
Myozitida rezistentní na léčbu
Treatment resistant myositis
When confronted with a dermatomyositis or polymyositis patient not responding to immunosuppressive treatment, physician must firstly ask whether the original diagnosis was correct. In this review we provide a guide to the clinical features and ancillary tests, which might be helpful in the differential diagnosis of myositis. A particular attention is paid to the role of autoantibody detection, as some of them are not only relatively specific for the disease but also associated with unique clinical features including resistance to treatment. Subsequently other possible explanations of treatment resistance are listed and a short overview of treatment options for resistant myositis patients is given.
Key words:
polymyositis, dermatomyositis, inclusion body myositis (IBM)
Authors:
H. Mann 1; J. Vencovský; Lundberg Ie 1 2
Authors‘ workplace:
Revmatologický ústav a Revmatologická klinika 1. LF UK, Praha, 2Rheumatology Unit, Department of Medicine
Karolinska University Hospital, in Solna, Karolinska Institutet, Stockholm Sweden
1
Published in:
Čes. Revmatol., 18, 2010, No. 2, p. 97-108.
Category:
Overview Reports
Overview
U nemocného s dermatomyozitidou nebo s polymyozitidou, která nereaguje na imunosupresivní léčbu je třeba nejprve ověřit, že původní diagnóza je správná. V přehledném článku jsme poskytli přehled klinických projevů a pomocných vyšetření, které mohou být užitečné v diferenciání diagnóze myozitidy. Zvláštní pozornost je věnována roli vyšetření autoprotilátek, protože některé z nich jsou nejen relativně specifické pro myozitidu, ale mohou být navíc spojeny s určitými klinickými projevy, včetně rezistence na léčbu. Uvádíme také další možná vysvětlení rezistence na léčbu a krátký přehled možností léčby pacientů s nedostatečnou odpovědí na léčbu.
Klíčová slova:
polymyozitida, dermatomyozitida, myozitida s inkluzními tělísky (IBM)
Kazuistika
27letý muž bez významné osobní a rodinné anamnézy byl hospitalizován pro svalovou slabost. V předchorobí neužíval žádné léky ani drogy a nebyl vystaven působení toxických látek. Zdravotní obtíže nemocného začaly před 10 měsíci dysfagií. Před 5 měsíci se objevila rychle progredující symetrická svalová slabost postihující převážně proximální svalové skupiny. Svaly obličeje postiženy nebyly. Nemocný neměl žádné kloubní obtíže, dušnost ani kašel a nevšiml si žádných kožních příznaků. Vzhledem k těžké svalové slabosti je pacient momentálně na JIP neurologického oddělení.
Při fyzikálním vyšetření nejsou přítomny kožní změny typické pro dermatomyozitidu, artritidy ani abnormální poslechový nález při vyšetření plic. Svalová síla je výrazně oslabená v proximálních svalových skupinách (mm. glutei 2/10, m. deltoideus 4/10) a normální distálně. Laboratorně je přítomna významná elevace svalových enzymů (myoglobin 1700, CK 52) a snížená hodnota kreatininu. Vyšetření antinukleárních autoprotilátek a dalších běžně vyšetřovaných autoprotilátek s myozitidou asociovaných nebo pro myozitidu specifických je negativní. Při EMG vyšetření je zachycena zvýšená inzerční aktivita s fibrilacemi a pozitivními ostrými vlnami a myopatické akční potenciály, nález byl hodnocený jako konzistentní s myozitidou. Ve vzorku svalové biopsie odebraném z m. deltoideus jsou patrné převážně nekrotické změny s jedním malým ložiskem perivaskulárného infiltrátu. Je přítomná difuzní exprese HLA-I v kolísající intenzitě. Pomocná vyšetření neprokázala postižení vnitřních orgánů.
Pro suspektní idiopatickou zánětlivou myopatii charakteru polymyozitidy bylo rozhodnuto o zahájení léčby glukokortikoidy. Po podání i.v. pulzů metylprednisolonu a nasazení prednisonu v dávce 1 mg/kg došlo k mírnému poklesu svalových enzymů, ale svalová síla se nezlepšila. Vzhledem k závažnému stavu nemocného byla léčba glukokortikoidy doplněna dvěma dalšími imunosupresivy – metotrexátem a cyklosporinem, ale ani tato kombinace nevedla k významnému zlepšení stavu nemocného. Jaký by měl být další postup?
