
Novinky v biologické terapii revmatoidní artritidy a výhled do budoucna
News in the biological therapy of rheumatoid arthritis and future prospects
In recent years, up-to-date knowledge of pathogenetic mechanisms in rheumatoid arthritis contributed to establishment of biological therapy in clinical practice. For patients non-responsive to traditional disease modifying antirheumatic drugs, there are TNF-α inhibitors available. In case of their contraindication, lack of effect or adverse events, the treatment with B-lymphocyte depleting agent (rituximab) or with a drug blocking T-lymphocyte costimulation (abatacept) has been approved in rheumatoid arthritis. The progress in biotechnology manufacturing contributed to the development of several other perspective agents which may form the basis for the immunotherapy of rheumatoid arthritis in the near future. For instance, new or modified TNF-α inhibitors (golimumab, certolizumab pegol), and new monoclonal antibodies against other cytokines (e.g. IL-1, IL-6, IL-12, IL-15, IL-17, IL-23), and agents directed at B-lymphocyte depletion (e.g. ocrelizumab, ofatumumab) are in various stages of development. Many pharmaceutical companies put great expectations in small molecules with possible peroral administration. Nowadays, more than a half of new anti-inflammatory agents in the preclinical and clinical trials are represented by small molecules, which are recognised as potentially very promising drugs in rheumatoid arthritis. In most cases, theese are inhibitors of proteins which mediate the signalling and transcription of proinflammatory genes inside the cells. The increasing numbers of cytokine inhibitors and modulators of immune processes, which are in the current clinical trials, will further expand the spectrum of efficient therapies for patients with rheumatoid arthritis in the future.
Key words:
rheumatoid arthritis, biological therapy, clinical trials, monoclonal antibodies, small molecules
:
L. Šenolt; J. Vencovský
:
Revmatologický ústav a 1. LF UK, Praha
:
Čes. Revmatol., 17, 2009, No. 1, p. 36-49.
:
Overview Reports
Nové poznatky patogenetických mechanismů revmatoidní artritidy přispěly v posledních letech k zavedení biologické terapie do klinické praxe. Pro pacienty refrakterní k základním lékům modifikujícím průběh choroby jsou dostupné inhibitory TNF-α. Při jejich kontraindikaci, nedostatečném účinku nebo nežádoucích projevech je pro léčbu revmatoidní artritidy schválena terapie blokující B-lymfocyty (rituximab) nebo T-lymfocyty (abatacept). Pokrok v biotechnologii výroby přispěl k rozvoji několika dalších perspektivních léků, které se mohou stát základem imunoterapie revmatoidní artritidy. Jedná se například o nové nebo modifikované inhibitory TNF-α (golimumab, certolizumab pegol) a nové monoklonální protilátky proti jiným cytokinům (např. IL-1, IL-6, IL-12, IL-15, IL-17, IL-23) a léky zaměřené na depleci B-lymfocytů (např. ocrelizumab, ofatumumab). Většina farmaceutických firem se zabývá vývojem tzv. malých molekul s možností perorální aplikace. V současné době představuje více než polovina nových protizánětlivých preparátů ve stadiu preklinických a klinických studií právě malé molekuly, které se považují za potenciálně velmi nadějné léky revmatoidní artritidy. Jsou to především inhibitory bílkovin, které uvnitř buněk zprostředkovávají přenos signálu a transkripci prozánětlivých genů. Předpokládá se, že rostoucí počet inhibitorů cytokinů nebo imunomodulačních pochodů a malých molekul, které jsou ve stadiu klinického výzkumu v budoucnu rozšíří spektrum účinné terapie pro pacienty s revmatoidní artritidou.
Klíčová slova:
revmatoidní artritida, biologická léčba, klinické studie, monoklonální protilátky, malé molekuly
Úvod
Revmatoidní artritida (RA) je chronické zánětlivé autoimunitní onemocnění, které vede k destruktivnímu postižení kloubů, ireverzibilním deformitám a funkčnímu omezení. Méně časté jsou systémové projevy, zvýšené je také riziko kardiovaskulárních onemocnění. Pokud pacienti nejsou včas a dostatečně léčeni, snižuje se výrazně kvalita života a zkracuje se průměrná doba přežití. Přestože není přesně známa příčina vzniku onemocnění, dochází v posledních letech k významné změně ve strategii léčby RA. Důraz se klade na časnou léčbu, běžná je kombinace více léků a mezníkem v revmatologii se stala biologická terapie. Její vznik umožnily nové poznatky o patogenezi RA, které přispívají k rozšiřování spektra účinné terapie. Centrální úloha tumor nekrotizujícího faktoru (TNF)-α v průběhu patogeneze RA vedla k úspěšnému zavedení terapie blokující TNF-α do klinické praxe. Tato léčba tlumí převážně aktivitu monocytů. Centrem pozornosti se v poslední době stala léčba zaměřená také proti B-lymfocytům a T-lymfocytům. Léčba je tak cíleně zaměřena proti molekulám nebo buňkám, které jsou specifické pro pochody spojené s patogenezí RA. Cílem tohoto sdělení je poskytnout přehled o potenciálně nových terapeutických cílech pro revmatoidní artritidu.
Etiopatogeneze revmatoidní artritidy
Synoviální tkáň je v průběhu RA prostoupena řadou buněk. Ty se v průběhu zánětu zachytí na povrchu aktivovaných endoteliálních buněk cév, které exprimují řadu adhezivních molekul. Další jejich pohyb je v synoviální tkáni řízen chemotakticky. Buňky vrozeného, evolučně staršího imunitního systému – granulocyty, makrofágy a dendritické buňky jsou aktivovány prostřednictvím specifických receptorů PRRs (pathogen recognition receptors), které váží struktury charakteristické pro patogenní mikroorganismy (PAMPs, pathogen-associated molecular pattern) nebo signály uvolňované z aktivovaných či apoptotických buněk (1). Následně jsou aktivovány buňky získaného, antigenně specifického, imunitního systému. Předpokládané „artritogenní“ peptidy jsou předkládány T-lymfocytům buňkami prezentujícími antigen, jakými jsou makrofágy, dendritické buňky nebo aktivované B-lymfocyty. T-lymfocyty rozpoznávají antigen v komplexu s HLA molekulou II. třídy na buňce prezentující antigen. K tomu, aby byly T-lymfocyty plně aktivovány, je třeba interakce mezi kostimulačními molekulami CD80 nebo CD86 na buňkách prezentujících antigen a CD28 na T-lymfocytech, tzv. kostimulační signál. Tento signál může být blokován molekulou CTLA4 (cytotoxic T-lymphocyte antigen 4), která má větší afinitu ke kostimulačním molekulám CD80/D86 než jakou má CD28 a slouží k inhibici nadměrné aktivace T-lymfocytů (2).
Přibližně 80 % RA pacientů představuje nosiče takzvaného sdíleného epitopu, což je krátký úsek aminokyselin vazebného místa HLA molekuly (4). Sdílený epitop je charakteristický pro některé HLA-DR geny (např. HLA-DR4 a HLA-DR1) a je pravděpodobně schopen prezentovat „artritogenní“ peptid T-lymfocytům a spustit tak imunitní reakci. Nedávno byla vyslovena hypotéza, že sdílený epitop je nezbytný k prezentaci specificky modifikovaných – citrulinovaných peptidů. Výsledkem je imunologická odpověď s tvorbou protilátek proti citrulinovaným peptidům (ACPA) (5). Hypotéza sdíleného epitopu je umocněna dokladem o významné asociaci mezi sdíleným epitopem a RA pouze u pacientů s pozitivitou ACPA, které jsou tímto odpovědné za asociaci sdíleného epitopu s prognosticky závažnější formou RA (6). V klinické praxi to znamená vztah mezi vyššími hladinami ACPA na počátku onemocnění a větším rizikem závažnějšího průběhu s rychlejší rentgenovou progresí (7). RA je pravděpodobně polygenní onemocnění a patogenetický význam byl popsán pro řadu dalších genů, například jednonukleotidový polymorfismus molekul PTPN22, nebo geny pro PADI4, TRAF1-C5 a STAT4 a možná i CTLA4 (8, 9). Důležitým předpokladem spuštění imunitní reakce a vzniku RA mohou být rizikové faktory zevního prostředí jakými jsou stres, kouření nebo infekce u geneticky vnímavých jedinců. Nedávno bylo zjištěno, že kouření může urychlit tvorbu citrulinovaných peptidů a zvýšit tak riziko vzniku ACPA pozitivní RA u nemocných s přítomností HLA-DRB1 sdíleného epitopu (10).
Revmatická synoviální tkáň je infiltrována aktivovanými T-lymfocyty, převážně CD4+ pomocnými (Th) lymfocyty. U geneticky vnímavých jedinců dochází působením některých cytokinů, např. interleukinu (IL)-12, k relativní převaze Th1 lymfocytů a jimi produkovaných cytokinů IL-2 a interferonu (IFN)-γ (11). Pod vlivem dalších cytokinů s převahou IL-23 může docházet k diferenciaci směrem k nově popsanému subsetu paměťových T-lymfocytů – Th17 (12). Th17 lymfocyty pak zvýšeně produkují zejména cytokin IL-17. Jedná se o cytokin s pleiotropními účinky na řadu buněk ve smyslu zvýšené tvorby prozánětlivých cytokinů (IL-1, IL-6, TNF-α apod.) a indukce osteoklastogeneze. Současně dochází k poklesu Th2 a regulačních lymfocytů Tregs CD4+ CD25+. Tím se vysvětluje nedostatečná inhibice prozánětlivé aktivity ostatních T-lymfocytů v revmatické synoviální tkáni. Vedle T-lymfocytů jsou v patologické kloubní výstelce aktivovány monocyty/makrofágy, B-lymfocyty, dendritické buňky, ale také rezidentní buňky kloubu – synoviální fibroblasty, chondrocyty a osteoklasty (13, 14). Aktivované makrofágy tvoří např. IL-1, IL-6 a TNF-α. Tyto prozánětlivé cytokiny se již v současné době staly terapeutickým cílem. Makrofágy zvýšeně produkují také IL-15, IL-18, IL-32 a další mediátory včetně chemotaktických molekul (15). Aktivované monocyty a makrofágy v synoviální tkáni mohou vlivem ligandu pro receptor aktivující transkripční faktor NF-κB (RANKL) diferencovat v osteoklasty, které vedou ke vzniku kostních erozí (16). B-lymfocyty mají schopnost diferencovat v plazmatické buňky produkující patogenní autoprotilátky (např. RF, ACPA). Ty mohou vytvářet imunitní komplexy a vést tak k aktivaci zánětlivých buněk a tvorbě prozánětlivých cytokinů. B-lymfocyty mohou fungovat také jako buňky prezentující antigen, mohou aktivovat T-lymfocyty a podporovat autoimunitní proces (14). Aktivovány jsou také synoviální fibroblasty, které si navíc mohou zachovávat invazivní potenciál nezávisle na přítomnosti imunitních buněk a prozánětlivých cytokinů (13). Výsledkem je proliferace a hyperplazie synoviální intimy, v intersticiu synoviální tkáně jsou aktivované endotelové buňky a dochází k angiogenezi a tvorbě lymfoidních folikulů. Vzniká tak agresivní synoviální tkáň, tzv. panus, který infiltruje kloubní struktury a navozuje postupně jejich destrukci.
