
Rituximab v terapii Wegenerovy granulomatózy refrakterní na standardní léčbu
Rituximab in the treatment of Wegener’s granulomatosis non-responsive to standard therapy
Wegener’s granulomatosis (WG) is a severe systemic disease characterised by the development of granulomatous inflammation, tissue necrosis and vasculitis of small - and medium-sized vessels. The pulmonary involvement with a risk of the development of alveolar haemorrhage, and renal involvement characterised by necrotizing glomerulonephritis with crescent formation endangering the patient by the development of rapidly progressive renal failure, belong to the most severe organ manifestations threatening the patient’s life. The standard induction therapy leads to remission in more than 90% of patients. This disease is marked by a substantial tendency to flare. Frequent flares of WG non-responsive to standard treatment can be alternatively managed by so called “rescue therapy“. We present a case report of a patient with WG non-responsive to standard induction therapy, treated with rituximab. Rituximab is a chimeric monoclonal antibody specific for cell membrane antigen CD20 of mature B-lymphocytes, administration of which leads to a depletion of peripheral B-lymphocytes. Results of some earlier published studies suggest a decrease of ANCA production and a possible induction of remission after the administration of rituximab to patients with ANCA associated vasculitis.
Key words:
rituximab, therapy, Wegener’s granulomatosis, inflammation
:
P. Němec 1; J. Vlček 1; V. Postránecká 2; A. Krpenský 3; M. Souček 1
:
II. interní klinika, FN u sv. Anny v Brně a Lékařská fakulta Masarykovy univerzity, Brno
1; Klinika zobrazovacích metod, FN u sv. Anny v Brně a Lékařská fakulta Masarykovy univerzity, Brno
2; I. patologicko-anatomický ústav, FN u sv. Anny v Brně a Lékařská fakulta Masarykovy univerzity, Brno
3
:
Čes. Revmatol., 17, 2009, No. 1, p. 51-61.
:
Case Report
Wegenerova granulomatóza (WG) je závažné systémové onemocnění charakterizované vývojem granulomatózního zánětu, tkáňové nekrózy a vaskulitidou tepen malého a středního kalibru. K nejzávažnějším orgánovým projevům, ohrožujícím život pacienta, patří plicní postižení s rizikem vývoje alveolárního krvácení a postižení ledvin charakterizované nekrotizující glomerulonefritidou s přítomností srpků ohrožující pacienta vývojem rychle progredujícího renálního selhání. Standardní indukční léčba je schopna navodit remisi onemocnění u více než 90 % pacientů. Onemocnění má značnou tendenci k relapsům. Relabující WG refrakterní na standardní terapii může být alternativně léčena tzv. rescue therapy. Předkladáme kazuistiku pacienta s WG refrakterní na standardní indukční terapii, který byl léčen rituximabem. Rituximab je chimerická monoklonální protilátka specifická pro povrchový antigen CD20 zralých B-lymfocytů, jejíž podání vede k depleci periferních B-lymfocytů. Výsledky některých dříve publikovaných studií naznačily, že podání rituximabu pacientům s ANCA asociovanými vaskulitidami snižuje produkci ANCA a může navodit remisi onemocnění.
Klíčová slova:
rituximab, terapie, Wegenerova granulomatóza, zánět
Úvod
Wegenerova granulomatóza (WG) je systémové onemocnění neznámé etiologie, charakterizované vývojem granulomatózního zánětu, tkáňové nekrózy a variabilním stupněm vaskulitického postižení cévní stěny malých a středně velkých tepen. Onemocnění poprvé popsal Heinz Klinger v roce 1931 (1). Zaznamenal případ 70letého muže s postižením dýchacích cest, pansinusitidou, hemoragickými krustami v dutině nosní a otitidou. V průběhu onemocnění se u pacienta vyvinula sedlovitá deformita nosu, glomerulonefritida a plicní infiltráty. Následně pacient zemřel na následky bronchopneumonie. Pitva a histologické vyšetření prokázalo nekrotizující granulomatózní vaskulitidu postihující horní i dolní cesty dýchací. O několik roků později popsal Friedrich Wegener tři případy tohoto onemocnění (2, 3).
Epidemiologické studie, zahrnující převážně kavkazoidní populaci, prokázaly roční incidenci onemocnění okolo 8,5 případů na milion obyvatel (4). Z hospitalizačních záznamů zdravotnických zařízení v USA vyplývá údaj o prevalenci choroby přibližně 30 případů na milion obyvatel. WG postihuje rovnoměrně obě pohlaví. Onemocnění se pravděpodobně vyskytuje celosvětově. Jsou rovněž popsány případy WG postihující Afroameričany, Hispánce a Asiaty. Je však patrná tendence k častějšímu výskytu onemocnění u kavkazoidní rasy pocházející z oblastí severní Evropy. WG se manifestuje převážně ve čtvrté a páté dekádě života. Postihuje ale i starší pacienty a přibližně 10 % případu se objevuje v dětském věku (5).
Etiologie tohoto onemocnění není doposud známá. Předpokládá se však, že se na jeho vývoji podílejí faktory genetické stejně jako zevní faktory prostředí včetně infekce některými mikroorganismy. Dominantní postižení kavkazoidní populace severních oblastí Evropy může ukazovat na genetickou predispozici. Na druhé straně onemocnění postihuje pouze výjimečně více členů jedné rodiny, což může svědčit pro skutečnost, že genetické riziko není jediným faktorem podílejícím se na vývoji onemocnění (6). Doposud nebyla prokázána asociace mezi WG a alelami hlavního histokompatibilního komplexu (7). Zevními spouštěcími faktory WG může být inhalace relativně málo rozpustných částic obsažených v materiálech jako je azbest, cement, křemičitý nebo dřevěný prach. Je známo, že tyto látky mohou vyvolat zánětlivou odpověď v dýchacích cestách včetně tvorby granulomů. Řada prací prokazuje u pacientů s WG subklinické změny sliznice horních a dolních cest dýchacích i bez přítomnosti klinických symptomů. Tyto nálezy potvrzují hypotézu, že právě dýchací cesty mohou být místem vstupu patogenních mikroorganismů přispívajících k vývoji onemocnění. Jednoznačný důkaz pro to však nebyl doposud podán.
Současná představa o patogenezi WG zahrnuje především zvýšenou aktivitu a proliferaci CD4+CD28 - T-lymfocytů, které ve zvýšené míře produkují tumor nekrotizující faktor α (TNFα) a ostatní prozánětlivé Th1 cytokiny (8). U pacientů s aktivní WG bývají prokazovány zvýšené sérové hladiny solubilního receptoru pro TNFα. Jeho hladiny se normalizují po dosažení remise onemocnění. Mononukleární buňky z periferní krve produkují pod vlivem interleukinu-12 (IL-12) ve zvýšené míře interferon-γ (8).
WG je řazena k tzv. ANCA asociovaným vaskulitidám, tedy k chorobám jež jsou spojeny s průkazem autoprotilátek namířených proti primárním granulům nacházejícím se v cytoplazmě neutrofilních granulocytů a monocytů (ANCA – antineutrophil cytoplasmic antibody). Tyto autoprotilátky byly poprvé popsány v roce 1982 a v souvislosti s WG v roce 1985 (9, 10). Imunofluorescenčně lze prokázet přítomnost tří typu ANCA: cytoplazmatické ANCA (c-ANCA), perinukleární (p-ANCA) a atypické (x-ANCA). U pacientů s vaskulitidou c-ANCA znak obvykle koresponduje s přítomností autoprotilátek namířených proti proteináze-3 (PR3) a p-ANCA znak s přítomností autoprotilátek proti myeloperoxidáze (MPO). Přítomnost c-ANCA (PR3+) autoprotilátek je výrazně asociována s WG, zatímco p-ANCA (MPO+) autoprotilátky bývají přítomny u pacientů s WG pouze v 10 % případů a jsou spíše asociovány s jinými chorobami, jakými jsou mikroskopická polyangitida, Churg-Straussové syndrom a imunokomplex negativní rychle progredující glomerulonefritida (11). Autoprotilátky x-ANCA (PR3 - MPO-) bývají prokazovaný u řady chorob, například u nespecifických střevních zánětů nebo infekcí. Přítomnost c-ANCA (PR3+) autoprotilátek bývá prokazována u 80–90 % pacientů s WG. Jejich titr však koreluje s aktivitou onemocnění jen asi v 24 % případů (12). Remise onemocnění, následující po zahájení indukční imunosupresivní léčby, bývá často, ale ne vždy provázena poklesem titru c-ANCA (PR3+). Přesto bývají ANCA prokazovány i u pacientů s WG v klinické remisi.