Úvod
Dospělé idiopatické zánětlivé myopatie (IZM), běžně označované jako myozitidy, jsou heterogenní skupinou onemocnění s pravděpodobně autoimunitní etiologií, které jsou charakterizovány proximální svalovou slabostí a nehnisavým zánětem kosterních svalů. IZM se tradičně dále dělí na polymyozitidu, dermatomyozitidu, myozitidu asociovanou s maligním onemocněním (CAM) a myozitidu v rámci překryvného syndromu s jiným onemocněním pojiva. Sporadická myozitida s inkluzními tělísky(s-IBM) bývá navzdory odlišným klinickým projevům a rezistenci na imunosupresivní léčbu také řazena mezi IZM (1). Kromě těchto byly popsány ještě další vzácné formy myozitidy (1).
Diagnóza zánětlivých myopatií
Nejčastěji užívaná klasifikační kritéria pro IZM, která byla navržena Bohanem a Peterem v r. 1975, jsou založena na kombinaci klinických projevů, elektromyografického nálezu, elevace svalových enzymů a výsledku svalové biopsie (2). Autoři uvádějí řadu nervosvalových onemocnění, která musí být vyloučena u nemocných s diagnózou IZM. Kritéria Bohana a Petera bývají kritizována především pro svou nízkou specificitu při určování diagnózy polymyozitidy, přesto zůstávají i v současné době standardním nástrojem běžně užívaným v klinické praxi a, pokud jsou podporována pozitivním bioptickým nálezem, jsou považována za dostatečná pro zařazení pacientů s IZM do klinických hodnocení podle doporučení mezinárodní společnosti IMACS (The International Myositis Assessment and Clinical Studies Group). Kritéria Bohana a Petera neumožňují diagnostikovat s-IBM, tato jednotka je obvykle diagnostikována na základě typického nálezu ve svalové biopsii (3). Používání kritérií Bohana a Petera může vést k chybné diagnóze polymyozitidy u nemocného s jinou myopatií spojenou se zánětlivými změnami ve svalové biopsii. Nejčastějším chybně diagnostikovaným onemocněním je pravděpodobně s-IBM. V současné době probíhá mezinárodní multidisciplinární spolupráce s cílem vytvořit nová klasifikační kritéria pro IZM.
Standardní léčba IZM v současnosti
Léčba IZM je založena na kombinaci farmakoterapie a rehabilitace. Stav většiny pacientů s polymyozitidou nebo s dermatomyozitidou se v průběhu léčby zlepší alespoň částečně, zatímco nemocní s s-IBM jsou většinou rezistentní na imunosupresivní léčbu i na fyzioterapii. Dále popisovaná doporučení se proto vztahují na léčbu polymyozitidy a dermatomyozitidy léčba s-IBM bude zmíněna zvlášť. Základem farmakoterapie IZM je imunosupresivní léčba. Vzhledem k tomu, že bylo provedeno jen velmi málo kontrolovaných klinických studií na malém počtu nemocných, je většina doporučení založena na výsledcích nezaslepených studií a kazuistických sérií. Léčbou volby zůstávají orálně podávané glukokortikoidy v úvodních dávkách mezi 0,75 mg až 1 mg/kg. V závažných případech mohou být indikovány i.v. pulzy metylprednisolonu. V současné době většina autorů doporučuje kombinovat glukokortikoidy s dalším imunosupresivním přípravkem. Nejčastěji je v této indikaci užíván metotrexát v dávce 15 až 25 mg týdně nebo azatioprin v dávce 2 mg/kg/den. Nedílnou součástí léčby IZM je rehabilitace, která by měla být zahájena současně s imunosupresivní léčbou (4). Aktivita onemocnění není kontraindikací fyzioterapie, ovšem je důležité aby byl zajištěn dohled odborného rehabilitačního pracovníka.
V průběhu léčby je nutné monitorovat aktivitu onemocnění a svalovou sílu. U pacientů s přetrvávající svalovou slabostí je důležité odlišit, zda je příčinou pokračující aktivita onemocnění nebo spíše svalové poškození. Změny léčby nelze provádět pouze na základě hodnot sérové kreatinkinázy. IMACS vypracoval základní soubor nástrojů, které hodnotí aktivitu onemocnění, rozsah poškození a kvalitu života u nemocných s myozitidou (5). K posouzení aktivity onemocnění slouží šest položek: hodnocení celkové aktivity onemocnění lékařem a pacientem, dotazník HAQ (Health Assessment Questionnaire), svalový test, hodnota svalových enzymů (dva z následujících: kreatinkináza, laktát dehydrogenáza, ALT, AST) a nástroje k hodnocení extramuskulární aktivity – MDAAT a MYOACT. Tyto nástroje byly původně vytvořeny pro užití v rámci klinických hodnocení, ale jsou užitečné i v běžné klinické praxi.