Perspektivní biologické léky
Biologická léčba představuje významný milník v terapii revmatoidní artritidy refrakterní ke standardní léčbě DMARDs. Jedná se o vysoce účinnou terapii, která snižuje klinickou aktivitu, významně zpomaluje progresi onemocnění a zlepšuje kvalitu života. V současné době existuje v České republice 5 registrovaných biologických léků (tab. 1). Jedná se o tři monoklonální protilátky blokující TNF-α, protilátku navozující depleci B-lymfocytů a lék inhibující kostimulaci (aktivaci T-lymfocytů). Biologická léčba může být na druhé straně provázena některými nežádoucími projevy (např. infekce, tumory, kardiální insuficience, demyelinizační onemocnění, hepatopatie), někteří pacienti nemusí odpovídat na léčbu dostatečně, mohou klinickou odpověď v průběhu léčby ztratit nebo na léčbu neodpovídají vůbec. Navíc u některých nemocných dojde po přerušení léčby k opětovnému vzplanutí nemoci. Proto je snahou farmaceutických firem modifikovat stávající léky nebo vývoj dalších biologických preparátů a léčebných strategií ve snaze zlevnit technologii výroby a zlepšit klinickou účinnost a případně navodit úplnou remisi onemocnění. Biologické léky, které jsou ve stadiu klinického zkoušení lze jednoduše rozdělit na monoklonální protilátky a malé molekuly (17). Ty v současné době nejvíce nadějné jsou shrnuty v tabulce 2.

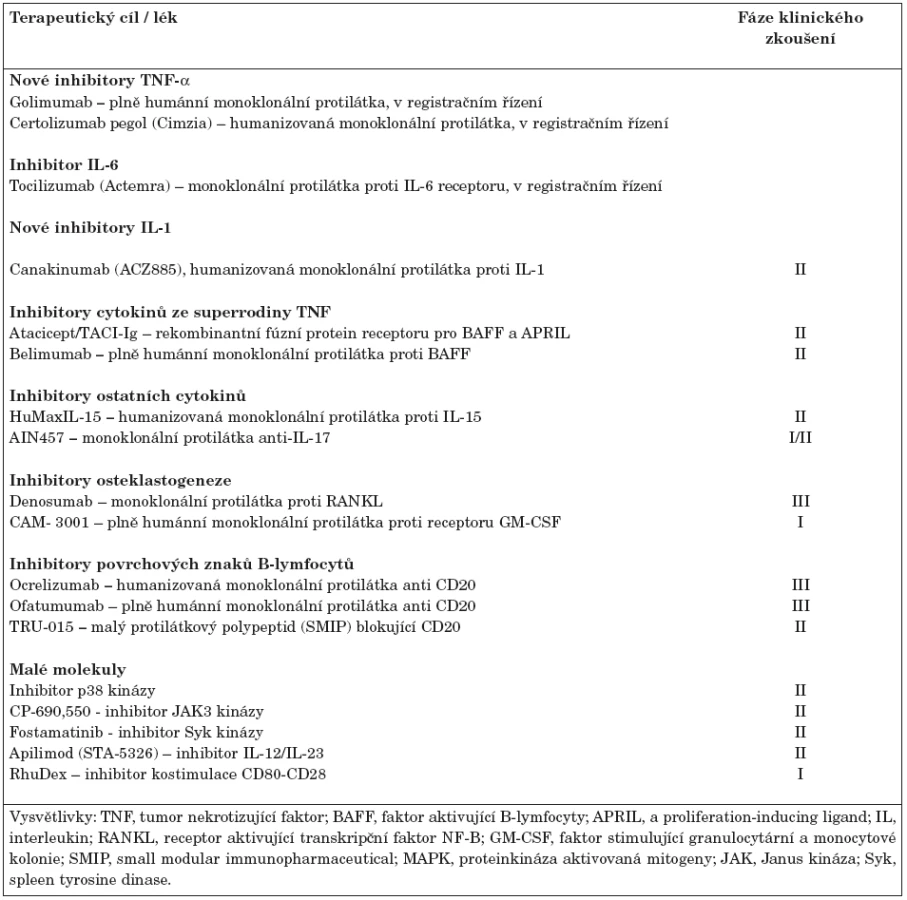
1. Monoklonální protilátky proti cytokinům a jejich receptorům
V revmatologii se v posledních letech stále více začala prosazovat terapie pomocí monoklonálních protilátek. Zpočátku se jednalo o protilátky chimerické, kde konstantní části lehkého a těžkého řetězce jsou lidské a variabilní oblasti odpovědné za vazbu na cílovou molekulu jsou myší. Snížení imunogenicity přinesly pokroky v technologii výroby a vznikly protilátky humanizované a nakonec i plně humánní. Monoklonální protilátky již představují základ imunoterapie některých revmatických onemocnění. První generace protilátek byly zaměřeny proti solubilním faktorům (cytokinům a receptorům), další proti molekulám na povrchu buněk. Mechanismus jejich působení tedy představuje neutralizaci cílového cytokinu nebo jeho receptoru, blokádu kostimulačních molekul a navození cytolýzy, apoptózy nebo snížení proliferace cílové buňky.
Inhibitory TNF-α byly pro léčbu RA registrovány v USA v roce 1999 a o rok později v Evropě. V současné době jsou dostupné tři preparáty: infliximab, chimerická monoklonální protilátka, adalimumab, plně humánní monoklonální protilátka a etanercept, solubilní receptor pro TNF-α. TNF-α inhibitory v kombinaci s metotrexátem podstatným způsobem zabraňují strukturální progresi, významně snižují aktivitu nemoci a zlepšují kvalitu života jedinců s RA. Na druhé straně se jedná o léčbu finančně nákladnou, přičemž stále většina pacientů nedosáhne klinické remise onemocnění. Proto je snahou farmaceutických firem modifikace současně používaných TNF-α inhibitorů za účelem jejich zefektivnění a zjednodušení výroby. V registračním řízení jsou další dva TNF-α inhibitory. Jedná se o golimumab(CNTO148), plně humánní monoklonální protilátku, která se podává v dávce 50–100 mg subkutánně každé 4 týdny. Předběžná data ukazují na stejnou účinnost i profil nežádoucích projevů jako u současně dostupných TNF-α inhibitorů (18). Dalším subkutánním inhibitorem TNF-α je humanizovaná monoklonální protilátka, certolizumab pegol(CDP870, cimzia), která významně snižuje aktivitu a zpomaluje rentgenovou progresi nemoci (19). Jedná se o Fab fragment anti-TNF-α, který je navázán na polyethylenglykol. Vazba na nosič významně prodlužuje biologický poločas a zlepšuje biologickou dostupnost monoklonální protilátky. Výhodou by měla být nižší imunogenicita a levnější technologie výroby.
Existují i snahy o lokální podání TNF neutralizující látky, což se děje v rámci postupů genové terapie, jdoucí napříč medicínskými obory. V poslední době je největší nadějí rekombinantní adeno asociovaný virus (AAV), který nese zároveň část genetické informace, kterou po inkorporaci do nitra buňky tato začne využívat a tvořit příslušný protein. V konkrétním případě u revmatoidní artritidy byl v iniciálním klinickém hodnocení fáze I/II do kloubu vpraven AAV sérotypu 2, který byl geneticky pozměněn tak, aby obsahoval gen kódující pro TNF receptor spojený s Fc částí IgG1, tedy vlastně etanercept. Látka se nazývá tgAAC94 a byla použita u 127 nemocných s RA. Hodnocení bylo načas zastaveno, protože došlo k jednomu úmrtí z důvodů disseminované histo-plazmózy, nicméně toto nakonec nebylo přisouzeno podávané léčbě. Výsledky jsou zpracovávány a předběžně ukazují na jinak velmi dobrý bezpečnostní profil a známky efektivity (http://ir.targen.com).
Inhibitory IL-1. Anakinra, rekombinantní antagonista receptoru pro IL-1 (IL-1Ra), vykazuje slabší léčebný efekt než inhibice TNF-α a je registrována ve Spojených státech amerických i v Evropě (20), ale v České republice není v současné době k dispozici. Byla zkoušena i plně humánní monoklonální protilátka proti receptoru IL-1 (AMG108), nicméně výsledky II. fáze klinického zkoušení nebyly příliš povzbudivé. Za účelem jednoduššího dávkování, snížení rizika lokálních nežádoucích reakcí, vyšší afinity k IL-1 a tudíž i lepší účinnosti než má anakinra byla nedávno byla vyvinuta humanizovaná monoklonální protilátka proti samotnému IL-1, canakinumab(ACZ885). Nadějné jsou první výsledky klinického zkoušení u pacientů se systémovou formou juvenilní idiopatické artritidy (21). V roce 2008 byla zahájena II. fáze klinického zkoušení u pacientů s RA (http://clinicaltrials.gov).