Začátek onemocnění bývá akutní, ale často se vyskytují i případy dlouhodobě „doutnající“ choroby, kde k manifestaci fulminantního průběhu onemocnění dochází až po řadě měsíců nebo roků. V úvodu se často objevují celkové příznaky jako únava, subfebrilie, nechutenství, hmotnostní úbytek nebo artralgie. Například artralgie postihují až 60 % pacientů s WG. U 90 % pacientů dochází k postižení horních cest dýchacích a uší. K příznakům postižení dutiny nosní patří bolesti a pocit ucpaného nosu, příznaky zánětu dutiny nosní, epistaxe a tvorba hemoragických krust. Postižení dutiny nosní může vyústit v tvorbu erozí a perforací nosního septa a ke vzniku sedlovité deformity nosu. K dalším příznakům patří sinusitida postihující kteroukoliv paranazální dutinu. Zánětlivé postižení ucha může vést ke kombinovanému postižení sluchu v důsledku poškození středního a vnitřního ucha. Poškození vestibulárního aparátu se může manifestovat nauzeou, vertigem a tinnitem. Postižení dolních cest dýchacích se může manifestovat tracheitidou a může vést až k fatálnímu postižení hlasivek a subglotického prostoru vyžadujícímu v některých případech provedení tracheotomie. Postižení v této oblasti může být klinicky němé, případně se může manifestovat chrapotem, bolestí v krku, kašlem nebo dušností. Postižení plic může zahrnovat prchavé plicní infiltráty nebo asymptomatické plicní uzly, ale i fulminantní alveolární hemoragii. Častými rentgenovými nálezy bývají mnohočetné, obvykle oboustranné plicní uzly, často s rozpadovou dutinou. Tento nález může vést k obavě mykobakteriální nebo houbové infekce. V 1/3 případů jsou tyto plicní uzly symptomatické (13). Plicní kapilaritida může vést k alveolárnímu krvácení, manifestujícímu se hemoptýzou, a k nálezu rychle se měnících plicních infiltrátů. Difuzní alveolární hemoragie je život ohrožující příhodou. U pacientů s WG bývá popisována hilová a/nebo mediastinální lymfadenopatie. Postižení oka v rámci WG se může manifestovat zánětlivým granulomatózním procesem vyskytujícím se retrobulbárně, projevujícím se exoftalmem, diplopií, případně ztrátou zraku v důsledku ischemie optického nervu a dále nekrotizující skleritidou, která rovněž může vyústit v poškození zraku v důsledku perforace rohovky. K dalším očním manifestacím choroby patří konjunktivitida, episkleritida, keratitida, uveitida, případně obturace slzných kanálků. Postižení ledvin patří k nejzávažnějším orgánovým projevům WG. V úvodu onemocnění bývá přítomno jen asi ve 20 % případů, ale v průběhu choroby se může objevit u téměř 80 % pacientů (13). Klinickým projevem postižení ledvin je rychle progredující glomerulonefritida s příznaky akutní ledvinné nedostatečnosti nebo selhání s narůstající hladinou sérového kreatininu, hematurií, proteinurií (obvykle ne nefrotickou). Bez adekvátní terapie může toto postižení vést během několika dnů až týdnů v nevratnému poškození ledvinných funkcí. K projevům vaskulitického postižení kůže v rámci WG patří hmatná purpura, vezikobulózní léze, papuly, kožní vředy, digitální infarkty nebo třískovité hemoragie. Postižení nervového systému se může manifestovat pod obrazem mononeuritis multiplex, periferní polyneuropatie, neuropatie hlavových nervů nebo meningitidy (14). Bylo rovněž popsáno vzácné postižení mozkové tkáně vaskulitidou. K neuroendokrinním projevům WG patří i diabetes insipidus nebo panhypopituitarismus (15). WG vzácně postihuje srdce, gastrointestinální a urogenitální trakt, prsní žlázu, příušní žlázu, případně plicní tepny.
K monitorování aktivity onemocnění se používají standardizované skórovaní systémy, z nichž se nejčastěji používá Birminghamské skóre aktivity vaskulitidy (BVAS) (16). Jiné skórovací systémy umožňují hodnotit chronické orgánové postižení v rámci vaskulitidy – Vasculitis Damage Index (VDI).
Kazuistika
16letý mladý muž byl poprvé vyšetřen v naší revmatologické ambulanci v říjnu 2005. Pacient s negativní rodinnou anamnézou s polyvalentní pylovou alergií a asthma bronchiale v osobní anamnéze si stěžoval na chrapot, hlenovou sekreci z nosu, opakovanou epistaxi, která se objevovala od července 2005. Dále udával výraznější dušnost při běžné zátěži. Při cíleném dotazování si vzpomněl na epizodu bolestí kolenních kloubů a zápěstí v období jara 2005.
ORL vyšetření prokázalo v dutině nosní neporušené septum, nosní průduchy s ulpívajícími hemoragickými krustami, prosáklou, fragilní sliznici, oboustranně ztluštělé a zarudlé hlasivky, edematózní prosáknutí spodní strany hlasivek, glottis zúženou na 3–4 mm, obleněnou hybnost pravé hlasivky a subglotický prostor s překrvenou sliznicí s hemoragickými krustami. Byla provedena laryngoskopie a tracheoskopie, která prokázala oboustranně ztluštělé, omezeně hybné hlasivky, edematózní prosáknutí sliznice zejména na dolní ploše hlasivek a subgloticky, kde vpravo nelezena drobná ulcerace. Další průběh průdušnice byl zcela klidný. Byly odebrány vzorky sliznice dutiny nosní k histologickému vyšetření, které prokázalo ložisko nekrotizující vaskulitidy se smíšenou buněčnou zánětlivou infiltrací. Bakteriologické kultivační vyšetření prokázalo bakteriální infekci (Staphylococcus aureus). Laboratorní biochemické vyšetření nevykazoval významnější odchylky od normy, hodnota C-reaktivního proteinu (CRP) byla 1,4 mg/l. Biochemické vyšetření moči, ledvinné funkce a proteinurie byly v normě. Hematologické vyšetření prokazovalo pouze mírnou leukocytózu (11,6 * 109/l) s hraniční neutrofilií (81,9 %) a lymfopenií (11,7 %). Sedimentace erytrocytů (FW) byla 5 mm/h. Imunologické vyšetření prokázalo zvýšenou hladinu celkového imunoglobulinu E (IgE) (234,00 IU/ml) a pozitivitu c-ANCA (PR3+) v titru 1 : 160. Bylo vysloveno podezření na c-ANCA asociovanou vaskulitidu, nejspíše WG.
Rentgenové vyšetření prokázalo přiměřenou velikost a transperenci frontálních a maxiálních dutin a dále normální nález při vyšetření hrudníku. Bylo provedeno vyšetření plic výpočetní tomografií s vysokým rozlišením (HRCT), které prokázalo minimální postižení plicního intersticia v podobě nerovností hlavních interlobií. V obou plicních křídlech byla patrná nevelká (do 20 mm), neostře ohraničená zastínění acinárního typu. Funkční vyšetření plic prokázalo lehkou obstrukční ventilační poruchu (mírný pokles FEV1/FVC, normální TLC), normální saturaci O2 (97 %) a normální difuzní plicní kapacitu (DLCO 108 %). Cytologické vyšetření bronchiálního aspirátu prokázalo převahu neutrofilních granulocytů a siderofágů. Bakteriologické a mykologické kultivační vyšetření bylo negativní.
Byla zahájena terapie methylprednisolonem v dávce 0,5 mg/kg/den a terapie cotrimoxazolem v dávce 960 mg každých 12 hodin. Pacient dále dostával vápník v dávce 1000 mg/den, vitamin D v dávce 400 IU/den a lanzoprazol. Pokračovala terapie antihistaminiky (levocetrizin) a bronchodilatancii (salmeterol). V lednu 2006 byl do terapie přidán azathioprin v dávce 2 mg/kg/den. Dávka methylprednisolonu byla postupně redukována.
Kontrolní HRCT, provedené v dubnu 2006, prokázalo vymizení vícečetných zastínění acinárního charakteru. Přetrvávaly pouze diskrétní změny v hlavních interlobiích. Funkční plicní vyšetření prokázalo normální ventilační parametry a normální DLCO. ORL vyšetření prokazovalo přítomnost pouze ojedinělých hemoragických krust v levém nosním průduchu. Oblast hlasivek a subglotický prostor byl bez patologického nálezu. Autoprotilátky c-ANCA (PR3+) byly negativní. Pokračovala udržovací terapie methylprednisolonem v dávce 4 mg/den a azathioprinem v dávce 2 mg/kg/den. Cotrimoxazol byl vysazen.
Při kontrolním HRCT vyšetření, provedeném v únoru 2007, se nově objevily diskrétní okrsky denzit mléčného skla vpravo v apikálním a ventrálním segmentu horního laloku parahilózně a v apikálním a mediobazálním segmentu dolního laloku a dále vlevo v apikálním a mediobazálním segmentu dolního laloku. Funkční plicní vyšetření prokazovalo normální ventilační parametry, normální saturaci O2, ale mírný pokles difuzní kapacity plic (DLCO 78 %). Byla provedena fibrobronchoskopie s bronchoalveolární laváží. Cytologické vyšetření prokazovalo přimněřenou celularitu se zvýšeným podílem neutrofilních granulocytů (9,5 %) a alveolárních siderofágů (21 %). Bakteriologické kultivační vyšetření bylo negativní. Vyšetření mykobakteriální infekce metodou polymerase chain reaction (PCR) a kultivací bylo negativní. Laboratorní biochemické vyšetření nevykazoval významnější odchylky od normy, hodnota CRP byla 0,8 mg/l. Biochemické vyšetření moči, ledvinné funkce a proteinurie byly v normě. Hematologické vyšetření prokazovalo pouze mírnou leukocytózu (13,6 * 109/l), s hraniční neutrofilií (78,3 %) a lymfopenií (14,2 %), FW byla 5 mm/h. Imunologické vyšetření neprokázalo přítomnost c-ANCA (PR3+). CT vyšetření paranazálních dutin prokázalo slizniční změny v levé maxilární dutině s hladinkou sekretu. ORL vyšetření prokázalo stacionární klidný nález v dutině nosní a v hrtanu. Dávka methylprednisolonu byla zvýšena na 16 mg/den, pokračovala terapie azathioprinem v dávce 2 mg/kg/den.
