outcome, relapses, prognostic factors. A single-center experience of 48 cases Ann Hematol: 2000; 79 : 66–72.
8. Olivencia - Simmons I. Wegener’s granulomatosis: symptoms, diagnosis and treatment. J Am Acad Nurse Pract 2007 Jun; 19 (6): 315–320.
9. Finkielman JD, Lee AS, Hummel AM, Viss MA, Jacob GL, Homburger HA, et al. WGET Research Group. ANCA are detectable in nearly all patients with active severe Wegener’s granulomatosis. Am J Med 2007 Jul; 120(7); 643.e9–14.
10. Ahmed M, Niffenegger JH, Jakobiec FA, Ben-Arie-Weintrob Y, Gion N, Androudi S, et al. Diagnosis of limited ophthalmic wegener granulomatosis: distinctive pathologic features with ANCA test confirmation. Int Ophthalmol Jun 23 (Epub ahead of print).
11. Lidar M, Carmel E, Kronenberg Y, Langevitz P. Hearing loss as the presenting feature of systemic vasculitis. Ann N Y Acad Sci. 2007 Jun; 1107 : 136–141.
12. O’Donnell JL, Hayman MW, Spellerberg MB, McLellan AD, Brooksbank K, Chapman PT, et al. Antineutrophil cytoplasmic antipody measurement: advantages and disadvantages of a capture PR3 ELISA and a direct PR3 ELISA. Pathology 200 Apr; 39(2): 258–263.
13. Murphy JM, Gomez-Anson B, Gillard JH, Antoun NM, Cross J, Elliot JD, et al. Wegener granulomatosis, MR imaging findings in brain and meninges. Radiology 1999 Dec; 213 (3): 3 : 794–799.
14. Kuross S, Davin T, Kjellstrand CM. Wegener’s granulomatosis with severe renal failure; clinical course and results of dialysis and transplantation. Clin.Nephrol 1981 Oct; 16 (4):172–180.
15. Hoffmann GS, Kerr GS, Leavitt RY, Hallahan CW, Lebovics RS, Travis WD, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; Mar 15; 116 (6): 488–498.
16. Hoffmann GS, Leavitt RY, Fleisher TA, Minor JR, Fauci AS. Treatment of Wegener’s granulomatosis with intermittent high-dose intravenous cyclophosphamide. Am J Med 1990 Oct; 89 (4): 403–410.
17. Specks U, DeRemee RA. Granulomatous vasculitis. Wegener’s granulomatosis and Churg-Strauss syndrome. Rheum Dis Clin North Am 1990 May; 16 (2): 377–397.
18. Haubitz M. ANCA – associated vasculitis: diagnosis, clinical characteristics and treatment. Vasa 2007 May; 36 (2): 81–89.
19. Villa-Forte A, Clark TM, Gomes M, Carev J, Mascha E, Karafa MT, et al. Substitution of methotrexate for cyclophosphamide in Wegener granulomatosis: a 12 - year single-practice experience. Medicine (Baltimore) 2007 Sep; 86 (5): 269–277.
Labels
Dermatology & STDs Paediatric rheumatology RheumatologyArticle was published in
Czech Rheumatology

2008 Issue 2
Most read in this issue
- Ošetření extenzorů zápěstí
- Wegenerova granulomatóza s trombotickou trombopenickou purpurou – neurologické projevy
- Skórovací systémy při hodnocení progrese revmatoidní artritidy
- Výsledky volumetrického měření patologických ložisek mozku u nemocných se systémovým lupus erythematodes