Inhibitory IL-6. Humanizovaná monoklonální protilátka proti receptoru pro IL-6 tocilizumab představuje velmi perspektivní preparát pro léčbu aktivní RA. IL-6 totiž představuje pleiotropní cytokin, který je schopen aktivovat řadu imunitních a rezidentních buněk synoviální tkáně a představuje zároveň důležitý faktor podporující tvorbu reaktantů akutní fáze v játrech. V současné době probíhá III. fáze klinického zkoušení u pacientů s aktivní RA. Výsledky několika studií poukazují na podstatné zlepšení nejen symptomů a příznaků, ale také zpomalení rentgenové progrese onemocnění a zlepšení funkčních schopností a kvality života jedinců s RA (22, 23). Tocilizumab byl klinicky vyvinut pro léčbu RA, juvenilní idiopatické artritidy, zejména její systémové formy, Crohnovy choroby, apod.. Podává se formou infuzí každé 4 týdny. Nejlepších výsledků bylo dosaženo při podání 8 mg/kg v kombinaci s MTX. V současné době probíhá registrační řízení. To se ale může protáhnout kvůli žádosti Amerického úřadu pro kontrolu potravin a léčiv (FDA), který si vyžádal další testy z důvodu doprovodných nežádoucích projevů ve smyslu elevace transamináz, hypercholesterolemie a postižení trávicího traktu. V roce 2008 byla zahájena II. fáze klinického zkoušení subkutánní monoklonální protilátky anti-IL-6 (CNTO 136) zjišťující vhodnou dávku a potvrzující účinnost a bezpečnost (http://clinicaltrials.gov).
Inhibitor IL-15. IL-15 je tvořen makrofágy, dendritickými buňkami, B-lymfocyty, synoviálními fibroblasty a může ovlivňovat řadu buněk imunitního systému (24). Blokování tohoto cytokinu vedlo na experimentálním modelu artritidy k podstatné redukci destrukce chrupavky a kosti. První otevřená, placebem kontrolovaná, dvojitě zaslepená studie na třiceti pacientech prokázala obdobnou účinnost humanizované monoklonální protilátky proti IL-15 (HuMaxIL-15) jakou mají TNF-α inhibitory (25). Proběhla již také II. fáze klinického zkoušení plně humánní monoklonální protilátky anti-IL15 (AMG 714) (http://clinicaltrials.gov). Zatím chybí bližší data o účinnosti této léčby na větším souboru pacientů. Shodnou receptorovou podjednotkou γ řetězce a tudíž některé funkce s IL-15 mají například cytokiny IL-2, IL-4, IL-7, IL-9 nebo IL-21. Na experimentální úrovni byla úspěšně zkoušena monoklonální protilátka proti alfa řetězci receptoru pro IL-2 (26). Rekombinantní monoklonální protilátky proti receptoru IL-2 (daclizumab a basiliximab) našly své uplatnění v transplantologii, ale chybí údaje o jejich terapeutickém uplatnění u pacientů s RA. Významné postavení v patogenezi zánětu a destruktivního procesu při RA mají cytokiny IL-7 a IL-21. Pozitivní výsledky jejich inhibice na experimentální úrovni jsou základem pro další nadějná klinická zkoušení (27, 28).
Inhibitor IL-17. IL-17 představuje klíčový produkt Th17 lymfocytů, ovlivňuje expresi několika prozánětlivých cytokinů a jeho inhibice je účinná v léčbě experimentálního modelu artritidy (29). Probíhá I./II. fáze klinického zkoušení podávání monoklonální protilátky anti-IL-17 (AIN457) nemocným s RA. Výsledky studie jsou v brzké době očekávané (http://clinicaltrials.gov).
Inhibitor IL-12/IL-23. Recentní klinická studie hodnotící účinek specifické subkutánně aplikované monoklonální protilátky proti podjednotce p40 cytokinů IL-12 a IL-23 - ustekinumab měla velmi dobré výsledky u pacientů s psoriázou (30). IL-23 je asociován se zánětlivými chorobami jakými jsou například nespecifické střevní záněty nebo revmatoidní artritida a to zejména pro stimulující účinek na Th1 imunitní odpověď (31). Cytokin IL-23 má jistou podobnost s IL-12, navíc je zodpovědný za proliferaci nově popsaného subsetu paměťových T-lymfocytů - Th17 buněk, které zvýšeně produkují zejména cytokin IL-17, ale také IL-6, TNF-α, GM-CSF a IL-22. Na experimentální úrovni byl popsán pozitivní účinek protilátky blokující IL-23 u kolagenem indukované artritidy ve smyslu potlačení zánětlivých a destruktivních změn (32), což bylo pozorováno již před více než 10 lety také při blokování IL-12 (33). Na druhé straně recentní práce ukazuje na nepřítomnost bioaktivní molekuly IL-23 v RA synoviální tkáni a téměř nedetekovatelné hladiny v synoviální tekutině a séru pacientů s RA (34). Jestli bude mít inhibice IL-12/IL-23 praktický význam v terapii RA jako se zdá, že může mít u jiných zánětlivých stavů nebo na myším modelu artritidy, ukáží právě zahajované klinická zkoušení.
Inhibitory cytokinů ze superrodiny IL-1 a TNF. V patogenezi RA hraje důležitou úlohu řada dalších cytokinů (35). Z cytokinů řazených do superrodiny IL-1 byl na experimentálním modelu artritidy pozorován pozitivní účinek při blokování např. IL-18, IL-32 nebo IL-33 (35). Pozornost řady odborníků se obrátila k členům superrodiny TNF, zejména k lymfotoxinu-beta, molekulám BAFF (neboli BLyS, B-lymphocyte stimulator) a APRIL. Ve II. fázi klinického zkoušení u RA byl nedávno baminercept(BG9924), inhibitor lymfotoxinu beta, cytokinu, který je tvořen lymfocyty a hraje zásadní úlohu během vývoje germinálních center v revmatické synoviální tkáni (36). Analýza data však nepotvrdila dostatečnou účinnost (http://clinicaltrials.gov). Aktivované buňky myeloidního původu a synoviální fibroblasty tvoří faktory BAFF (B-cell activating factor) a APRIL (a proliferation-inducing ligand) podporující maturaci, proliferaci a oddalující apoptózu B-lymfocytů (37). Atacicept představuje rekombinantní fúzní protein receptoru pro BAFF a APRIL (TACI-Ig). Výsledky I. fáze klinického zkoušení ukazují dobrý efekt na humorální systém s poklesem periferních B-lymfocytů, RF a ACPA protilátek. Během tří měsíčního sledování byl pozorován na malém souboru nemocných pozitivní klinický účinek, nicméně nebyl provázen významným poklesem reaktantů akutní fáze (38). Belimumabje plně humánní monoklonální protilátka proti BAFF, která snižuje humorální i klinickou aktivitu u pacientů se systémovým lupus erythematodes (SLE), ale výsledky II. fáze klinického zkoušení u pacientů s RA nejsou zatím příliš povzbudivé (39).
Inhibitory osteoklastogeneze. V poslední době je veliký zájem o ovlivnění molekul regulujících osteoklastogenezi. Zkoušena je např. protilátka proti faktoru stimulujícímu kolonie monocytů (M-CSF). Předpokladem je fakt, že blokování účinku M-CSF může snižovat diferenciaci osteoklastů a tím potažmo destrukci kloubu (35). V Německu nyní probíhá I. fáze klinického zkoušení plně humánní monoklonální protilátky proti receptoru pro faktor stimulující granulocytární a monocytové kolonie (GM-CSF) (CAM - 3001) (http://clinicaltrials.gov). Dobrý efekt tohoto přístupu podporuje experimentální model, kde blokování GM-CSF významně snižuje otok kloubu a destrukci chrupavky (29). Navíc v kombinaci s protilátkou anti-IL-17 byl efekt léčby ještě výraznější. Ve III. fázi klinického zkoušení u postmenopauzální osteoporózy je účinný inhibitor osteoklastogeneze, denosumab, monoklonální protilátka proti RANKL (40). Subkutánní podání denosumabu po 6 měsících vedlo po jednom roce k významnému zpomalení rentgenové progrese u pacientů s aktivní RA (41). Léčba byla dobře tolerována, nicméně nebyl pozorován větší efekt na aktivitu nemoci.
Inhibitory chemokinů. Chemokiny jsou malé molekuly zvýšeně exprimované RA synoviální tkání, které jsou odpovědné za migraci leukocytů do míst zánětu. Na experimentální úrovni vedlo blokování několika specifických chemokinů nebo jejich receptorů ke snížení klinické aktivity artritidy (42). V preklinické fázi byl například úspěšně zkoušen inhibitor CCL-2/MCP-1 (monocyte chemoattractant protein-1) (43). Bohužel, časné fáze klinických zkoušení na pacientech s aktivní RA dostatečný klinický účinek monoklonální protilátky proti CCL-2/MCP-1 (44) nebo protilátky proti jeho receptoru CCR-2 (45) neprokázaly. Na adjuvantním modelu artritidy byly také úspěšně zkoušeny další inhibitory chemokinových receptorů, například CXCR3 (46) nebo CCR6 (47). Zejména posledně jmenovaná monoklonální protilátka významně snižovala migraci artritogenních Th17 pozitivních lymfocytů do míst zánětu. Významné potlačení artritidy na experimentální úrovni bylo pozorováno po blokování chemokinových receptorů CCR1 a CCR5, které váží chemokiny CCL3 (macrophage inflammatory protein 1α, MIP-1α) a CCL5 (RANTES) (48).
Inhibitory angiogeneze. V časné fázi RA se uplatňuje novotvorba cév, která umožňuje chemotaktickou migraci zánětlivých buněk do synoviální tkáně. Na tomto procesu se podílí řada chemokinů a prozánětlivých cytokinů, z niž asi nejdůležitější je vaskulární endoteliální růstový faktor (VEGF) (49). Monoklonální protilátka proti VEGF, Bevacizumab (Avastin), našla již své uplatnění v terapii několika maligních onemocnění a významně tak rozšířila léčebné možnosti v onkologii (50). V preklinické fázi bylo nedávno prokázáno profylaktické působení monoklonální protilátky proti VEGF beta u myšího modelu (51). Pokud ale byla protilátka podána až v průběhu etablované choroby, artritida ovlivněna významně nebyla.