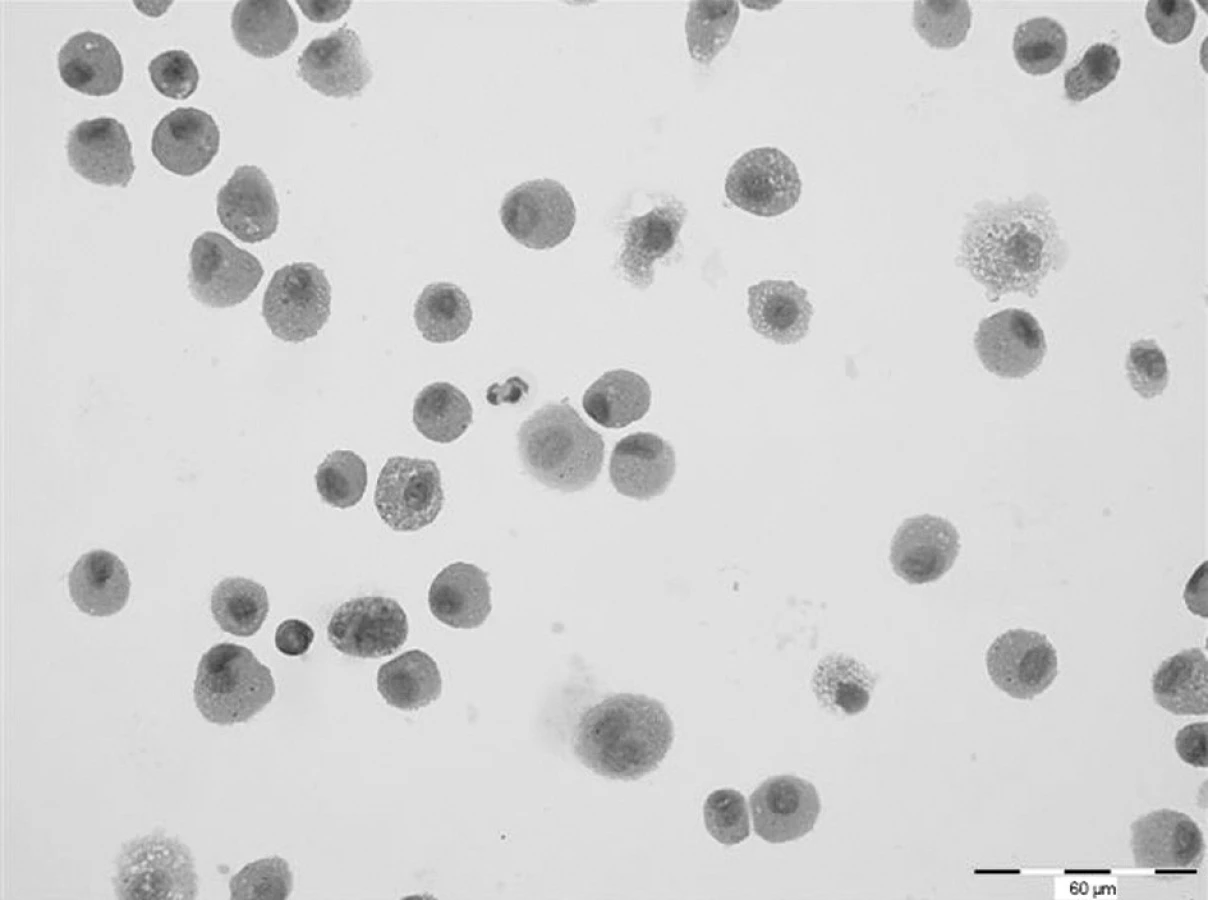
Další HRCT vyšetření plic bylo provedeno s říjnu 2007. V obou plicních křídlech přetrvávaly změny charakteru mléčného skla, jejichž rozsah se zvětšil. V levém plicním křídle v dolním laloku na rozhraní ventrobazálního a laterobazálního segmentu se nově objevilo cystické vzdušné ložisko velikosti 8 mm se silnější pravidelnou stěnou. Obdobné ložisko bylo nalezeno i v pravé plíci v laterobazálním segmentu dolního laloku. Spirometrické vyšetření nevykazovalo odchylku od normy. Rovněž difuzní kapacita plic byla v rozsahu normy (DLCO 87 %). Cytologické vyšetření z bronchoalveolárního aspirátu však prokázalo prakticky 100% podíl alveolárních siderofágů (obr. 1). Laboratorní biochemické vyšetření nevykazoval významnější odchylky od normy, hodnota CRP byla zvýšena (30,4 mg/l). Biochemické vyšetření moči prokazovalo proteinurii a v močovém sedimentu byly přítomny erytrocyty. Ledvinné funkce nevykazovaly odchylku od normy, ale byl zvýšen odpad bílkovin do moči na hodnotu 556 mg/24 h. Hematologické vyšetření nově prokazovalo normocytární, normochromní anemii (erytrocyty 3,40 * 1012/l, hemoglobin 95 g/l, hematokrit 29,30 %) a mírnou leukocytózu (13,4x109/l) s přetrvávající mírnou lymfopenií (16,4 %). Hodnota FW byla zvýšena na 67 mm/h. Imunologické vyšetření prokázalo opět přítomnost c-ANCA (PR3+) v titru 1 : 160. Byla zahájena standardní indukční terapie i.v. cyklofosfamidem (CFA) v dávce 10 mg/kg tělesné hmotnosti. Další dva pulzy cyklofosfamidu byly podány ve čtyřtýdenních intervalech. Současně s CFA byly aplikovány tři i.v. pulzy methylprednisolonu v celkové dávce 1500 mg, po kterých následovalo p.o. podávání methylprednisolonu v dávce 16 mg/den. Před zahájením terapie CFA bylo zamraženo pacientovo sperma s důvodu rizika gonadotoxicity CFA.
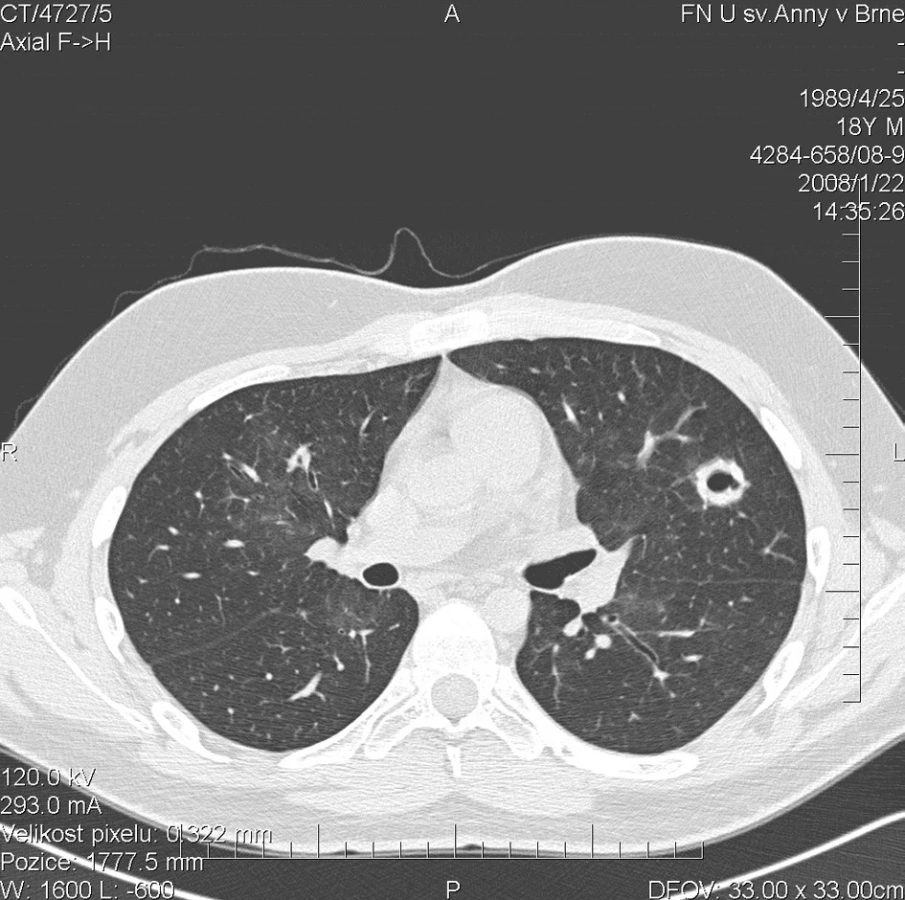
Kontrolní HRCT plic, provedené v lednu 2008, však prokázalo další progresi plicního postižení. Dříve popsané denzity mléčného skla přetrvávaly v nezměněném rozsahu. Navíc se nově objevily hrubé uzlovité ložiskové infiltráty v obou plicních křídlech s rozpadovými dutinami velikosti až 25 mm (obr. 2). Cytologické vyšetření bronchoalveolárního aspirátu se prakticky nezměnilo a opět prokázalo výrazné zastoupení alveolárních siderofágů. Laboratorní biochemické vyšetření nevykazovalo významnější odchylky od normy, hodnota CRP byla zvýšena na 90,5 mg/l. Došlo k mírnému zlepšení parametrů krevního obrazu (erytrocyty 4,03 * 1012/l, hemoglobin 118 g/l, hematokrit 33,8 %). Imunologické vyšetření prokazovalo přítomnost c-ANCA (PR3+) v titru 1 : 20. Byl podán 4. pulz CFA a současně tři i.v. pulzy methylprednisolonu v celkové dávce 3000 mg, po kterých následovala terapie p.o. methylprednisolonem v dávce 64 mg/den. Vzhledem k pozitivitě ANCA byla terapie dále rozšířena o 15 plazmaferéz. Po ukončení série plazmaferéz byly c-ANCA (PR3+) negativní.