Idiopatická zánětlivá myopatie rezistentní na léčbu
U nemocného, který neodpovídá na léčbu musíme zvažovat několik možných vysvětlení: 1) nesprávná diagnóza, 2) nedostatečné dávkování léčiv nebo přítomnost steroidní myopatie, 3) přetrvávající svalová slabost v důsledku svalového poškození nebo atrofie, 4) okultní malignita (zvláště u dermatomyozitidy) a 5) skutečná rezistence na léčbu.
Je diagnóza správná?
První otázka, na kterou si musíme odpovědět, když pacient neodpovídá na léčbu, je zda je diagnóza určena správně. Je třeba si uvědomit, že IZM jsou vzácná a poměrně heterogenní skupina onemocnění s velmi širokou diferenciální diagnózou (tab. 1). Poněkud jednodušší je situace u pacientů s dermatomyozitidou, u nichž typické kožní projevy mohou být patognomonické (6). Kožní projevy mohou předcházet svalovému postižení o několik měsíců až let. V některých případech se může dermatomyozitida manifestovat jako čistě kožní onemocnění bez svalového zánětu (tzv. amyopatická forma) a jindy mohou být změny typické pro dermatomyozitidu přítomny v bioptickém nálezu u nemocného bez kožních projevů (dermatomyositis sine dermatitis) (7). Diagnóza polymyozitidy je mnohem složitější, protože neexistuje žádný specifický příznak nebo nález, s možnou výjimkou několika specifických autoprotilátek, a proto poměrně často bývá diagnóza určena chybně. Je důležité vědět, že i u nemocných se svalovými dystrofiemi nebo s jinými nezánětlivými myopatiemi může dojít v průběhu léčby glukokortikoidy k částečnému zlepšení, a proto nelze odpověď na léčbu použít jako průkaz autoimunitní etiologie onemocnění. Chybně indikovaná léčba glukokortikoidy u nemocného s nezánětlivou myopatií vystavuje pacienta zbytečnému riziku vedlejších účinků a v případě deficitu dysferlinu může vést k nevratnému zhoršení svalové slabosti (8). U rezistentních nemocných je nezbytné pečlivě revidovat anamnézu, klinické projevy a výsledky provedených vyšetření (tab. 2) se zvláštním důrazem na nejcennější informaci – výsledek svalové biopsie. Často je k potvrzení nebo odmítnutí původní diagnózy nebo k posouzení aktivity onemocnění potřeba indikovat vyšetření nová a často i zopakovat svalovou biopsii.


Anamnéza
Pozitivní rodinná anamnéza svalového onemocnění není u nemocných s IZM častá a měla by vzbudit podezření na možnou svalovou dystrofii (x vázaná, autozomálně recesivní nebo dominantní dědičnost), mitochondriální (dědičnost v mateřské linii) nebo metabolickou myopatii (většinou autozomálně recesivní dědičnost). Nemocní s IZM často trpí dalšími autoimunitními chorobami, ale tento fakt nepomůže odlišit polymyozitidu od s-IBM, protože zvýšená frekvence autoimunitních chorob byla popsána u obou (9). Důležitý je také věk v době prvních projevů, protože polymyozitida se prakticky nevyskytuje u mladších 18 let a s-IBM obvykle postihuje osoby nad 50 let věku.
Řada infekcí a toxinů může způsobovat příznaky podobné IZM, a proto je třeba aktivně pátrat po anamnéze toxické expozice, cestách do endemických oblastí nebo po jiných relevantních rizikových faktorech. Toxické myopatie jsou často navozené léčivy (tab. 3) (10). Nejčastěji užívanými léčivy v běžné praxi, která mají potenciál způsobovat myopatii jsou hypolipidemika. Některá léčiva mohou způsobovat kožní změny připomínající dermatomyozitidu bez svalového postižení (11).