Ovlivnění apoptózy. Revmatická synoviální tkáň je infiltrována mnoha zánětlivými buňkami a synoviálními fibroblasty. Tyto buňky jsou aktivovány a mají narušený mechanismus apoptózy – programovaného zániku buněk. Současná biologická terapie je zaměřena proti makrofágům (TNF inhibitory), T-lymfocytům (abatacept) a B-lymfocytům (rituximab), přičemž aktivované synoviální fibroblasty mohou této léčbě unikat. Lze tak předpokládat, že aktivované synovialocyty mohou být v některých případech důvodem nedostatečné účinnosti současné léčby. Jednou z možností jak tyto buňky ovlivnit je navození apoptózy, která může být teoreticky indukována aktivací tzv. receptorů smrti (FAS, TRAIL-R a TNF-R) nebo ovlivněním apoptotických molekul (52, 53). Rezistence buněk revmatické synovie k apoptóze je vysvětlována působením prozánětlivého prostředí na zvýšenou tvorbu anti-apoptotických molekul FLIP (Fas-associated death domain-like interleukin (IL)-1beta-converting enzyme-inhibitory protein), sentrin a transkripčních faktorů NF-κB, Stat-3, p53 nebo signální dráhy PI3K/AKT (52, 53). Invazivní charakter RA synoviálních fibroblastů může navíc podporovat nedostatečná exprese tumor supresorového proteinu zvaného PTEN synoviální tkání v místech kloubní invaze, což pravděpodobně umožňuje delší přežívání agresivních fibroblastů (54). Nedávno bylo na experimentálním modelu artritidy pomocí genového inženýrství poprvé prokázáno, že stimulace exprese PTEN dokáže zlepšit aktivitu nemoci (55). Již před více než 10 lety byla na „in vivo“ modelu artritidy představena nová strategie léčby RA procesem apoptózy navozené vazbou na receptor FAS (56). V návaznosti na to byla nedávno vyvinuta první monoklonální protilátka anti-FAS IgM (ARG098), která v současné době podstupuje I. fázi klinického zkoušení u pacientů s RA. Preparát se podává v jednorázové intraartikulární aplikaci do aktivního kolenního kloubu (http://clinicaltrials.gov). Předběžné výsledky studie by mohly být známy koncem roku 2009.
Inhibitory T-lymfocytů. CD4+ T-lymfocyty tvoří většinu buněk lymfocytárního infiltrátu revmatické synoviální tkáně a jsou považovány za klíčové buňky regulující mnoho mechanismů patogeneze RA (11). Klinické studie zaměřené na depleci T-lymfocytů nebyly dosud příliš úspěšné. Příkladem neúspěšného preparátu je alemtuzumab (anti-CD52, Campath-1H), který neprokázal dobrý klinický účinek a byl naopak provázen řadou závažných nežádoucích reakcí spojených s těžkou CD4+ T-lymfopenií a vyrážkou (57). Výhodou nebyla ani parciální deplece CD4 lymfocytů, kterou navozují chimerické monoklonální protilátky proti CD4, keliximab nebo IgG4 verze předchozí protilátky clenoliximab(58, 59). Pro časté kožní vyrážky bylo klinické zkoušení těchto preparátů také přerušeno. Předpokládá se, že ovlivnění znaku CD4 může postihovat také regulační T-lymfocyty, což by mohlo vysvětlit nedostatečnou účinnosti anti-CD4 terapie. Firma Genetech vyvinula nedávno humanizovanou monoklonální protilátku anti-CD4 (MTRX1011A), která blokuje aktivaci T-efektorových buněk, přičemž ale nepostihuje T-regulační buňky. V současné době se nabírají RA pacienti do první fáze klinického zkoušení (http://clinicaltrials.gov). Úspěšnější je preparát snižující aktivaci a proliferaci T-lymfocytů pomocí ovlivnění kostimulace. Jedná se o abatacept, fúzní protein tvořený extracelulární doménou humánního CTLA4, který má vyšší afinitu ke kostimulačním molekulám antigen prezentujících buněk než má molekula CD28. Po vazbě CTLA4-Ig na CD80/86 je tak blokována přirozená cesta kostimulace CD80/86-CD28 a nedojde tak k aktivaci T-lymfocytů. Abatacept je podáván intravenózně v týden 0, 2, 4 a poté každý 4. týden. Použití abataceptu pro léčbu RA bylo v Evropě schváleno v polovině roku 2007, léčba je vyhrazena pro pacienty refrakterní alespoň k jednomu TNF-α inhibitoru. V USA je abatacept schválen pro použití i jako první biologický lék po selhání jednoho nebo více DMARDs. Klinické studie prokázaly velmi dobrou klinickou účinnost abataceptu u aktivních RA pacientů refrakterních k terapii DMARDs nebo TNF-α inhibitory (60–62). Abatacept má indikaci v kombinaci s metotrexátem a tato kombinace významně zpomaluje rentgenovou progresi onemocnění. Léčba abataceptem zatím nebyla provázena výskytem oportunních infekcí ani vyšším počtem malignit. Při srovnání s infliximabem vykazoval abatacept přinejmenším srovnatelnou účinnost po 1. roce hodnocení a navíc byl provázen méně nežádoucími projevy. Protože nebyl pozorován aditivní efekt a naopak se čtyřnásobně zvýšil počet infekčních komplikací, není doporučeno podávat abatacept v kombinaci s TNF-α inhibitory nebo IL-1 inhibitory (63).
Inhibitory B-lymfocytů. Rituximab je chimerická monoklonální protilátka proti antigenu CD20, který je přítomen na maturovaných B-lymfocytech a pre-B-lymfocytech. Již delší dobu je používán pro léčbu pacientů s CD20+ non-Hodgkinským lymfomem (64). B-lymfocyty mohou kromě tvorby protilátek a cytokinů fungovat jako buňky prezentující antigen, proto se předpokládá že mají důležitý význam v patogenezi RA (14). Od roku 2007 je rituximab schválen pro léčbu RA refrakterní alespoň k jednomu TNF-α inhibitoru (65). Podává se v dávce 1g intravenózně dvakrát v odstupu 2 týdnů a poté se opakuje nejdříve za 6 měsíců. Rituximab je klinicky účinný a vede ke zpomalení rentgenové progrese. Pozitivní efekt léčby je pozorován především u pacientů s pozitivním RF nebo ACPA protilátkami oproti seronegativním pacientům (66). Pokroky v technologii výroby vedly k vývoji humanizovaných a plně humánních protilátek, které jsou ve III. fázi klinického zkoušení. Ocrelizumab je humanizovaná monoklonální protilátka (67) a ofatumumab(HuMax-CD20) je plně humánní monoklonální protilátka proti CD20 molekule (68). Výsledky časných fází ukazují na dobrý efekt obou preparátů u RA pacientů refrakterních k MTX i TNF-α inhibitorům. Dobrý klinický účinek je provázen významnou deplecí CD-19 B-lymfocytů a oproti rituximabu je minimální imunogenicita. V současné době probíhá I. fáze klinického zkoušení plně humánní monoklonální protilátky anti-CD19 (MDX-1342) u pacientů s aktivní RA (http://clinicaltrials.gov). U pacientů se středním až těžkým průběhem systémového lupus erythematodes je ve III. fázi klinického zkoušení monoklonální protilátka anti-CD22 (epratuzumab), kteráby cílenoudeplecí B-lymfocytů měla redukovat subset patologických B-buněk a zachovávat populaci neagresivních lymfocytů (69). U pacientů s RA zatím údaje o jeho zkoušení nejsou.
Nově byly vyvinuty jednořetězcové protilátkové polypeptidy známé jako SMIP (small modular immunopharmaceutical), které mají velikosti přibližně 1/2-1/3 standardní monoklonální protilátky. Přepokládá se, že tyto menší molekuly mají lepší tkáňovou penetraci, lepší účinnost a bezpečnostní profil. Firma Trubion Pharmaceuticals vyvinula SMIP, který váže a specificky blokuje CD20 molekulu (TRU-015), navozuje periferní depleci B-lymfocytů a má redukovanou schopnost vazby komplementu (http://www.trubion.com). Počáteční klinické zkoušení ukázalo dobrý bezpečnostní profil a klinickou účinnost u RA pacientů. V současné době probíhá II. fáze klinického zkoušení (http://clinicaltrials.gov). Její výsledky by měly ukázat výhody tohoto preparátu oproti ostatním lékům zaměřeným proti B-lymfocytům.
2. Malé molekuly
Současná biologická léčba je aplikována parenterálně, je spojována se zvýšeným rizikem některých nežádoucích projevů a je finančně velmi nákladná. To vede ke snaze získat preparáty v orálně dostupné formě, které by měly být levnější, alespoň stejně nebo více účinné jako současná biologická léčba a výhodou by bylo nižší riziko vedlejších nežádoucích projevů. Budoucnost léčby RA je tak v posledních několika letech spatřována v tzv. malých molekulách, které mají molekulovou hmotnost menší než 1kDa (17). Více než polovina nových protizánětlivých preparátů ve stadiu preklinických a klinických studií představuje právě malé molekuly. Je třeba si také uvědomit, že většina současně používaných DMARDs představuje malé molekuly, které ale většinou nemají známý mechanismus působení. Nově vyvíjené malé molekuly cíleně ovlivňují různé mezibuněčné faktory nebo buněčné struktury – receptory, intracelulární signální dráhy a enzymy.
Inhibitory proteolytických enzymů
Matrixové metaloproteinázy (MMPs) se významně podílejí na destrukci kloubních struktur během RA. Nicméně testované inhibitory MMPs pro nedostatečnou účinnost a muskuloskeletální nežádoucí projevy v klinickém zkoušení zatím selhávají. Velké naděje se vkládaly do duálních inhibitorů MMPs a enzymů konvertujících neaktivní formy TNF-α (TACE). Nicméně klinické zkoušení aprastatu (TMI 005), duálního inhibitoru TACE a MMP-13, bylo zastaveno koncem roku 2006 pro nedostatečnou účinnost již ve II. fázi (70). Perspektivním preparátem v klinickém zkoušení pro léčbu osteoporózy jsou inhibitory cysteinové proteázy cathepsinu K (71). Nedávno bylo prokázáno, že inhibice cathepsinu K zabraňuje nejenom osteklastické resorpci kosti, ale navíc také autoimunitnímu zánětu na experimentálním modelu artritidy (72). Ve II. fázi klinického výzkumu u pacientů s RA jsou zatím pouze inhibitory cathepsinu S, u kterých se předpokládá imunomodulační potenciál.