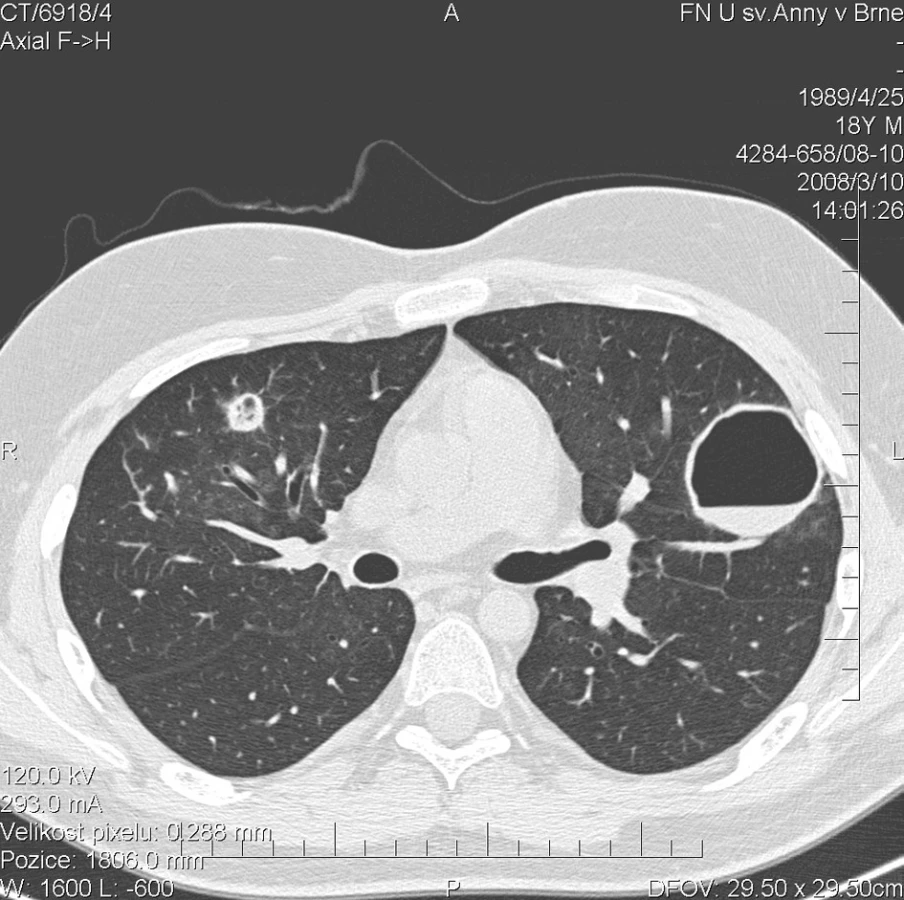
Kontrolní HRCT plic, provedené v březnu 2008, ovšem prokázalo další progresi plicního postižení. V obou plicní křídlech bylo popsáno celkem 17 uzlovitých ložisek, některé s rozpadovými dutinami, o velikosti až 42 mm (obr. 3). Cytologické vyšetření bronchoalveolárního aspirátu prokazovalo 98% zastoupení alveolárních siderofágů. Bakteriologické kultivační vyšetření bronchoalveolárního aspirátu prokázalo infekci bakterií Pseudomonas aeruginosa. Mikroskopické, PCR a kultivační vyšetření Mycobacterium tuberculosis bylo negativní. Kultivace aspirátu na přítomnost Pneumocystis jiroveci, Mycoplasma pneumoniae a Legionella pneumonophilla byla negativní. Rovněž mikroskopie a kultivace orgánových mykóz byla negativní. Sérologické vyšetření kandidového mananového antigenu ELISA metodou bylo negativní. Laboratorní biochemické vyšetření nevykazovalo významnější odchylky od normy, hodnota CRP byla 1,1 mg/l. Biochemické vyšetření moči prokazovalo proteinurii a v močovém sedimentu byly přítomny erytrocyty. Ledvinné funkce nevykazovaly odchylku od normy, odpad bílkovin do moči klesl na hodnotu 286 mg/24 h. Přetrvávala mírná normocytární, normochromní anemie (erytrocyty 3,81 * 1012/l, hemoglobin 118 g/l, hematokrit 34,9 %). Autoprotilátky c-ANCA (PR3+) nebyly přítomny. Byl podán 5. pulz CFA a současně byla zahájena terapie ciprofloxacinem v dávce 500 mg po 12 h. Dávka methylprednisolonu byla postupně snížena na 48 mg/den.
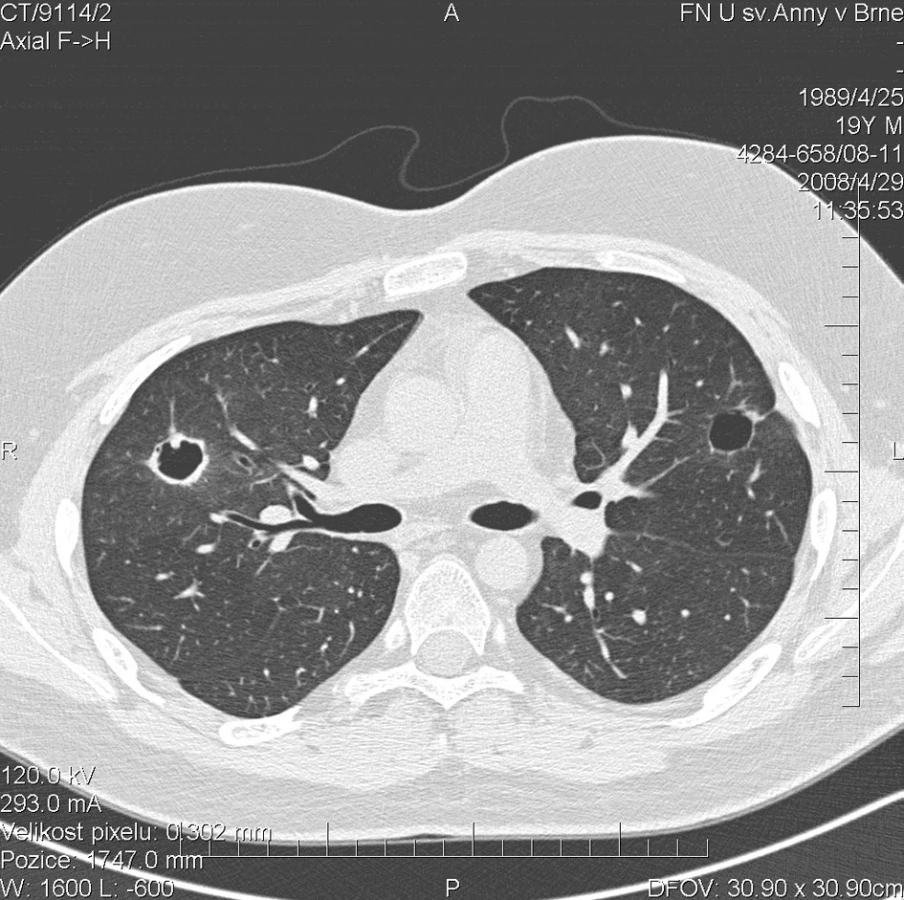
Další kontrolní HRCT plic, provedené v dubnu 2008, prokázalo zmenšení rozsahu a dokonce vymizení některých z uzlovitých ložisek v obou plicních křídlech (obr. 4). Cytologické vyšetření bronchoalveolárního apirátu však stále prokazovalo alveolární hemoragii s 99% zastoupením alveolárních siderofágů. Laboratorní biochemické vyšetření nevykazovalo významnější odchylky od normy, hodnota CRP byla 0,3 mg/l. Došlo k úpravě krevního obrazu (erytrocyty 4,43 * 1012/l, hemoglobin 141 g/l, hematokrit 41,2 %), přetrvávala leukocytóza (14,6 * 109/l) s neutrofílií (83,1 %) a lymfopenií (8,3 %). FW byla 6 mm/h. Autoprotilátky c-ANCA (PR3+) nebyly prokázány. Biochemické vyšetření moči a mikroskopie močovém sedimentu byly negativní. Aktivita onemocnění dle BVAS byla 8. Pokračovala terapie methylprednisolonem v dávce 40 mg/den.
Z důvodu přetrvávající aktivity onemocnění, zejména cytologického průkazu alveolární hemoragie i přes použití standardní indukční terapie, bylo rozhodnuto zahájit terapii rituximabem. V červnu 2008 byl rituximab podán celkem ve 4 i.v. infuzích s odstupem jednoho týdne v dávce 375 mg/m2 plochy povrchu těla. Před každou infuzí rituximabu byla aplikována 1 ampulka bisulepinu i.v. a infuze methylprednisolonu v dávce 125 mg. Aplikace rituximabu nebyla provázena žádnou akutní infuzní komplikací. Průtokovou cytoflowmetrií byl monitorován počet CD19+ lymfocytů. Z úvodních 17 % před aplikací rituximabu, klesl jejich počet již po první infuzi rituximabu na nulu. Autoprotilátky c-ANCA (PR3+) byly opakovaně negativní. Pokračovala terapie methylprednisolonem v dávce 32 mg/den. Byla zahájena terapie cotrimoxazolem v dáce 960 mg po 12 h.
Kontrolní HRCT plic bylo provedeno v srpnu 2008. Prokázalo výraznou redukci plicního postižení. Dříve prokazovaná uzlovitá ložiska v pravém plicním křídle zcela vymizela. Vlevo přetrvávalo pouze solitární ložisko ve ventrálním segmentu horního laloku (obr. 5). Vyšetření celotělovou pletyzmografií prokázalo kombinovanou ventilační poruchu s převahou restrikce lehkého až středně těžkého stupně a středně těžké snížení DLCO (54 %). Výsledky vyšetření mohly být negativně ovlivněny dekondicí pacienta s projevy svalové adynamie a rozvojem atrofie kosterního svalstva. Tento stav se vyvíjel plíživě v průběhu předchozích 8 měsíců a pravděpodobně se na jeho vývoji podílela i dlouhodobá terapie glukokortikoidy. Došlo k poklesu aktivity vaskulitidy (BVAS =1). Dávka methylprednisolonu byla snížena na 8 mg/den. Do terapie byl přidán azathioprin v dávce 1 mg/kg/den.