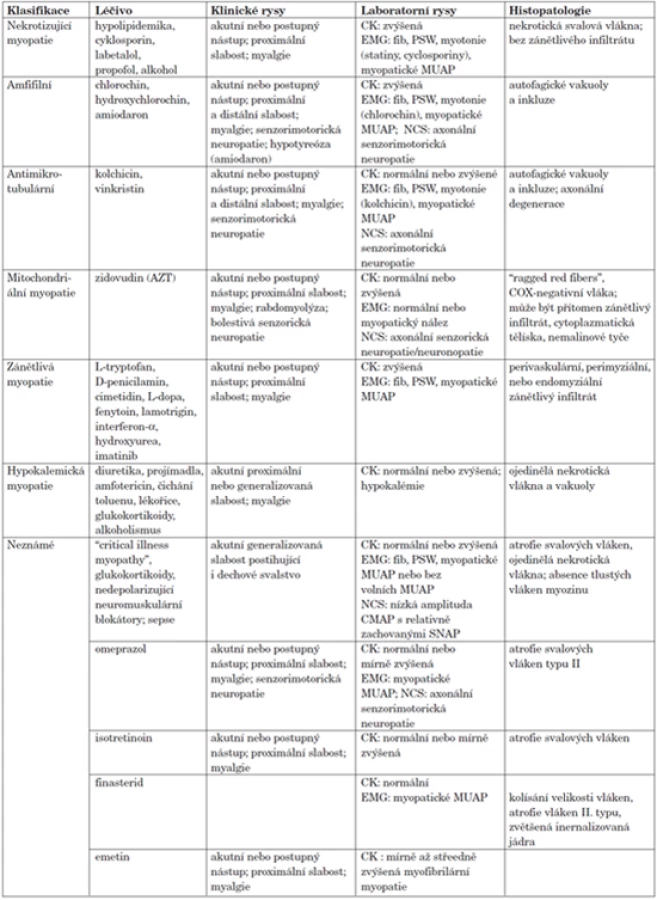
Svalové projevy
IZM typicky začínají subakutně nebo chronicky a hlavním projevem bývá převážně proximální symetrická svalová slabost. S výjimkou s-IBM nebývají postiženy svaly obličeje. Dlouhodobé onemocnění může vést ke vzniku svalových atrofií. Myalgie nejsou typickým příznakem IZM (12). Pro s-IBM je charakteristická pomalu progredující svalová slabost, která může být asymetrická. Selektivní postižení musculus quadriceps femoris a flexorů zápěstí a prstů spojené s výraznou atrofií v této distribuci je často patrné již v časných fázích onemocnění (4).
Náhlý začátek svalové slabosti, rabdomyolýza, klidová svalová bolest nebo těžké svalové křeče nejsou typické pro IZM a měly by podnítit párání po možné toxické myopatii nebo jiné diagnóze (10, 13). Pseudohypertrofie svalů nebo rychlý rozvoj svalové atrofie jsou typickými projevy svalových dystrofií (8). Dystrofinopatie se obvykle projevují v dětském věku, ale u nemocných s Beckerovou dystrofií (neúplný deficit dystrofinu) se mohou objevit příznaky po 30. roce věku a ženy-přenašečky genu pro Duchennovu svalovou dystrofii mohou mít svalovou slabost až v dospělosti. U některých pacientů s facioscapulohumerální svalovou dystrofií (FSHD) se mohou objevit příznaky dokonce až v 5. dekádě života. FSHD může být spojena se zánětlivým infiltrátem ve svalové biopsii, ale distribuce svalové slabosti s postižením svalů obličeje se výrazně liší od projevů polymyozitidy. Pletencové svalové dystrofie se mohou také poprvé projevovat v dospělém věku.
V závažných případech mohou IZM postihovat i dýchací svaly. Slabost dýchacích svalů disproporcionální v porovnání s postižením jiných svalových skupin, která vede k respiračnímu selhání, může být projevem deficitu kyselé maltázy. Myotonie není projevem IZM, a pokud je přítomná u dospělého pacienta s proximální svalovou slabostí, měla by vzbudit podezření na myotonickou dystrofii typu 2. Viditelné fascikulace jsou způsobené postižením dolního motoneuronu a mohou být patrné u pacientů s amyotrofickou laterální sklerózou.