Inhibitory intracelulárních signálních molekul
Oči odborníků se v současné době upírají na některé signální transdukční dráhy – malé molekuly bílkovin, které uvnitř buňky zprostředkovávají složitý přenos signálu. Po vazbě extracelulárního faktoru (např. cytokinu nebo patogenu) na buněčný receptor jsou aktivovány cytoplasmatické enzymy, tzv. kinázy, které následně ovlivňují transkripční faktory a ty po vazbě na DNA regulují expresi cílových genů (73). Změněná aktivita některých signálních transdukčních drah může podporovat zánětlivý stav a následný rozvoj autoimunitního onemocnění, kterým je revmatoidní artritida. Na podkladě nových patogenetických poznatků a moderní technologie výroby se v posledních letech rozvíjí nová éra léčby RA (74, 75). Klinicky jsou s různým úspěchem zkoušeny perorální léky, které zasahují do regulace složitých signálních drah (obr. 1).
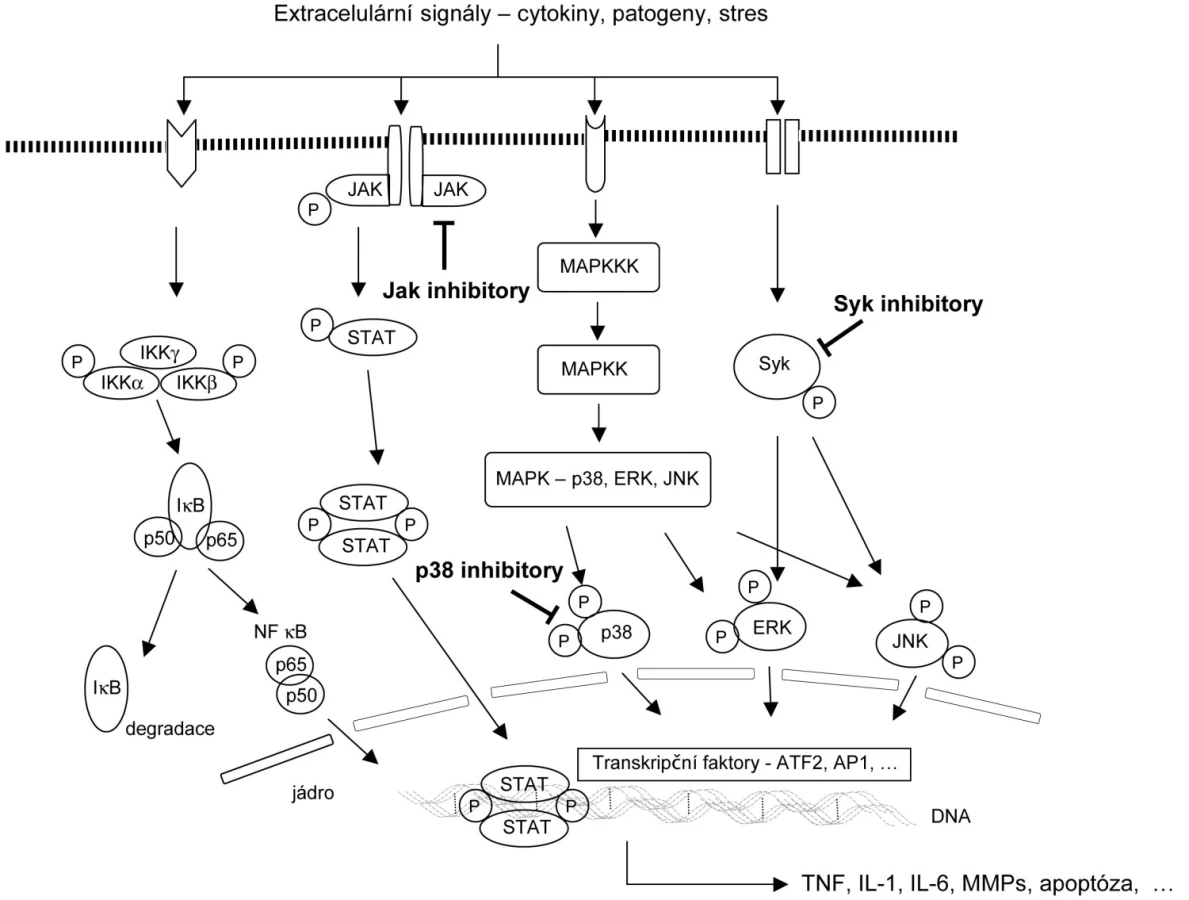
Inhibitory MAP-kinázy p38. Jednou z důležitých molekul signální transdukce je mitogenem aktivovaná proteinkináza (MAP) p38. Tento enzym se podílí na regulaci syntézy klíčových prozánětlivých cytokinů (IL-1, TNF-α, apod.) a uplatňuje se i v prozánětlivé odpovědi na tyto cytokiny (76). Na podkladě velmi slibných preklinických dat jsou vyvíjeny řadou farmaceutických firem inhibitory MAP-kináz p38 ovlivňují převážně její alfa podjednotku. Většina preparátů je ve II. fázi klinického zkoušení, ale účinek léčby zatím nedosahuje očekávaných předpokladů (77). Vysvětlením může být nemožnost použít vyšší dávkování s ohledem na gastrointestinální, kardiovaskulární nebo neurologické nežádoucí projevy. Racionální vysvětlení může podat i fakt, že MAP-kináza p38 zprostředkovává přibližně jen třetinu TNF - indukované genové exprese RA synoviálních fibroblastů (78). Pravděpodobně proto zatím nedosahuje inhibice MAP-kinázy p38 léčebného efektu inhibitorů TNF-α.
Inhibitory signální dráhy JAK/STAT. Tyrosinové Janus kinázy (JAK) mohou být aktivovány působením IFN-γ a cytokinů (např. L-2, IL-6, IL-12, IL-15), které mají významné postavení v patogenezi RA (79). V současné době jsou popsány 4 Janus kinázy: JAK1, JAK2, JAK3 a TYK2. Dendritické buňky RA synoviální tkáně silně exprimují JAK3, STAT4 a STAT6 a tato exprese významně korelovala s hladinou sérového revmatoidního faktoru (80). Proto se stala cílem řady výzkumných projektů právě inhibice této signální kaskády. Zpočátku byl studován imunosupresivní účinek inhibitoru JAK3 při prevenci odhojení transplantátu, přičemž nebyl provázen toxicitou pozorovanou u ostatních imunosupresivních léků (81). Později se stal výhodou jeho protizánětlivý účinek a firma Pfizer vyvinula inhibitor JAK3 (CP-690,550), který má za sebou již II. fázi klinického zkoušení u pacientů se střední až vysokou aktivitou RA (82). Výsledky této studie jsou zatím velmi povzbudivé, po 12 týdnech podávání preparátu bylo dosaženo remise onemocnění přibližně u třetiny jedinců. Po dobu 6 měsíců byla pozorována relativně dobrá tolerance a bezpečnostní profil léčby. V současné době probíhá multicentrická studie porovnávající účinek pěti různých dávkových schémat inhibitoru JAK-3 oproti TNF inhibitoru (adalimumab) a základní chorobu modifikující léčbě u nemocných s aktivní RA (http://clinicaltrials.gov). Podobná klinická data, ale na malém souboru pacientů bylo dosaženo s použitím inhibitoru MAP kinázy JAK 1&2 (83).
Inhibitory Syk kinázy. Intracelulární tyrosinová kináza Syk (Spleen tyrosine kinase) má významnou imunomodulační aktivitu. Je stimulována aktivací Fc receptorů a receptorů B-lymfocytů. Představuje nadřazenou signální dráhu ostatním MAP-kinázám a její aktivace hraje klíčovou roli při TNF - indukované expresi prozánětlivých cytokinů a proteolytických enzymů synoviálními fibroblasty (84). Velmi dobré výsledky s ohledem na potlačení klinické aktivity nemoci bylo dosaženo právě s použitím inhibitoru Syk kinázy (Fostamatinib disodium ) (85). Po 12 týdnech perorálního podávání 150mg léku dvakrát denně byla pozorována remise nemoci téměř u poloviny jedinců, nicméně u této relativně vyšší dávky stoupal výskyt některý nežádoucích projevů. Nejčastěji se jednalo o průjmy, zvýšený krevní tlak, neutropenii nebo elevaci jaterních enzymů.
Inhibitory MEK kináz. MEK-1/2 kináza má důležité uplatnění při růstu nádorových buněk a jejich inhibice se stala předmětem výzkumu v onkologii (86). Je nadřazená MAP-kináze ERK a obě pak představují důležité intracelulární enzymy regulující tvorbu MMPs a prozánětlivých cytokinů IL-1, IL-6 a TNF-α (86). V současné době probíhá II. fáze klinické studie hodnotící efekt tří různých dávek inhibitoru MEK-1/2 kinázy (ARRY-162) u pacientů s aktivní RA (87). Preklinické zkoušky vykázaly dobrou účinnost (88).
Inhibitor tyrosin kináz c-Kit a c-Abl. Imatinib mesylát představuje duální inhibitor tyrosin kinázy c-Kit asociované s receptorem pro PDGF (platelet-derived growth factor) a c-Abl asociované se signální dráhou TGF-β (transforming growth factor-β). Tento lék je používán při terapii chronické myeloidní leukémie a první klinická data ukazují na velmi dobrý antifibrotický účinek u systémová sklerodermie (89). V současné době také probíhá II. fáze klinického zkoušení účinnosti a bezpečnosti u pacientů s RA (90).