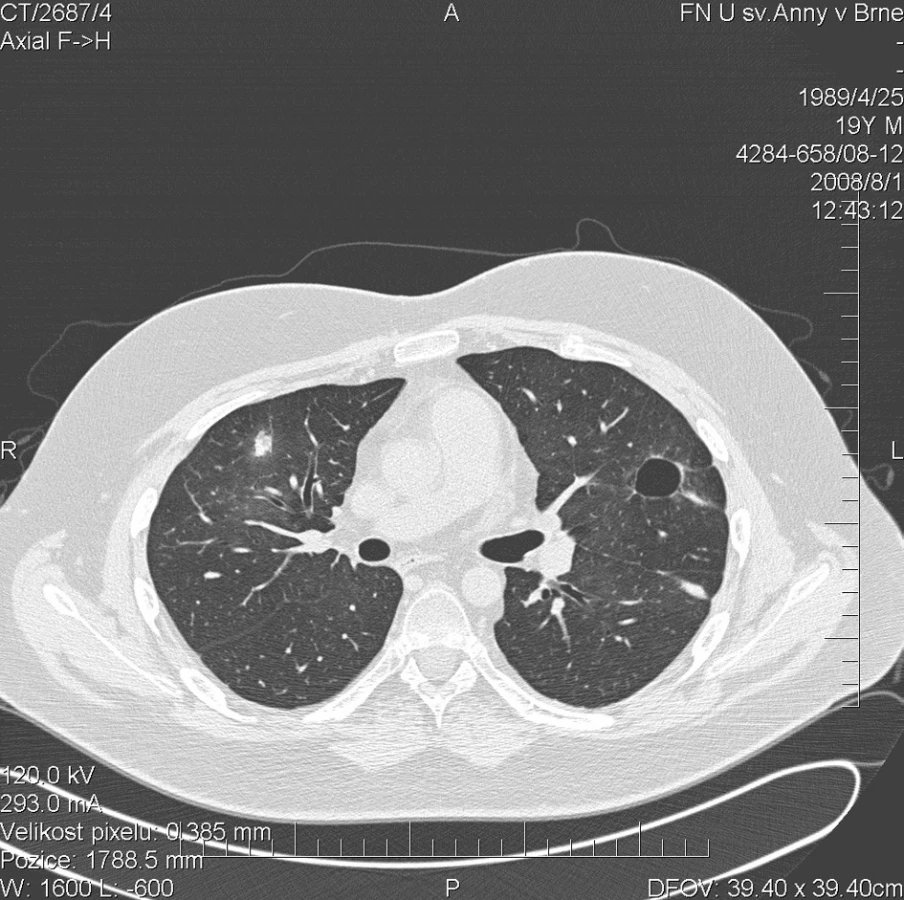
Zatím poslední vyšetření proběhlo v říjnu 2008. Subjektivně si pacient stěžoval pouze na občasnou epistaxi. Po zahájení pohybové rehabilitace došlo ke zlepšení svalové síly. Funkční vyšetření plic prokázalo mírné zlepšení ventilačních parametrů, lehkou kombinovanou ventilační poruchu, lehkou redukci DLCO 64 % a normální saturaci O2 96 %. Laboratorní biochemické vyšetření nevykazovalo významnější odchylky od normy, hodnota CRP byla 0,1 mg/l. Krevní obraz byl normální (erytrocyty 4,58 * 1012/l, hemoglobin 144 g/l, hematokrit 40,6 %), FW byla 4 mm/h. Autoprotilátky c-ANCA (PR3+) nebyly prokázány. Biochemické vyšetření moči a mikroskopie močovém sedimentu byly negativní. Nebyl zaznamenán výskyt nežádoucích účinků. Pokračovala terapie methylprednisolonem v dávce 8 mg/den a azathioprinem v dávce 2 mg/kg/den. Terapie cotrimoxazolem byla ukončena.
Komentář
WG je závažné systémové onemocnění charakterizované vývojem granulomatózního zánětu, tkáňové nekrózy a vaskulitidou tepen malého a středního kalibru. Je řazena mezi tzv. ANCA - asociované vaskulitidy, které jsou charakterizované přítomností jen minimálních depozit v cévní stěně (pauciimunní vaskulitidou) a přítomností c-ANCA (PR3+) autoprotilátek v krevní plazmě. K nejzávažnějším orgánovým projevům, ohrožujícím život pacienta, patří plicní postižení s rizikem vývoje alveolárního krvácení a postižení ledvin charakterizované nekrotizující glomerulonefritidou s přítomností srpků, které může vést během několika týdnů k vývoji terminálního ledvinného selhání.
Léčba WG je závislá na aktivitě onemocnění a liší se podle toho, zda se jedná o závažné, nebo limitované onemocnění. K závažným projevům WG patří rychle progredující glomerulonefritida, alveolární hemoragie, ischemické postižení trávicího traktu, nekrotizující skleritida nebo vaskulitická neuropatie. Data z 50. let minulého století, tedy z období kdy nebyla dostupná efektivní terapie WG, ukazují na převážně fatální průběh onemocnění. Walton v roce 1958 zmiňuje střední přežívání pouze 5 měsíců a 82% mortalitu během prvního roku (17). Léčba závažné WG vyžaduje včasné zahájení agresivní terapie. Standardní léčbu závažných forem WG lze rozdělit na léčbu indukční, jejímž cílem je navodit remisi onemocnění, a léčbu udržovací, jejímž cílem je remisi onemocnění udržet.
Indukční léčba WG obvykle zahrnuje podání vysokých dávek glukokortikoidů a CFA (18, 19). Je schopna navodit remisi onemocnění obvykle do 3 měsíců u více než 90 % nemocných. Z glukokortikoidů se nejčastěji používá prednison v úvodní dávce 1 mg/kg/den. Tato dávka je postupně redukována, tak aby byl pacient na konci třetího měsíce léčen přibližně 20 mg prednisonu denně. Jinou možnosti je použití methylprednisolonu v ekvivalentních dávkách. V případě výskytu život ohrožujících projevů onemocnění, lze perorální podání glukokortikoidů nahradit opakovaným parenterálním podáním methylprednisolonu v dávkách 500 až 1000 mg pro infuzi, po kterých následuje p.o. léčba methylprednisolonem nebo prednisonem.
Léčba CFA je buď kontinuální perorální s úvodní dávkou obvykle 2 mg/kg/den (s redukcí dávky u pacientů s renálním selháním) trvající po dobu 3–6 měsíců, nebo pulzní v dávce 10 mg/kg v i.v. pulzech, které jsou opakovány ve třítýdenních až čtyřtýdenních intervalech po dobu 3–6 měsíců (20). Pulzní terapie CFA je považována za šetrnější s menším výskytem nežádoucích účinků a současně kumulativní dávka CFA dosažená při tomto způsobu podávání je obvykle nižší.
Do jedné prospektivní, randomizované studie bylo zařazeno celkem 50 pacientů s nově diagnostikovanou WG (21). Všem pacientům byl podán parenterálně methylprednisolon ve třech pulzech a CFA v dávce 0,7 g/m2 plochy povrchu těla. Následně byli pacienti randomizováni do dvou skupin. V první skupině byli léčení prednisonem v úvodní dávce 1 mg/kg/den a pulzním CFA a v druhé skupině opět prednisonem ve stejné dávce a perorálně podáváným CFA. Terapie trvala 1 rok. Studie ukázala, že pulzně podávaný CFA je přinejmenším stejně účinný v navození remise WG jako perorální CFA. Navíc byla parenterální terapie CFA spojena s menším výskytem nežádoucích účinků. Parenterální CFA však byl horší v udržení remise WG a prevenci vzniku relapsů.
Zatím ne zcela jasné postavení v indukční terapii WG má plazmaferéza. Její použítí se nabízí vzhledem k tomu, že v patogenezi tohoto onemocnění hrají pravděpodobně roli cirkulující c-ANCA. Plazmaferéza by tedy mohla být doplňkovou terapií u pacientů s alveolárním krvácením a u pacientů s akutním selháním ledvin vyžadujícím hemodialýzu.
Po dosažení remise onemocnění, nejdříve však po 3 měsících od zahájení léčby, je možno indukční léčbu ukončit a přejít na léčbu udržovací. K udržení remise a k prevenci relapsů WG se nejčastěji používají nízké dávky prednisonu v dávce 5–10 mg denně po dobu dalších minimálně 12–18 měsíců případně kombinace s jiným imunosupresivním lékem. Nejčastěji je doporučován azathioprin v dávce 2 mg/kg/den minimálně po dobu 18 měsíců (22). Je možné i pokračování pulzní parenterální aplikace CFA v prodloužených intervalech 6 týdnů až 3 měsíce. K udržovací terapii WG je možno rovněž použít cyklosporin A, mykofenolát mofetyl, metotrexát nebo leflunomid, v případě izolovaného postižení horních cest dýchacích i cotrimoxazol (23–31). Při použití výše uvedených imunosupresivních léků došlo k výraznému zlepšení prognózy pacientů s WG. Desetileté přežívání narostlo na více než 80 %. Problémem však stále zůstávají časté relapsy onemocnění (22).