Záchvatovité svalové příznaky, které se projeví pouze v okamžiku, kdy úroveň fyzické aktivity nebo stav výživy vyžaduje použití defektní metabolické dráhy k zajištění metabolismu svalů, jsou charakteristické pro metabolické myopatie. U mnoha takto nemocných dominuje únava nad skutečnou svalovou slabostí. Bolesti hlavy nebo přítomnost nauzey v průběhu symptomatické epizody svědčí pro možnou metabolickou příčinu. U nemocných s deficitem myofosforylázy (McArdleova choroba) typicky dochází k výraznému zlepšení fyzické výdrže po krátkém odpočinku. Tento jev se v angličtině nazývá „second wind“. Špatná tolerance fyzické zátěže doprovázená slabostí zhoršovanou příjmem cukrů („out of wind“ fenomén) bývá projevem deficitu fosfofruktokinázy (14).
Mimosvalové postižení
IZM jsou systémová onemocnění a u některých pacientů (např. u pacientů s antisyntetázovým syndromem (ASS) (tab. 4) může mimosvalové postižení dominovat klinickému obrazu onemocnění. Typ mimosvalového postižení může významně pomoci při formulování diferenciální diagnózy u pacientů s myozitidou rezistentní na léčbu. Přítomnost celkových příznaků zánětu jako je horečka a únava svědčí pro zánětlivou etiologii. Intersticiální plicní postižení a artritida jsou poměrně časté u nemocných s IZM a pokud jsou přítomné, klesá pravděpodobnost jiné etiologie myopatie. Dermatomyozitida je spojena s charakterickými kožními projevy. U nemocných s ASS mohou být přítomny hyperkeratotické léze na dlaních nebo laterálních stranách prstů, které se pro svůj vzhled nazývají „prsty“ nebo „ruce mechanika“. Neurologické příznaky nejsou projevem IZM a mohou být přítomny u pacientů s mitochondriální myopatií nebo s glykogenózou. Katarakty jsou častou komplikací léčby glukokortikoidy u nemocných s IZM, ale předčasný vznik katarakt je příznakem mitochondriálního onemocnění nebo myotonické dystrofie. Mezi gastrointestinální příznaky IZM patří dysfagie, která může způsobovat aspirační pneumonitidu. Hepatosplenomegalie a pseudoobstrukce tlustého střeva nebo postižení ledvin bývá projevem mitochondriální myopatie. Přestože jsou kardiovaskulární komplikace závažnou příčinou mortality u nemocných s IZM, klinicky významné postižení srdce je poměrně vzácné. U nemocných s IZM se mohou vyskytovat převodní poruchy, arytmie a městnavé srdeční selhání, pravděpodobně způsobené myokarditidou (14). Kardiomyopatie může být projevem svalové dystrofie, glykogenózy nebo mitochondriální myopatie.

Je důležité připomenout, že zvláště dermatomyozitida je spojená se zvýšeným rizikem přítomnosti maligního onemocnění, které nemusí být v době diagnózy zjevné. U všech nemocných se proto doporučuje skrínink maligních onemocnění přiměřený věku a rizikovým faktorům, a zvláště u starších osob nebo u pacientů s nedostatečnou odpovědí na léčbu je v průběhu prvních let trvání choroby nutná zvýšená ostražitost.
Svalové enzymy
U převážné většiny pacientů s dermatomyozitidou nebo s polymyozitidou je přítomna elevace sérové hladiny CK, obvykle do 50násobku horní hranice normy. Elevace CK není specifická pro IZM a může být přítomna rovněž u pacientů se svalovou dystrofií, metabolickou a mitochondriální myopatií, hypotyreózou a u nemocných s toxickými nebo léky indukovanými myopatiemi. U některých pacientů s IZM mohou být hodnoty CK normální navzdory přítomnosti aktivního zánětu ve svalové tkáni.
Dalšími běžně vyšetřovanými svalovými enzymy jsou laktátdehydrogenáza (LDH) a myoglobin. LDH je pravděpodobně nejvíce užitečným enzymem při monitorování aktivity IZM u nemocných s prokázanou diagnózou (14). Přítomnost myoglobinurie nebo zvýšení CK nad 100násobek horní hranice normy není u IZM obvyklé a svědčí spíše pro jinou etiologii myopatie.
Autoprotilátky
Vyšetřování přítomnosti autoprotilátek je v diagnostice IZM velmi přínosné, protože pozitivní výsledek směřuje naše uvažování směrem k systémovému autoimunitnímu onemocnění. Naopak zcela negativní výsledek vyšetření autoprotilátek u nemocného bez typických mimosvalových projevů IZM svědčí spíše pro nezánětlivou myopatii. Přítomnost některých autoprotilátek je navíc spojena s určitým souborem příznaků nebo typem orgánového postižení a jejich detekce tak může pomoci při posouzení prognózy nemocného.