Inhibitory transkripčních faktorů. Transkripční faktory hrají velice důležitou úlohu v průběhu regulace funkce imunitních buněk. Kontrolují expresi mnoha cytokinů, prozánětlivých mediátorů nebo mediátorů regulujících apoptózu buněk synoviální membrány. Nejvíce dokladů o uplatnění v patogenezi RA mají transkripční faktory NFκB (nuclear factor κB), NFAT (nuclear factor for activation of T-cells) a AP-1 (Activator protein) (91). Posledně jmenovaný transkripční faktor sestává ze dvou proteinových rodin Fos a Jun vázajících se na DNA. AP-1 ovlivňuje expresi prozánětlivých cytokinů, MMPs a reguluje aktivitu kostních buněk (92). První preklinická data na experimentálním modelu artritidy ukázaly schopnost inhibitoru c-Fos/AP-1 (T-5224) zabránit synovialitidě, osteklastogenezi a následné destrukci kloubu (93). Aktivaci transkripčního faktoru NF-κB zprostředkovává jemu nadřazená kináza Iκ (IKK). Ta je tvořena dvěmi katalytickými jednotkami a třetí jednotkou regulační, zvanou NEMO (NF-κB essential modulator). Nedávno byl zkonstruován permeabilní NBD peptid, který dobře proniká buněčnou membránou, blokuje regulační aktivitu NEMO a tím aktivaci NF-κB. Preklinické testy prokázaly, že NBD peptid snižuje prozánětlivou aktivitu synoviálních fibroblastů i makrofágů a jeho intraartikulární podání do kloubu potkanů s adjuvantní artritidou významně tlumí synovialitidu a destrukci kloubu (94). Transkripční faktory NFAT jsou přítomny v RA synoviální tkáni a regulují expresi genů různých zánětlivých mediátorů, včetně diferenciace osteoklastů. Specifická anti-NFAT léčba tak může rozšířit škálu léků pro RA nebo jiné zánětlivé stavy (95).
Protože transkripční faktory mají pleiotropní úlohu v mnoha biologických funkcích, může být jejich inhibice provázena řadou nežádoucích projevů. Je třeba provést další klinické studie na větších souborech pacientů, zhodnotit závažnost nežádoucích projevů, porovnat účinnost inhibitorů MAP-kináz oproti biologické terapii a eventuálně toleranci a účinnost kombinace této léčby s biologickou terapií. Pro ověření účinnosti a bezpečnosti inhibitorů transkripčních faktorů bude třeba provést více preklinických a klinických studií.
Inhibitory cytokinů a chemokinů
V klinické praxi jsou již dobře etablovány inhibitory TNF - α probíhají studie hodnotící bezpečnost a účinnost monoklonálních protilátek proti dalším cytokinům (např. IL-1, IL-6, IL-12, IL-15, IL-17 nebo IL-23). Za účelem snížení rizika vedlejších projevů parenterální aplikace jsou vyvíjeny malé molekuly s možností perorálního podávání. Firma Synta Pharmaceuticals vyvinula malou molekulu - inhibitor IL-12/IL-23 (Apilimod mesylate). Na podkladě preklinických studií se předpokládá ovlivnění Th1 dependentní imunitní odpovědi – snížení produkce Th1 cytokinů a zpomalení progrese RA. Lék je úspěšně testován u nemocných s Crohnovou chorobou a u pacientů s RA probíhá II. fáze klinického zkoušení (96). Ve II. fázi klinického zkoušení u pacientů s RA byl selektivní antagonista chemokinového receptoru CCR5, maraviroc. Chemokinový receptor CCR5 slouží jako koreceptor pro vstup viru HIV do buňky a jeho blokování se používá při léčbě HIV a AIDS (97). Preparát byl dobře tolerován, bez vážných nežádoucích projevů, ale pro nedostatečnou účinnost u nemocných s RA byla studie koncem roku 2008 ukončena (http://clinicaltrials.gov). Na preklinické úrovni experimentálního modelu artritidy byly se srovnatelnou účinností jakou mají TNF inhibitory zkoušeny malé molekuly blokující chemokiny CXCR1 a CXCR2 (98).
Inhibitory povrchových znaků buněk
Při selhání TNF-α inhibitorů jsou v současné době k dispozici další biologické léky. Ty působí buď pomocí blokování povrchového znaku CD20 na B-lymfocytech (rituximab) nebo inhibicí kostimulačních molekul CD80/86 na buňkách prezentujících antigen, čímž tlumí aktivaci T-lymfocytů (abatacept). Mezi první orálně dostupné modulátory kostimulace T-lymfocytů patří RhuDex – malá molekula blokující znak CD80 na buňkách prezentujících antigen. Tím je blokována přirozená cesta kostimulace CD80-CD28 a následná aktivace a proliferace T-lymfocytů. Proběhla již I. fáze klinického zkoušení. Do studie bylo dosud zařazeno 80 pacientů, ale v červenci 2008 byla studie pozastavena kvůli úmrtí jednoho dobrovolníka na infarkt myokardu v průběhu klinické studie. Podle biopsie se předpokládala přítomnost těžkého postižení myokardu ještě před zahájením studie, ale firma byla vyzvána doplnit „in vitro“ testy k vyloučení možné lékové interakce podporující aterogenezi (http://medigene.com). Další orálně dostupný regulátor funkce T-lymfocytů (TAK-783) byl vyvinut firmou Takeda, nyní probíhá II. fázi klinického zkoušení u pacientů s RA (http://clinicaltrials.gov).
Kombinace léků se synergickým účinkem
Firma CombinatoRx využívá kombinace léků, které vlivem synergického účinku mohou lépe ovlivnit patologický stav na více úrovních a vykazovat silnější léčebný účinek. Příkladem neočekávaného úspěchu byla II. fáze klinické studie, kde kombinace nízké dávky prednisonu (3 mg) a antiagregačního dipyridamolu (CRx-102) u pacientů s RA vedla po 42 dnech k výraznější odpovědi ACR20 oproti kontrolní skupině (63 vs. 30%). Předpokládá se, že dipyridamol zvyšuje protizánětlivý a imunomodulační účinek glukokortikoidů bez zvýšení rizika nežádoucích projevů (http://www.combinatorx.com).
Závěr
V posledním desetiletí došlo k dramatickému pokroku v léčbě revmatoidní artritidy. Registrováno je již pět biologický léků (etanercept, infliximab, adalimumab, rituximab a abatacept) a blíží se registrace dalších tří (certolizumab, golimumab a tocilizumab). Biologická léčba se tak stala neodmyslitelně významným přínosem imunoterapie RA pacientů refrakterních ke standardní antirevmatické terapii. Navíc se mění i strategie léčby. Včasná intenzivní léčba metotrexátem, kontrola pacientů po 1 měsíci a promptní úprava dávky léku podle jeho efektu je celkově účinnější než standardní léčba s kontrolami po 3 měsících (99).
Dosud ale žádná biologická léčba nedokáže navodit kompletní remisi u většiny RA pacientů. Proto se s rostoucími poznatky o patogenezi autoimunitního zánětlivého procesu rozšiřuje spektrum nových molekul, z nichž se některé v brzké budoucnosti stanou dalším cílem léčby RA. Ta se tak rozšiřuje o nové monoklonální protilátky proti TNF-α (certolizumab pegol, golimumab), IL-1 (canakinumab), IL-6 (tocilizumab) nebo blokátory B-lymfocytů (ocrelizumab, ofatumumab, TRU-015, belimumab, atacicept). Intenzivně jsou zkoumány malé molekuly, převážně inhibitory MAP-kináz zprostředkovávající nitrobuněčný přenos signálu. Největší naděje jsou vkládány do inhibitorů kináz JAK3 a Syk. Jejich výhodou je perorální aplikace. Protože nitrobuněčné signální dráhy mají pleiotropní účinky, může být jejich inhibice provázena nečekanými nežádoucími projevy, proto je pro další studie nezbytné těsnější zaměření na bezpečnostní profil.
Rostoucí počet inhibitorů cytokinů a imunomodulačních pochodů rozšiřuje léčebné možnosti pro pacienty s revmatoidní artritidou. Navíc moderní technologie výroby vylepšují vlastnosti již zavedených biologických léků a měla by navíc umožnit levnější vývoj malých molekul. Ty jsou cíleně namířeny na molekulární podstatu patogeneze onemocnění. Budoucnost léčby se může odvíjet od nových vědeckých poznatků genového inženýrství, možnosti ovlivnění genové transkripce pomocí RNA interference pomocí malých interferujících RNA (siRNA) nebo microRNA (miRNA) a možnosti epigenetických změn (DNA metylace, modifikace histonů), které mohou ovlivnit přepis genové informace ve smyslu regulace zánětu a autoimunitního stavu v průběhu revmatických onemocnění (100, 101).
Poděkování: Tato práce vznikla za podpory výzkumných záměrů č. 00023728 Ministerstva zdravotnictví České republiky.
MUDr. Ladislav Šenolt
Revmatologický ústav
Na Slupi 4
128 50 Praha 2
e-mail: seno@revma.cz
Sources
1. Malmstrom V, Trollmo C, Klareskog L. The additive role of innate and adaptive immunity in the development of arthritis. Am J Med Sci 2004; 327 : 196–201.
2. Frauwirth KA, Thompson CB. Activation and inhibition of lymphocytes by costimulation. J Clin Invest 2002; 109 : 295–9.
3. Lundberg K, Nijenhuis S, Vossenaar ER, et al. Citrullinated proteins have increased immunogenicity and arthritogenicity and their presence in arthritic joints correlates with disease severity. Arthritis Res Ther 2005; 7: R458–67.
4. Gregersen PK, Silver J, Winchester RJ. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum 1987; 30 : 1205–13.
5. Hill JA, Southwood S, Sette A, Jevnikar AM, Bell DA, Cairns E. Cutting edge: the conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-DRB1*0401 MHC class II molecule. J Immunol 2003; 171 : 538–41.
6. Kaltenhäuser S, Pierer M, Arnold S, et al. Antibodies against cyclic citrullinated peptide are associated with the DRB1 shared epitope and predict joint erosion in rheumatoid arthritis. Rheumatology (Oxford). 2007; 46 : 100–4.
7. Vencovský J, Machácek S, Sedová L, Kafková J, Gatterová J, Pesáková V, Růzicková S. Autoantibodies can be prognostic markers of an erosive disease in early rheumatoid arthritis. Ann Rheum Dis 2003; 62 : 427–30.
8. Gutierrez-Roelens I, Lauwerys BR. Genetic susceptibility to autoimmune disorders: clues from gene association and gene expression studies. Curr Mol Med 2008; 8 : 551–61.
9. Andersson AK, Li C, Brennan FM. Recent developments in the immunobiology of rheumatoid arthritis. Arthritis Res Ther 2008; 10 : 204.
10. Klareskog L, Stolt P, Lundberg K, et al. A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum 2006; 54 : 38–46.
11. Cope AP, Schulze-Koops H, Aringer M. The central role of T cells in rheumatoid arthritis. Clin Exp Rheumatol 2007; 25: S4–11.