Rezistence onemocnění na standardní indukční terapii je relativně vzácná a vyskytuje se u méně než 10 % nemocných. U těchto pacientů mohou být použity některé alternativní léčebné postupy tzv. rescue therapy. Nejrozsáhlejší zkušenosti jsou s podáním vysokých i.v. dávek imunoglobulinů v dávce 2 g/kg rozdělené do 2–5 dní a následným opakováním v měsíčních intervalech v dávce 0,4 g/kg (32, 33). Tato terapie je účinná v navození remise, ale poměrně časté jsou relapsy onemocnění. V odborné literatuře se objevuji kazuistická sdělení nebo pozorování na malých souborech pacientů prokazující úspěšné použití antitymocytárního globulinu, humanizovaných monoklonálních protilátek anti-CD52, anti-CD4, chimerické monoklonální protilátky anti-TNFα (infliximab), solubilního receptoru (etanercept) nebo látky 5-desoxyspergualin v léčbě WG (34–40). V mnoha případech však byla léčba těmito preparáty zatížena výskytem infekčních komplikací a krátkým trváním efektu léčby (41).
Role B-lymfocytů v patogenezi autoimunitních chorob včetně WG již byla popsána (42–44). Byla rovněž popsána korelace mezi počtem aktivovaných periferních B-lymfocytů, aktivitou WG a rozsahem orgánového postižení (45). B-lymfocyty zřejmě hrají důležitou roli v patogenezi WG zejména jako producenti ANCA autoprotilátek, které mají řadu prozánětlivých účinků přispívajících k rozvoji vaskulitidy a tkáňovému poškození v rámci WG (45–49).
Rituximab je chimerická lidská/myší monoklonální protilátka specifická pro povrchový antigen CD20 B-lymfocytů vytvořená metodami genetického inženýrství (50, 51). Je tvořena humánní IgG1 konstantní oblastí a variabilní oblastí izolovanou z myších anti-CD20 protilátek. CD20 je povrchová molekula exprimovaná pouze na B-lymfocytech (zejména pre B-lymfocytech a zralých B-lymfocytech) během procesu jejich dozrávání. CD20 není přítomna na kmenových buňkách ani na pro B-lymfocytech a plazmatických buňkách, což znamená, že po podání rituximabu nedochází k poškození kmenových buněk a populace B-lymfocytů může být obnovena a rovněž nedochází k zástavě produkce sérových imunoglobulinů plazmatickými buňkami. Rituximab se selektivně váže na povrchový antigen CD20 pouze těch B-lymfocytů, které tuto molekulu na svém povrchu exprimují. Vede k depleci B-lymfocytů z periferní cirkulace aktivací komplementu, buňkami zprostředkovanou cytotoxicitou a indukcí apopotózy B-lymfocytů (52). Rituximab byl poprvé schválen v roce 1997 v USA a celosvětově v roce 1998 k léčbě non-Hodgkinských lymfomů. Od roku 2007 je rituximab registrován v České republice k terapii revmatoidní artritidy.
Doposud bylo v odborné literatuře publikováno několik práci zabývajících se podáním rituximabu pacientům s WG refrakterní na standardní indukční terapii. Jednou z prvních publikovaných prací na toto téma bylo kazuistické sdělení týkající se pacienta s c-ANCA pozitivní WG, jehož onemocnění iniciálně odpovídalo na terapii glukokortikoidy a CFA (53). Následně se však objevila myelotoxicita CFA. Terapie musela být ukončena a CFA byl nahrazen azathioprinem a mykofenolátem, které však nebyly schopny udržet remisi onemocnění. Pacientovi byl proto aplikován rituximab v podobě 4 infuzí v dávce 375 mg/m2 plochy povrchu těla současně s vysokou dávkou methylprednisolonu. Po skončení terapie u pacienta došlo k vymizení c-ANCA, což bylo provázeno klinickou remisí choroby. Autoprotilátky c-ANCA se znovu objevily 11 měsíců po skončení terapie. Pacientovi byla podána další série infuzí rituximabu tentokrát bez podání glukokortikoidů. Osm měsíců po této druhé sérii byl pacient bez projevů WG.
Následně bylo publikováno několik prací zahrnující otevřené prospektivní nebo retrospektivní studie s malým počtem pacientů (9–11) s ANCA asociovanou vaskulitidou zahrnující c-ANCA pozitivní WG a rovněž p-ANCA pozitivní mikroskopickou polyangitidu refrakterní na standardní terapii (54–57). Pacienti byli léčeni rituxumabem v dávce 375 mg/m2 plochy povrchu těla v podobě dvou nebo čtyř v týdenních intervalech po sobě jdoucích infuzí nebo rituximabem v dávce 1000 mg v podobě dvou infuzí aplikovaných s odstupem 14 dní. Aktivita onemocnění byla hodnocena pomocí BVAS. V těchto studiích byla kompletní nebo parciální remise onemocnění zaznamenána u všech pacientů. U přibližně 1/4 pacientů došlo k relapsu onemocnění. Opakované podání rituximabu v těchto případech vedlo znovu k remisi WG. Terapie rituximabem byla spojena s poklesem titru nebo vymizením ANCA. Léčba byla obecně dobře tolerována. V jednom případě se vyskytla mírná akutní infuzní reakce. Výsledky těchto prací naznačily, že podání rituximabu pacientům s ANCA asociovanými vaskulitidami, které je spojeno s deplecí CD20+ B-lymfocytů, snižuje produkci ANCA a může vést k navození remise onemocnění.
Některé z později publikovaných prací však nepřinesly jednoznačné výsledky. Například v retrospektivní studii Brihayeet al. zahrnující osm pacientů s refrakterní nebo relabující WG byl rituximab aplikován sedmi pacientům v podobě čtyř infuzí v dávce 375 mg/m2 v týdenních intervalech a jednomu pacientovi v dávce 1000 mg v den 1 a v den 15 (58). Aktivita onemocnění byla hodnocena pomocí BVAS skóre před a 6 měsíců po podání rituximabu. Medián skóre BVAS před léčbou byl 14,3 (4–30). 6 měsíců po podání rituximabu mělo pět pacientů BVAS = 0. U třech pacientů bylo dosaženo kompletní a u třech částečné remise onemocnění. Dva pacienti neodpovídali na terapii. U dvou pacientů přetrvával nález plicních uzlů. U jednoho pacienta došlo po roce k relapsu onemocnění. Opakované podání rituximabu vedlo u tohoto pacienta opět k remisi onemocnění. Tato práce však naznačila možný rozdílný efekt terapie rituximabem na vaskulitické a na granulomatózní projevy onemocnění. Zatímco vaskulitické projevy choroby ustupovaly v řádu dní nebo týdnů, granulomatózní projevy regredovaly pomalu během několika měsíců.
V práci Aries et al. byly zveřejněny výsledky otevřené prospektivní studie s osmi pacienty s WG refrakterní na standardní terapii (59). Standardní terapie zahrnovala methylprednisolon, CFA, metotrexát, mykofenolát mofetyl a rovněž anti-TNFα preparáty (infliximab a etanercept). WG se manifestovala retroorbitálním granulomatózním procesem u pěti nemocných, plicními uzly a progredující granulomatózní sinusitidou u jednoho nemocného a subglotickou stenózou u dvou nemocných. Rituximab byl přidán ke standardní terapii CFA (n = 5), metotrexátem (n = 2) nebo mykofenolátem (n = 1) v dávce 375 mg/m2plochy povrchu těla každý 4. týden. Před každou infuzí rituximabu bylo podáno 100 mg methylprednisolonu a antihistaminika 30–60 minut před infuzí ke zmírnění akutních infuzních reakcí. Aktivita WG byla monitorována pomocí BVAS a pomocí dotazníku Disease Extent Index (DEI). Remise byla definována podle BVAS skóre jako absence nových nebo zhoršujících se příznaků choroby. Relaps onemocnění byl pak definován jako první nebo opětovná manifestace jednoho z příznaků choroby obsaženého v BVAS. Jako velký relaps bylo označeno život ohrožující nebo závažné postižení orgánů (plíce, mozek, oko, motorické nervy, střevo nebo ledviny). Léčba rituximabem byla u všech pacientů dobře tolerována. Čtyři týdny po poslední infuzi rituximabu byly dva pacienti v celkové a jeden v částečné remisi dle BVAS skóre. U třech pacientů se aktivita onemocnění nezměnila a u zbylých dvou pacientů došlo dokonce k nárůstu aktivity onemocnění. Jednalo se o pacienty s retroorbitálním postižením a subglotickou stenózou. U tří z pěti pacientů s retroorbitálními granulomy byla zaznamenána další progrese postižení, dokumentovaná MRI. U všech pacientů došlo k poklesu periferních B-lymfocytů pod měritelnou úroveň. K poklesu titru ANCA však došlo pouze u dvou pacientů. Po ukončení terapie rituximabem poklesla hladina CRP z mediánu 38 mg/l na hodnotu 14 mg/l a hodnota FW ze 70 mm/h na 32 mm/h. Dávka glukokortikoidů mohla být snížena u třech pacientů, neměnila se u jednoho a musela být zvýšena u čtyř pacientů. Pět pacientů refrakterních na terapii rituximabem bylo následně úspěšně léčeno pulzní terapií azathioprinem. Autoři se domnívají, že za relativním neúspěchem terapie rituximabem v této studii stojí převaha granulomatózního postižení u sledovaných pacientů s WG na rozdíl od dříve publikovaných prací, kde dominantním postižením byla vaskulitida a především glomerulonefritida. Autoři předpokládají, že jimi sledovaný a léčený soubor, zahrnující zejména pacienty s granulomatózním postižením, nereagující na terapii rituximabem, ale ani na terapii anti-TNFα preparáty, může představovat skupinu obtížně léčitelných pacientů s patogenezí choroby odlišnou od pacientů s převážně vaskulitickým postižením.