Tradičně se autoprotilátky dělí na protilátky specifické pro IZM a ty, které jsou s IZM jen asociované a lze je nalézt i u jiných nemocí, jako např. u systémového lupus erytematodes, sklerodermie nebo u smíšeného onemocnění pojiva (MCTD). V posledních letech bylo popsáno několik nových protilátek specifických pro myozitidu. Některé z nich jsou již vyšetřovány rutinně, ale jiné mohou být detekovány zatím pouze na několika specializovaných výzkumných pracovištích.
Detekce autoprotilátek
Při vyšetřování autoprotilátek specifických pro nebo asociovaných s IZM se používají různé metody. Nepřímá imunofluorescence na HEp-2 buňkách detekuje antinukleární a/nebo anticytoplazmatické autoprotilátky. Tyto autoprotilátky musí být následně dourčeny specifickými testy, např. technikou ELISA s purifikovanými nebo rekombinantními antigeny, line nebo dot-blot immunoeseji s autoantigeny nanesenými na nitrocelulózovém papíru, imunodifuzí nebo protisměrnou imunoelektroforézou. Některé autoprotilátky lze však prokázat pouze imunoprecipitačními metodami. Tato komplikovaná vyšetření jsou prováděna pouze v několika málo specializovaných laboratořích. Ukazuje se, že výsledky různých běžně dostupných testů se mohou lišit a poskytovat tak falešně pozitivní nebo falešně negativní výsledky. Senzitivita a specificita daného testu by měla být ověřena, ideálně porovnáním s výsledkem imunoprecipitace. Při interpretaci výsledků vyšetření autoprotilátek u nemocných s podezřením na IZM je třeba vždy velké opatrnosti, k odhalení skutečné pozitivity je někdy třeba použít kombinace různých detekčních metod.
Antisyntetázové protilátky
Přítomnost jedné z antisyntetázových protilátek (tab. 5) je silně asociovaná s antisyntetázovým syndromem (ASS) (16). Zatím bylo popsáno osm autoprotilátek proti různým syntetázám, z nichž 5 již lze detekovat komerčně dostupnými testy (tab. 5). Daleko nejčastější jsou protilátky prot histidyl-tRNA syntetáze (anti-Jo-1), které jsou přítomné u 20–25 % pacientů s IZM (17). Nejnověji byly popsány anti-fenylalanyl-tRNA syntetáza (anti-Zo) a anti-tyrosyl tRNA syntetáza (anti-YRS), každá zatím jen u jednoho nemocného a obě byly spojené s přítomností antisyntetázového syndromu (18, 19). ASS obvykle začíná subakutně, prvním nebo hlavním projevem je často intersticiální plicní proces (ILD), který může dominovat nad postižením svalů. Tento scénář je zvláště častý u nemocných s anti-PL-12, anti-OJ a anti-AsnRS protilátkami. I u pacientů s pozitivitou anti-Jo-1 protilátek se intersticiální plicní proces projeví zhruba v jedné třetině případů dříve než myozitida (20). Pacienti často mívají symetrickou polyartritidu kloubů ruky a zápěstí, která se podobá revmatoidní artritidě, ale obvykle nepostihuje klouby nohou. ASS odpovídá na léčbu středně dobře a má tendenci k exacerbacím při snižování dávek imunosupresiv.