12. Zheng Y, Danilenko DM, Valdez P, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature 2007; 445 : 648–51.
13. Šenolt L. Význam synoviálních fibroblastů v patogenezi revmatoidní artritidy. Čes Revmatol 2006; 2 : 65–70.
14. Silverman GJ, Carson DA. Roles of B cells in rheumatoid arthritis. Arthritis Res Ther 2003;5 Suppl 4:S1–6.
15. Conti P, Youinou P, Theoharides TC. Modulation of autoimmunity by the latest interleukins (with special emphasis on IL-32). Autoimmun Rev 2007; 6 : 131–7.
16. O’ Gradaigh D, Ireland D, Bord S, Compston JE. Joint erosion in rheumatoid arthritis: interactions between tumour necrosis factor alpha, interleukin 1, and receptor activator of nuclear factor kappaB ligand (RANKL) regulate osteoclasts. Ann Rheum Dis 2004; 63 : 354–9.
17. Stanczyk J, Ospelt C, Gay S. Is there a future for small molecule drugs in the treatment of rheumatic diseases? Curr Opin Rheumatol 2008; 20 : 257–62.
18. Zhou H, Jang H, Fleischmann RM, et al. Pharmacokinetics and safety of golimumab, a fully human anti-TNF-alpha monoclonal antibody, in subjects with rheumatoid arthritis. J Clin Pharmacol 2007; 47 : 383–96.
19. Keystone E, Heijde DV, Mason D Jr, et al. Certolizumab pegol plus methotrexate is significantly more effective than placebo plus methotrexate in active rheumatoid arthritis: Findings of a fifty-two-week, phase III, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum 2008; 58 : 3319–29.
20. Cohen S, Hurd E, Cush J, et al. Treatment of rheumatoid arthritis with anakinra, a recombinant human interleukin-1 receptor antagonist, in combination with methotrexate: results of a twenty-four-week, multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum 2002; 46 : 614–24.
21. Ruperto N, Quartier P, Wulffraat N, et al. ACZ885 (canakinumab), A New Il-1 Beta Blocking Monoclonal Antibody Has A Beneficial Effect In Children With Systemic Juvenile Idiopathic Arthritis (sjia). In: Abstracts of the 2008 annual scientific meeting of the American college of Rheumatology; San Francisco, CA.
22. Nishimoto N, Hashimoto J, Miyasaka N, et al. Study of active controlled monotherapy used for rheumatoid arthritis, an IL-6 inhibitor (SAMURAI): evidence of clinical and radiographic benefit from an x ray reader-blinded randomised controlled trial of tocilizumab. Ann Rheum Dis 2007; 66 : 1162–7.
23. Genovese MC, McKay JD, Nasonov EL, et al. Interleukin-6 receptor inhibition with tocilizumab reduces disease activity in rheumatoid arthritis with inadequate response to disease-modifying antirheumatic drugs: the tocilizumab in combination with traditional disease-modifying antirheumatic drug therapy study. Arthritis Rheum 2008; 58 : 2968–80.
24. Carroll HP, Paunovic V, Gadina M. Signalling, inflammation and arthritis: Crossed signals: the role of interleukin-15 and -18 in autoimmunity. Rheumatology (Oxford) 2008; 47 : 1269–77.
25. Baslund B, Tvede N, Danneskiold-Samsoe B, et al. Targeting interleukin-15 in patients with rheumatoid arthritis: a proof-of-concept study. Arthritis Rheum 2005; 52 : 2686–92.
26. Brok HP, Tekoppele JM, Hakimi J, et al. Prophylactic and therapeutic effects of a humanized monoclonal antibody against the IL-2 receptor (DACLIZUMAB) on collagen-induced arthritis (CIA) in rhesus monkeys. Clin Exp Immunol 2001; 124 : 134–41.
27. Young DA, Hegen M, Ma HL, et al. Blockade of the interleukin-21/interleukin-21 receptor pathway ameliorates disease in animal models of rheumatoid arthritis. Arthritis Rheum 2007; 56 : 1152–63.
28. Churchman SM, Ponchel F. Interleukin-7 in rheumatoid arthritis. Rheumatology (Oxford) 2008; 47 : 753–9.
29. Plater-Zyberk C, Joosten LA, Helsen MM, Koenders MI, Baeuerle PA, van den Berg WB. Combined blockade of GM-CSF and IL-17 pathways potently suppresses chronic destructive arthritis in a TNF{alpha}independent mouse model. Ann Rheum Dis 2008 [Epub ahead of print].
30. Leonardi CL, Kimball AB, Papp KA, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet 2008 17; 371 : 1665–74.
31. Tan ZY, Bealgey KW, Fang Y, Gong YM, Bao S. Interleukin-23: Immunological roles and clinical implications. Int J Biochem Cell Biol 2008 [Epub ahead of print]
32. Yago T, Nanke Y, Kawamoto M, et al. IL-23 induces human osteoclastogenesis via IL-17 in vitro, and anti-IL-23 antibody attenuates collagen-induced arthritis in rats. Arthritis Res Ther 2007; 9: R96.
33. Malfait AM, Butler DM, Presky DH, Maini RN, Brennan FM, Feldmann M. Blockade of IL-12 during the induction of collagen-induced arthritis (CIA)markedly attenuates the severity of the arthritis. Clin Exp Immunol 1998; 111 : 377–83.
34. Brentano F, Ospelt C, Stanczyk J, Gay RE, Gay S, Kyburz D. Abundant expression of the IL-23 subunit p19, but low levels of bioactive IL-23 in the rheumatoid synovium. Ann Rheum Dis 2008 [Epub ahead of print].
35. Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest 2008; 118 : 3537–45.
36. Braun A, Takemura S, Vallejo AN, Goronzy JJ, Weyand CM. Lymphotoxin beta-mediated stimulation of synoviocytes in rheumatoid arthritis. Arthritis Rheum 2004; 50 : 2140–50.
37. Roschke V, Sosnovtseva S, Ward CD, et al. BLyS and APRIL form biologically active heterotrimers that are expressed in patients with systemic immune-based rheumatic diseases. J Immunol 2002; 169 : 4314–21.
38. Tak PP, Thurlings RM, Rossier C, et al. Atacicept in patients with rheumatoid arthritis: results of a multicenter, phase Ib, double-blind, placebo-controlled, dose-escalating, single - and repeated-dose study. Arthritis Rheum 2008; 58 : 61–72.
39. Ding C. Belimumab, an anti-BLyS human monoclonal antibody for potential treatment of inflammatory autoimmune diseases. Expert Opin Biol Ther 2008; 8 : 1805–14.
40. Hamdy NA. Denosumab: RANKL inhibition in the management of bone loss. Drugs Today (Barc) 2008; 44 : 7–21.
41. Cohen SB, Dore RK, Lane NE, et al. Denosumab treatment effects on structural damage, bone mineral density, and bone turnover in rheumatoid arthritis: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, phase II clinical trial. Arthritis Rheum 2008; 58 : 1299–309.
42. Tak PP. Chemokine inhibition in inflammatory arthritis. Best Pract Res Clin Rheumatol 2006; 20 : 929–39.
43. Shahrara S, Proudfoot AE, Park CC, et al. Inhibition of monocyte chemoattractant protein-1 ameliorates rat adjuvant-induced arthritis. J Immunol 2008; 180 : 3447–56.
44. Haringman JJ, Gerlag DM, Smeets TJ, et al. A randomized controlled trial with an anti-CCL2 (anti-monocyte chemotactic protein 1) monoclonal antibody in patients with rheumatoid arthritis. Arthritis Rheum 2006; 54 : 2387–92.
45. Vergunst CE, Gerlag DM, Lopatinskaya L, et al. Modulation of CCR2 in rheumatoid arthritis: a double-blind, randomized, placebo-controlled clinical trial. Arthritis Rheum 2008; 58 : 1931–9.
46. Mohan K, Issekutz TB. Blockade of chemokine receptor CXCR3 inhibits T cell recruitment to inflamed joints and decreases the severity of adjuvant arthritis. J Immunol 2007; 179 : 8463–9.
47. Hirota K, Yoshitomi H, Hashimoto M, et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med 2007; 204 : 2803–12.
48. Shahrara S, Proudfoot AE, Woods JM, et al. Amelioration of rat adjuvant-induced arthritis by Met-RANTES. Arthritis Rheum 2005; 52 : 1907–19.
49. Lainer-Carr D, Brahn E. Angiogenesis inhibition as a therapeutic approach for inflammatory synovitis. Nat Clin Pract Rheumatol 2007; 3 : 434–42.
50. Grothey A, Ellis LM. Targeting angiogenesis driven by vascular endothelial growth factors using antibody-based therapies. Cancer J 2008;14 : 170–7.
51. Mould AW, Scotney P, Greco SA, Hayward NK, Nash A, Kay GF. Prophylactic but not therapeutic activity of a monoclonal antibody that neutralizes the binding of VEGF-B to VEGFR-1 in a murine collagen-induced arthritis model. Rheumatology (Oxford) 2008; 47 : 263–6.
52. Baier A, Meineckel I, Gay S, Pap T. Apoptosis in rheumatoid arthritis. Curr Opin Rheumatol 2003; 15 : 274–9.
53. Pope RM. Apoptosis as a therapeutic tool in rheumatoid arthritis. Nat Rev Immunol 2002; 2 : 527–35.
54. Pap T, Franz JK, Hummel KM, Jeisy E, Gay R, Gay S. Activation of synovial fibroblasts in rheumatoid arthritis: lack of Expression of the tumour suppressor PTEN at sites of invasive growth and destruction. Arthritis Res 2000; 2 : 59–64.
55. Wang CR, Shiau AL, Chen SY, et al. Amelioration of collagen-induced arthritis in rats by adenovirus-mediated PTEN gene transfer. Arthritis Rheum 2008; 58 : 1650-6.
56. Sakai K, Matsuno H, Morita I, et al. Potential withdrawal of rheumatoid synovium by the induction of apoptosis using a novel in vivo model of rheumatoid arthritis. Arthritis Rheum 1998; 41 : 1251–7.
57. Schnitzer TJ, Yocum DE, Michalska M, et al. Subcutaneous administration of CAMPATH-1H: clinical and biological outcomes. J Rheumatol 1997; 24 : 1031–6.
58. Mason U, Aldrich J, Breedveld F, et al. CD4 coating, but not CD4 depletion, is a predictor of efficacy with primatized monoclonal anti-CD4 treatment of active rheumatoid arthritis. J Rheumatol 2002; 29 : 220–9.