Granulomatózní léze u pacientů s WG jsou tvořeny aktivovanými tkáňovými makrofágy, obrovskými buňkami, neutrofilními granulocyty, CD4+CD28 - T-lymfocyty a B-lymfocyty (60, 61). Imunohistochemické studie prokázaly, že tyto CD4+CD28 - T-lymfocyty jsou hlavním zdrojem interferonu-γ a TNFα (62). Prostřednictvím těchto cytokinů mohou CD4+CD28 - T-lymfocyty podporovat migraci monocytů do místa zánětu a jejich aktivaci a následnou tvorbu granulomů. V granulomatózních lézích ze vzorků nosní sliznice pacientů s WG byla popsána infiltrace CD20+ B-lymfocyty vytvářejícími folikulární ložiska podobně jako v lymfatických uzlinách (63). Tyto CD20+ B-lymfocyty mohou být potenciálními producenty c-ANCA. Doposud však neexistuje přímá evidence, že by tyto B-lymfocyty a/nebo ANCA autoprotilátky hráli významnou roli v patogenezi granulomatózního zánětlivého procesu u WG. Na zvířecím modelu vedl lokální zánětlivý proces vyvolaný podáním intradermální injekce TNFα k vytvoření zánětu podkožní tukové tkáně (panikulitidě) za přítomnosti PR3+ ANCA, avšak nebyl pozorován vývoj granulomatózního zánětlivého procesu (64). Dalším důvodem relativního neúspěchu terapie rituximabem mohlo být přetrvávání hladiny ANCA a pravděpodobně nedostatečná deplece B-lymfocytů, případně existence dlouho žijících plazmatických buněk neexprimujících na svém povrchu znak CD20 (65–68). V poslední době narůstá evidence, že dlouho žijící plazmatické buňky s délkou přežívání několik měsíců až roků jsou významným zdrojem autoprotilátkové produkce v rámci různých autoimutních onemocnění (69, 70). Právě přítomnost ANCA autoprotilátek navzdory terapii rituximabem u pacientů s WG může svědčit pro přítomnost těchto dlouho žijících plazmatických buněk (71).
Závěr
Prezentujeme případ pacienta c-ANCA (PR3+) pozitivní WG s relabujícím aktivním onemocněním, manifestujícím se alveolární hemoragií, mnohočetnými plicními uzly s rozpadovými dutinami a postižením dutiny nosní. Onemocnění bylo refrakterní na standardní indukční terapii vysokými dávkami methylprednisolonu, pulzní terapii CFA a plazmaferézu. Při této terapii sice došlo k vymizení ANCA, ale přetrvávala vysoká aktivita onemocnění. Terapie rituximabem v podobě 4 i.v. infuzí aplikovaným v týdenním intervalu v dávce 375 mg/m2plochy povrchu těla vedla u pacienta k remisi onemocnění, zejména k výrazné redukci postižení plicního parenchymu. Léčba rituximabem byla dobře tolerována. V následujícím téměř 6měsíčním období sledování nebyl zaznamenán relaps onemocnění ani výskyt nežádoucích účinků. Tato terapie může být pravděpodobně použita u pacientů s vysoce aktivní WG s postižením životně důležitých orgánů. Dříve publikované práce naznačují příznivý efekt rituximabu zejména v léčbě vaskulitických projevů onemocnění.
MUDr. Petr Němec, Ph.D.
Revmatologická ambulance
II. interní klinika
FN u sv. Anny v Brně a LF MU
Pekařská 53
656 91 Brno
e-mail.: petr.nemec@fnusa.cz
Sources
1. Klinger H. Grenzformen der periarteritis nodosa. Frankfurt Z Pathol 1931; 42 : 455–80.
2. Wegener F. Uber generalisierte, septische Gefasserkrankungen. Verh Dtsch Ges Pathol 1936; 29 : 201–10.
3. Wegener F. Uber eine eigenartige rhinogene granulomatose mit besonderer beteiligung des arteriensystems und der nieren. Beitr Pathol Anat Allg Pathol 1939; 36 : 36–68.
4. Watts RA, Carruthers DM, Scott DG. Epidemiology of systemic vasculitis: changing incidence or definition? Semin Arthritis Rheum 1995; 25 : 28–34.
5. Stone HS, Hoffman GS. Wegeneręs granulomatosis and lymphomatoid granulomatosis. In.: Hochberg MC, Silman AJ, Smolen JS, Weinblatt ME, Weisman MH (eds). Rheumatology. 3rd ed. Edinburgh: Mosby; 2003. p. 1635–48.
6. Rottem M, Cotch MF, Fauci AS, Hoffman GS. Familial vasculitis: report of 2 families. J Rheumatol 1994; 21 : 561–3.
7. Cotch MF, Fauci AS, Hoffman GS. HLA typing in patients with Wegener granulomatosis. Ann Intern Med 1995; 122 : 635.
8. Lúdvíksson BR, Sneller MC, Chua KS, Talar-Williams C, Langford CA, Ehrhardt RO, et al. Active Wegener‘s granulomatosis is associated with HLA-DR+ CD4+ T cells exhibiting an unbalanced Th1-type T cell cytokine pattern: reversal with IL-10. J Immunol 1998; 160 : 3602–9.
9. Davies DJ, Moran JE, Niall JF, Ryan GB. Segmental necrotising glomerulonephritis with antineutrophil antibody: possible arbovirus aetiology? Br Med J (Clin Res Ed) 1982; 285 : 606.
10. van der Woude FJ, Rasmussen N, Lobatto S, Wiik A, Permin H, van Es LA, et al. Autoantibodies against neutrophils and monocytes: tool for diagnosis and marker of disease activity in Wegener‘s granulomatosis. Lancet 1985; 1 : 425–9.
11. Hagen EC, Daha MR, Hermans J, Andrassy K, Csernok E, Gaskin G, et al. Diagnostic value of standardized assays for anti-neutrophil cytoplasmic antibodies in idiopathic systemic vasculitis. EC/BCR Project for ANCA Assay Standardization. Kidney Int 1998; 53 : 743–53.
12. Kerr GS, Fleisher TA, Hallahan CW, Leavitt RY, Fauci AS, Hoffman GS. Limited prognostic value of changes in antineutrophil cytoplasmic antibody titer in patients with Wegener‘s granulomatosis. Arthritis Rheum 1993; 36 : 365–71.
13. Hoffman GS, Kerr GS, Leavitt RY, Hallahan CW, Lebovics RS, Travis WD, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116 : 488–98.
14. Nishino H, Rubino FA, Parisi JE. The spectrum of neurologic involvement in Wegener‘s granulomatosis. Neurology 1993; 43 : 1334–7.
15. Roberts GA, Eren E, Sinclair H, Pelling M, Burns A, Bradford R, et al. Two cases of Wegener‘s granulomatosis involving the pituitary. Clin Endocrinol (Oxf) 1995; 42 : 323–8.
16. Luqmani RA, Bacon PA, Moots RJ, Janssen BA, Pall A, Emery P, et al. Birmingham Vasculitis Activity Score (BVAS) in systemic necrotizing vasculitis. QJM 1994; 87 : 671–8.
17. Walton EW. Giant-cell granuloma of the respiratory tract (Wegener‘s granulomatosis). Br Med J 1958; 2 : 265–70.
18. Jayne D. Current attitudes to the therapy of vasculitis. Kidney Blood Press Res 2003; 26 : 231–9. Review.
19. Jayne D. How to induce remission in primary systemic vasculitis. Best Pract Res Clin Rheumatol 2005; 19 : 293–305. Review.
20. Tesař V. Terapie ANCA-pozitivní systémové vaskulitidy. In.: Pavelka K, et al. Farmakoterapie revmatických onemocnění. Praha: Grada Publishing; 2005: p. 287–90.
21. Guillevin L, Cordier JF, Lhote F, Cohen P, Jarrousse B, Royer I, et al. A prospective, multicenter, randomized trial comparing steroids and pulse cyclophosphamide versus steroids and oral cyclophosphamide in the treatment of generalized Wegener‘s granulomatosis. Arthritis Rheum 1997; 40 : 2187–98.
22. Pagnoux C. Wegener‘s granulomatosis and microscopic polyangiitis Rev Prat 2008; 58 : 522–32. Review.
23. Regan MJ, Hellmann DB, Stone JH. Treatment of Wegener‘s granulomatosis. Rheum Dis Clin North Am 2001; 27 : 863–86.
24. Langford CA, Talar-Williams C, Barron KS, Sneller MC. A staged approach to the treatment of Wegener‘s granulomatosis: induction of remission with glucocorticoids and daily cyclophosphamide switching to methotrexate for remission maintenance. Arthritis Rheum 1999; 42 : 2666–73.
25. Sneller MC, Hoffman GS, Talar-Williams C, Kerr GS, Hallahan CW, Fauci AS. An analysis of forty-two Wegener‘s granulomatosis patients treated with methotrexate and prednisone. Arthritis Rheum 1995; 38 : 608–13.
26. Nowack R, Göbel U, Klooker P, Hergesell O, Andrassy K, van der Woude FJ. Mycophenolate mofetil for maintenance therapy of Wegener‘s granulomatosis and microscopic polyangiitis: a pilot study in 11 patients with renal involvement. J Am Soc Nephrol 1999; 10 : 1965–71.
27. Ghez D, Westeel PF, Henry I, Pruna A, Fournier A, Lassoued K. Control of a relapse and induction of long-term remission of Wegener‘s granulomatosis by cyclosporine. Am J Kidney Dis 2002; 40: E6.
28. Inoue K, Kondo M, Inoue M, Ishino H, Kamitsuji Y, Sano H. Successful treatment with combination therapy of cyclophosphamide and cyclosporin for late recurrence of Wegener granulomatosis. Arch Intern Med 2000; 160 : 393–4.
29. Metzler C, Miehle N, Manger K, Iking-Konert C, de Groot K, Hellmich B, et al. Elevated relapse rate under oral methotrexate versus leflunomide for maintenance of remission in Wegener‘s granulomatosis. Rheumatology (Oxford) 2007; 46 : 1087–91.