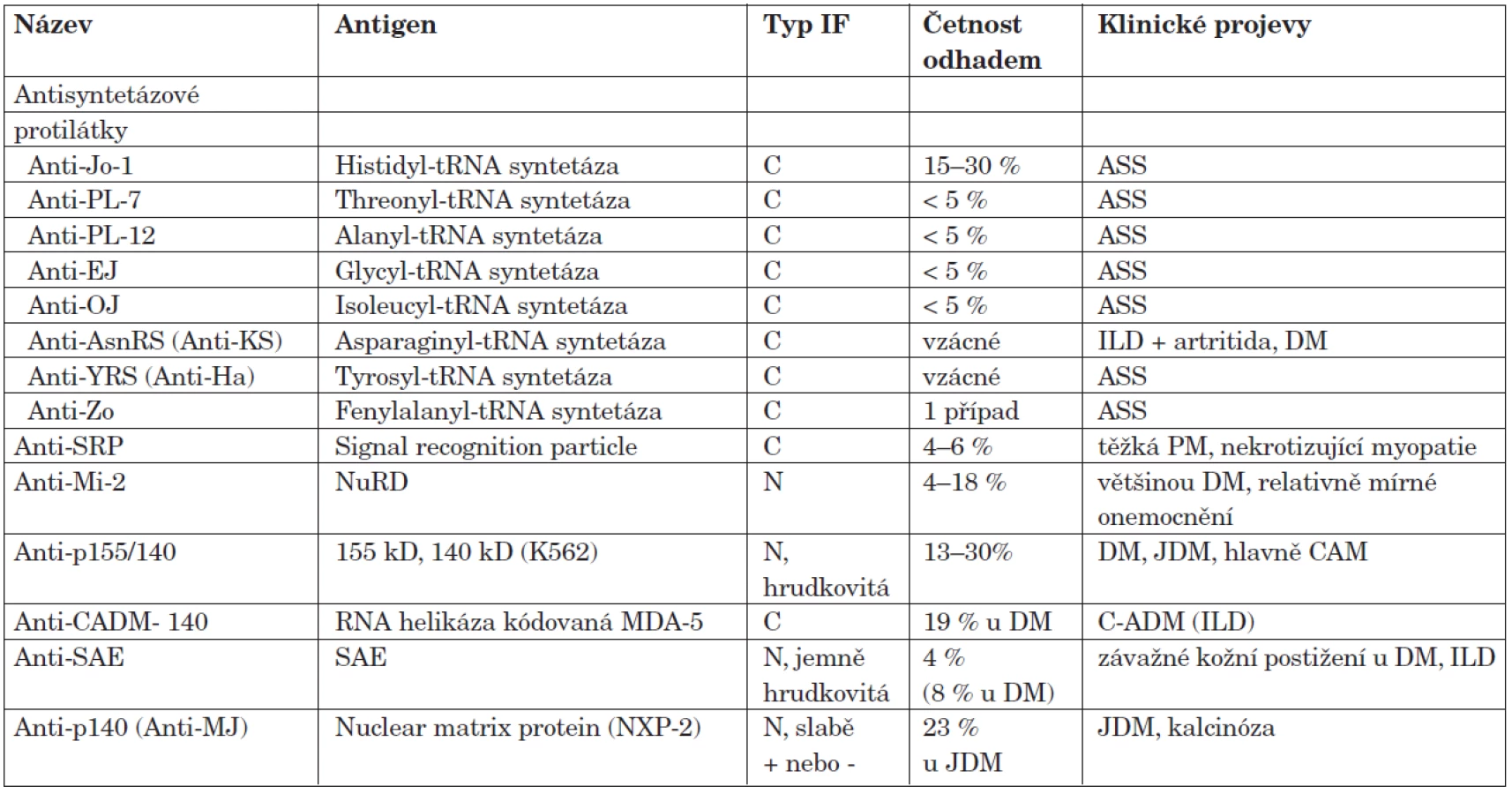
Sérové hladiny protilátek anti-Jo-1 do určité míry korelují s aktivitou onemocnění (21). Zatím ale není jasné, zda lze tohoto poznatku prakticky využít v klinické praxi. Anti-Jo-1 autoprotilátky jsou poměrně specifické pro polymyozitidu a dermatomyozitidu, i když byly v jedné laboratoři prokázány i u několika pacientů s s-IBM (17) a proto jejich přítomnost tuto diagnózu nevylučuje.
Anti-SRP
Nemocní s pozitivitou anti-SRP protilátek jsou ve srovnání s ostatními pacienty s polymyozitidou obvykle rezistentnější k léčbě glukokortikoidy a imunosupresivy. Anti-SRP autoprotilátky jsou přítomné převážně u pacientů s polymyozitidou, i když byly publikovány ojedinělé případy nemocných se systémovou sklerodermií nebo s antisyntetázovým syndromem (22). Pacienti s anti-SRP protilátkami obvykle mají závažnou svalovou slabost a atrofie svalů s disabilitou, která rychle progreduje v průběhu několika málo měsíců(23). Nemocní často trpí závažnou dysfagií. Původní zprávy o častém postižení srdce u nemocných s anti-SRP protilátkami se v dalších studiích nepotvrdily (22). Intersticiální plicní proces je přítomen u téměř čtvrtiny pacientů. Sérové hodnoty CK bývají velmi vysoké a ve svalová biopsii jsou přítomna četná nekrotická svalová vlákna s jen mírným nebo bez endomyziálního zánětu (22, 23).