59. Hepburn TW, Totoritis MC, Davis CB. Antibody-mediated stripping of CD4 from lymphocyte cell surface in patients with rheumatoid arthritis. Rheumatology (Oxford) 2003; 42 : 54–61.
60. Schiff M, Keiserman M, Codding C, et al. Efficacy and safety of abatacept or infliximab versus placebo in ATTEST: a phase III, multicenter, randomized, double-blind, placebo-controlled study in patients with rheumatoid arthritis and an inadequate response to methotrexate. Ann Rheum Dis 2008; 67 : 1096–103.
61. Genant HK, Peterfy CG, Westhovens R, et al. Abatacept inhibits structural damage progression in rheumatoid arthritis: results from the long-term extension of the AIM trial. Ann Rheum Dis 2008; 67 : 1084–9.
62. Genovese MC, Schiff M, Luggen M, et al. Efficacy and safety of the selective co-stimulation modulator abatacept following 2 years of treatment in patients with rheumatoid arthritis and an inadequate response to anti-tumour necrosis factor therapy. Ann Rheum Dis 2008; 67 : 547–54.
63. Weinblatt M, Combe B, Covucci A, Aranda R, Becker JC, Keystone E. Safety of the selective costimulation modulator abatacept in rheumatoid arthritis patients receiving background biologic and nonbiologic disease-modifying antirheumatic drugs: A one-year randomized, placebo-controlled study. Arthritis Rheum 2006; 54 : 2807–16.
64. Johnson PW, Glennie MJ. Rituximab: mechanisms and applications. Br J Cancer 2001; 85 : 1619–23.
65. Smolen JS, Keystone EC, Emery P, et al. Consensus statement on the use of rituximab in patients with rheumatoid arthritis. Ann Rheum Dis 2007; 66 : 143–50.
66. Cohen SB, Emery P, Greenwald MW, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum 2006; 54 : 2793–806.
67. Genovese MC, Kaine JL, Lowenstein MB, et al. Ocrelizumab, a humanized anti-CD20 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: A phase I/II randomized, blinded, placebo-controlled, dose-ranging study. Arthritis Rheum 2008; 58 : 2652–61.
68. Robak T. Ofatumumab, a human monoclonal antibody for lymphoid malignancies and autoimmune disorders. Curr Opin Mol Ther 2008; 10 : 294–309.
69. Jacobi AM, Goldenberg DM, Hiepe F, Radbruch A, Burmester GR, Dörner T. Differential effects of epratuzumab on peripheral blood B cells of patients with systemic lupus erythematosus versus normal controls. Ann Rheum Dis. 2008; 67 : 450–7.
70. Thabet MM, Huizinga TW. Drug evaluation: apratastat, a novel TACE/MMP inhibitor for rheumatoid arthritis. Curr Opin Investig Drugs 2006; 7 : 1014–9.
71. Stoch SA, Wagner JA. Cathepsin K inhibitors: a novel target for osteoporosis therapy. Clin Pharmacol Ther 2008; 83 : 172–6.
72. Asagiri M, Hirai T, Kunigami T, et al. Cathepsin K-dependent toll-like receptor 9 signaling revealed in experimental arthritis. Science 2008; 319 : 624–7.
73. Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002; 298 : 1911–2.
74. Sweeney SE, Firestein GS. Primer: signal transduction in rheumatic disease-a clinician’s guide. Nat Clin Pract Rheumatol 2007; 3 : 651–60.
75. Šenolt L, Vencovský J, Pavelka K. Transdukční signální dráhy – cíl terapie revmatoidní artritidy budoucnosti? Čes Revmatol 2005; 2 : 58–66.
76. Schett G, Zwerina J, Firestein G. The p38 mitogen-activated protein kinase (MAPK) pathway in rheumatoid arthritis. Ann Rheum Dis 2008; 67 : 909–16.
77. Genovese MC, Cohen SB, Wofsy D, et al. A Randomized, Double-Blind, Placebo-Controlled Phase 2 Study of an Oral p38@ MAPK Inhibitor, SCIO-469, in Patients with Active Rheumatoid Arthritis. In: Abstracts of the 2008 annual scientific meeting of the American college of Rheumatology; San Francisco, CA.
78. Zer C, Sachs G, Shin JM. Identification of genomic targets downstream of p38 mitogen-activated protein kinase pathway mediating tumor necrosis factor-alpha signaling. Physiol Genomics 2007; 31 : 343–51.
79. Walker JG, Smith MD. The Jak-STAT pathway in rheumatoid arthritis. J Rheumatol 2005; 32 : 1650-3.
80. Walker JG, Ahern MJ, Coleman M, et al. Characterisation of a dendritic cell subset in synovial tissue which strongly expresses Jak/STAT transcription factors from patients with rheumatoid arthritis. Ann Rheum Dis 2007; 66 : 992–9.
81. Changelian PS, Flanagan ME, Ball DJ, et al. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science 2003; 302 : 75–8.
82. Kremer J, Cohen S, Wilkinson B, et al. The Oral Jak Inhibitor CP-690,550 (CP) in Combination with Methotrexate (MTX) is Efficacious, Safe and Well Tolerated in Patients with Active Rheumatoid Arthiritis (RA) with an Inadequate Response to Methotrexate Alone. In: Abstracts of the 2008 annual scientific meeting of the American college of Rheumatology; San Francisco, CA.
83. Williams W, Scherle P, Shi J, et al. A Randomized Placebo-Controlled Study of INCB018424, a Selective Janus Kinase1& 2 (JAK1&2) Inhibitor in Rheumatoid Arthritis (RA) In: Abstracts of the 2008 annual scientific meeting of the American college of Rheumatology; San Francisco, CA. Presentation 1189.
84. Cha HS, Boyle DL, Inoue T, et al. A novel spleen tyrosine kinase inhibitor blocks c-Jun N-terminal kinase-mediated gene expression in synoviocytes. J Pharmacol Exp Ther 2006; 317 : 571-8.
85. Weinblatt ME, Kavanaugh A, Burgos-Vargas R, et al. Treatment of rheumatoid arthritis with a syk kinase inhibitor: A twelve-week, randomized, placebo-controlled trial. Arthritis Rheum 2008; 58 : 3309–18.
86. Friday BB, Adjei AA. Advances in targeting the Ras/Raf/MEK/Erk mitogen-activated protein dinase cascade with MEK inhibitors for cancer therapy. Clin Cancer Res 2008; 14 : 342–6.
87. Carter L, Brown S, Klopfenstein N, et al. ARRY-162, A Novel MEK Inhibitor: Results of a 14-Day Phase 1a Study in Healthy Subjects and a 28-Day Phase 1b Study in Rheumatoid Arthritis patiens. In: Abstracts of the 2008 annual scientific meeting of the American college of Rheumatology; San Francisco, CA. Presentation: 358.
88. Thiel MJ, Schaefer CJ, Lesch ME, et al. Central role of the MEK/ERK MAP kinase pathway in a mouse model of rheumatoid arthritis: potential proinflammatory mechanisms. Arthritis Rheum 2007; 56 : 3347–57.
89. Distler JH, Distler O. Intracellular tyrosine kinases as novel targets for anti-fibrotic therapy in systemic sclerosis. Rheumatology (Oxford). 2008 Oct;47 Suppl 5:v10-1.
90. Paniagua RT, Robinson WH. Imatinib for the treatment of rheumatic diseases. Nat Clin Pract Rheumatol 2007; 3 : 190–1.
91. Okamoto H, Cujec TP, Yamanaka H, Kamatani N. Molecular aspects of rheumatoid arthritis: role of transcription factors. FEBS J 2008; 275 : 4463–70.
92. Zenz R, Eferl R, Scheinecker C, et al. Activator protein 1 (Fos/Jun) functions in inflammatory bone and skin disease. Arthritis Res Ther 2008; 10 : 201.
93. Aikawa Y, Morimoto K, Yamamoto T, et al. Treatment of arthritis with a selective inhibitor of c-Fos/activator protein-1. Nat Biotechnol 2008; 26 : 817–23.
94. Tas SW, Vervoordeldonk MJ, Hajji N, May MJ, Ghosh S, Tak PP. Local treatment with the selective IkappaB kinase beta inhibitor NEMO-binding domain peptide ameliorates synovial inflammation. Arthritis Res Ther 2006; 8: R86.
95. Pessler F, Dai L, Cron RQ, Schumacher HR. NFAT transcription factors-new players in the pathogenesis of inflammatory arthropathies? Autoimun Rev 2006; 5 : 106–10.
96. Wada Y, Lu R, Zhou D, et al. Selective abrogation of Th1 response by STA-5326, a potent IL-12/IL-23 inhibitor Blood. 2007; 109 : 1156–64.
97. Meanwell NA, Kadow JF. Maraviroc, a chemokine CCR5 receptor antagonist for the treatment of HIV infection and AIDS. Curr Opin Investig Drugs 2007; 8 : 669–81.
98. Barsante MM, Cunha TM, Allegretti M, et al. Blockade of the chemokine receptor CXCR2 ameliorates adjuvant-induced arthritis in rats. Br J Pharmacol 2008; 153 : 992–1002.
99. Verstappen SM, Jacobs JW, van der Veen MJ, et al. Intensive treatment with methotrexate in early rheumatoid arthritis: aiming for remission. Computer Assisted Management in Early Rheumatoid Arthritis (CAMERA, an open-label strategy trial). Ann Rheum Dis 2007; 66 : 1443–9.
1 00. Stanczyk J, Pedrioli DM, Brentano F, et al. Altered expression of MicroRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum 2008; 58 : 1001–9.
101. Huber LC, Stanczyk J, Jüngel A, Gay S. Epigenetics in inflammatory rheumatic diseases. Arthritis Rheum 2007; 56 : 3523–31.
Labels
Dermatology & STDs Paediatric rheumatology RheumatologyArticle was published in
Czech Rheumatology

2009 Issue 1
Most read in this issue
- Interleukin 6 in rheumatic diseases
- News in the biological therapy of rheumatoid arthritis and future prospects
- Rituximab in the treatment of Wegener’s granulomatosis non-responsive to standard therapy
- Collagen and elastin degradation products as potential activity markers of scleroderma