30. White ES, Lynch JP. Pharmacological therapy for Wegener‘s granulomatosis. Drugs 2006; 66 : 1209–28. Review.
31. Specks U. Methotrexate for Wegener‘s granulomatosis: what is the evidence? Arthritis Rheum 2005; 52 : 2237–42. Review.
32. Langford CA, Hoffman GS. Intravenous immunoglobulin in Wegener‘s granulomatosis and microscopic polyangiitis. Arthritis Rheum 2008; 58 : 2211–2.
33. Richter C, Schnabel A, Csernok E, De Groot K, Reinhold-Keller E, Gross WL. Treatment of anti-neutrophil cytoplasmic antibody (ANCA)-associated systemic vasculitis with high-dose intravenous immunoglobulin. Clin Exp Immunol 1995; 101 : 2–7.
34. Lockwood CM, Thiru S, Stewart S, Hale G, Isaacs J, Wraight P, et al. Treatment of refractory Wegener‘s granulomatosis with humanized monoclonal antibodies. QJM 1996; 89 : 903–12.
35. Lamprecht P, Voswinkel J, Lilienthal T, Nolle B, Heller M, Gross WL, et al. Effectiveness of TNF-alpha blockade with infliximab in refractory Wegener’s granulomatosis. Rheumatology (Oxford) 2002; 41 : 1303–7.
36. Langford CA. Drug insight: anti-tumor necrosis factor therapies for the vasculitic diseases. Nat Clin Pract Rheumatol. 2008; 4 : 364–70. Review.
37. Stone JH, Wegener‘s Granulomatosis Etanercept Trial Research Group. Limited versus severe Wegener‘s granulomatosis: baseline data on patients in the Wegener‘s granulomatosis etanercept trial. Arthritis Rheum 2003; 48 : 2299–309.
38. Stone JH, Uhlfelder ML, Hellmann DB, Crook S, Bedocs NM, Hoffman GS. Etanercept combined with conventional treatment in Wegener‘s granulomatosis: a six-month open-label trial to evaluate safety. Arthritis Rheum 2001; 44 : 1149–54.
39. Flossmann O, Baslund B, Bruchfeld A, Cohen Tervaert JW, Hall C, et al. Deoxyspergualin in relapsing and refractory Wegener‘s granulomatosis. Ann Rheum Dis 2008. [Epub ahead of print]
40. Birck R, Warnatz K, Lorenz HM, Choi M, Haubitz M, Grunke M, et al. 15-Deoxyspergualin in patients with refractory ANCA-associated systemic vasculitis: a six-month open-label trial to evaluate safety and efficacy. J Am Soc Nephrol 2003; 14 : 440–7.
41. Hagen EC, de Keizer RJ, Andrassy K, van Boven WP, Bruijn JA, van Es LA, et al. Compassionate treatment of Wegener’s granulomatosis with rabbit anti-thymocyte globin. Clin Nephrol 1995; 43 : 351–9.
42. Davidson A, Diamond B. Autoimmune diseases. N Engl J Med 2001; 345 : 340–350.
43. Martin F, Chan AC. Pathogenic roles of B cells in human autoimmunity: insights from the clinic. Immunity 2004; 20 : 517–27.
44. Stevenson HC, Fauci AS. Activation of human B lymphocytes. XII. Differential effects of in vitro cyclophosphamide on human lymphocyte subpopulations involved in B-cell activation. Immunology 1980; 39 : 391–7.
45. Clayton AR, Savage CO. Production of antineutrophil cytoplasm antibodies derived from circulating B cells in patients with systemic vasculitis. Clin Exp Immunol 2003; 132 : 174–9.
46. Russell KA, Specks U. Are antineutrophil cytoplasmic antibodies pathogenic? Experimental approaches to understand the antineutrophil cytoplasmic antibody phenomenon. Rheum Dis Clin North Am 2001; 27 : 815–32.
47. Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, Aratani Y, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest 2002; 110 : 955–63.
49. Pfister H, Ollert M, Frohlich LF, Quintanilla-Martinez L, Colby TV, Specks U, et al. Antineutrophil cytoplasmic autoantibodies against the murine homolog of proteinase 3 (Wegener autoantigen) are pathogenic in vivo. Blood 2004; 104 : 1411–8.
50. Reff ME, Carner K, Chambers KS, Chinn PC, Leonard JE, Raab R, et al. Depletion of B-cells in vivo by a chimeric mouse human monoclonal antibody to C020. Blood 1994; 83 : 435–45.
51. Němec P. Rituximab (MabThera) a new biological medicine in rheumatoid arthritis therapy. Vnitr Lek 2007; 53 : 1199–210.
52. Maloney DG, Smith B, Appelbaum FR. The antitumor effect of monoclonal anti-CD20 antibody (mAb) therapy includes direct anti-proliferative activity and induction of apoptosis in CD20 positive non-Hodgkin‘s lymphoma (NHL) cell lines. Blood 1996; 88 (Suppl. 1): 637a.
53. Specks U, Fervenza FC, McDonald TJ, Hogan MCE. Response of Wegener’s granulomatosis to anti-CD20 chimeric monoclonal antibody therapy. Arthritis Rheum 2001; 44 : 2836–40.
54. Stasi R, Stipa E, Del Poeta G, Amadori S, Newland AC, Provan D. Long-term observation of patients with anti-neutrophil cytoplasmic antibody-associated vasculitis treated with rituximab. Rheumatology (Oxford) 2006; 45 : 1432–6.
55. Eriksson P. Nine patients with anti-neutrophil cytoplasmic antibody-positive vasculitis successfully treated with rituximab. J Intern Med 2005; 257 : 540–8.
56. Keogh KA, Wylam ME, Stone JH, Specks U. Induction of remission by B lymphocyte depletion in eleven patients with refractory antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2005; 52 : 262–8.
57. Keogh KA, Ytterberg SR, Fervenza FC, Carlson KA, Schroeder DR, Specks U. Rituximab for refractory Wegener‘s granulomatosis: report of a prospective, open-label pilot trial. Am J Respir Crit Care Med 2006; 173 : 180–7.
58. Brihaye B, Aouba A, Pagnoux C, Cohen P, Lacassin F, Guillevin L. Adjunction of rituximab to steroids and immunosuppressants for refractory/relapsing Wegener‘s granulomatosis: a study on 8 patients. Clin Exp Rheumatol 2007; 25 (Suppl 44): S23-7.
59. Aries PM, Hellmich B, Voswinkel J, Both M, Nölle B, Holl-Ulrich K, et al. Lack of efficacy of rituximab in Wegener‘s granulomatosis with refractory granulomatous manifestations. Ann Rheum Dis 2006; 65 : 853–8.
60. Csernok E, Trabandt A, Muller A, Wang GC, Moosig F, Paulsen J, et al. Cytokine profiles in Wegener’s granulomatosis: predominance of type 1 (Th1) in granulomatous inflammation. Arthritis Rheum 1999; 42 : 742–50.
61. Lamprecht P, Moosig F, Csernok E, Schnabel A, Mueller A, Gross WL. CD28 negative T cells are enriched in granulomas lesions of the respiratory tract in Wegener’s granulomatosis. Thorax 2001; 56 : 751–7.
62. Komocsi A, Lamprecht P, Csernok E, Mueller A, Holl-Ulrich K, Seitzer U, et al. Peripheral blood and granuloma CD4+CD28 - T cells are the major source of interferon-gamma and tumor necrosis factor-alpha in Wegeners granulomatosis. Am J Pathol 2002; 160 : 1717–24.
63. Voswinkel J, Kraemer J, Mueller A, Herlyn K, Lamprecht P, Gross WL, et al. B lymphocytes infiltrating Wegener’s granuloma: the immunoglobulin VH gene repertoire from granulomatous tissues displays an antigen-driven maturation and suggests a microbial trigger. Arthritis Res 2004; 6 : 24.
64. Pfister H, Ollert M, Frohlich LF, Quintanilla-Martinez L, Colby TV, Specks U, et al. Antineutrophil cytoplasmic autoantibodies against the murine homolog of proteinase 3 (Wegener autoantigen) are pathogenic in vivo. Blood 2004; 104 : 1411–18.
65. O’Connor BP, Gleeson MW, Noelle RJ, Erickson LD. The rise and fall of long-lived humoral immunity: terminal differentiation of plasma cells in health and disease. Immunol Rev 2003; 194 : 61–76.
66. Manz RA, Radbruch A. Plasma cells for a lifetime ? Eur J Immunol 2002; 32 : 923–7.
67. Manz RA, Thiel A, Radbruch A. A lifetime of plasma cells in the bone marrow. Nature 1997; 388 : 133–4.
68. Slifka MK, Anita R, Whitmire JK, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity 1998; 8 : 362–72.
69. Hoyer BF, Moser K, Hauser AE, Peddinghaus A, Voigt C, Eilat D, et al. Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in NZB/W mice. J Exp Med 2004; 199 : 1577–84.
70. Arce S, Cassese G, Hauser AE, Dorner T, Odenthal M, Manz R, et al. The role of long-lived plasma-cells in autoimmunity. Immunobiology 2002; 206 : 558–62.
71. Cambridge G, Leandro MJ, Edwards JC, Ehrenstein MR, Salden M, Bodman-Smith M, et al. Serologic changes following B lymphocyte depletion therapy for rheumatoid arthritis. Arthritis Rheum 2003; 48 : 2146–54.
Labels
Dermatology & STDs Paediatric rheumatology RheumatologyArticle was published in
Czech Rheumatology

2009 Issue 1
Most read in this issue
- Interleukin 6 in rheumatic diseases
- News in the biological therapy of rheumatoid arthritis and future prospects
- Rituximab in the treatment of Wegener’s granulomatosis non-responsive to standard therapy
- Collagen and elastin degradation products as potential activity markers of scleroderma