Anti-Mi-2
Pacienti s pozitivitou anti-Mi-2 protilátek obvykle dobře reagují na léčbu. Tito nemocní mívají klasickou dermatomyozitidu s typickými kožními projevy včetně Gottronových známek nebo papul, heliotropního exantému, šálového erytému a s periungválními změnami. Ačkoli anti-Mi-2 protilátky jsou obvykle asociovány s dermatomyozitidou, bývají přítomny pouze u některých pacientů s touto chorobou. Navíc, v závislosti na použité detekční metodě, byly zachyceny i u polymyozitidy a, při použití testu ELISA proti fragmentům Mi-2, dokonce i u s-IBM (24).
Anti-PM/Scl
Pacienti s anti-PM/Scl protilátkamí mívají myozitidu nebo sklerodermii, většinou s mírným kožním postižením. U 50–70 % pacientů s anti-PM/Scl protilátkami jsou přítomné klinické příznaky obou těchto onemocnění a 24 % pacientů s tímto překryvným syndromem má anti-PM/Scl autoprotilátky. Kromě asociace s myozitidou a/nebo sklerodermií byla u těchto nemocných popsána vyšší frekvence Raynaudova fenoménu, artritidy a artralgií, intersticiálního plicního postižení, Sjögrenova syndromu, dysfagie a kalcinózy (25). Plicní postižení bývá méně závažné než u ASS a pacienti mají obvykle dobrou prognózu (25). Anti-PM/Scl protilátky jsou zaměřeny převážně proti dvěma molekulám o hmotnosti 100 kDa (100 %) a 75 kDa (60 %). Nedávno byla vyvinuta citlivá metoda ELISA schopná detekovat hlavní alfa helikální epitop PM/Scl-100 (PM-1), která měla ve vyšetřované skupině senzitivitu 55 % u pacientů s překryvným syndromem polymyozitida/sklerodermie, 7,5 % u nemocných s polymyozitidou a 7,9 % u pacientů se sklerodermií.
Anti-Ku
Nemocní s anti-Ku protilátkami obvykle dobře odpovídají na léčbu glukokortikoidy a mají dobrou prognózu. Anti-Ku mohou být přítomny také u jiných nemocí pojivové tkáně (26). Velká variace v uváděné frekvenci výskytu je pravděpodobně závislá na použitých metodách. Nejčastější klinické projevy pozorované u těchto pacientů jsou postižení plic, myozitida, Raynaudův fenomén, sicca syndrom a sklerodaktylie (26).
Anti-U1RNP
Pacienti s anti-nRNP protilátkami obvykle mají myozitidu buď jako součást MCTD, nebo v rámci překryvného syndromu se SLE nebo se sklerodermií. Anti-U1RNP pozitivní myozitida většinou odpovídá na léčbu glukokortikoidy a bývá asociována s dalšími příznaky, jako jsou polyartritida, Raynaudův fenomén, sklerodaktylie a fibrotizující alveolitida.
Nové autoprotilátky u myozitidy
V posledních třech letech bylo popsáno několik nových autoprotilátek specifických pro myozitidu. Je zajímavé, že většina z nich se vyskytuje u nemocných s dermatomyozitidou a u každé z nich byla zjištěna asociace se specifickými klinickými projevy.
Anti-p155/140 (anti-TIF1γ)
Anti-p155/140 protilátky byly popsány zároveň dvěma nezávislými skupinami u dospělých (27, 28) a u dětských pacientů s dermatomyozitidou (27) (JDM). Detekce těchto autoprotilátek je v současnosti možná pouze radioaktivní imunoprecipitací a proto nebyly tyto autoprotilátky popsány dříve, přestože způsobují hrudkovitou jadernou fluorescenci na Hep-2 buňkách. Anti-p155/140 jsou, po anti-Jo-1, pravděpodobně druhou nejčastější specifickou autoprotilátkou u dospělých s myozitidou a zároveň vůbec nejčastější u JDM. Anti-p155/140 byly detekovány u 29 % (27) nebo u 23 % (28) nemocných s JDM, u kterých je přítomnost této autoprotilátky asociována se zvýšenou frekvencí kožních lézí (29). U dospělých nemocných je tato autoprotilátka specifická pro dermatomyozitidu a je silně asociovaná s maligním nádorovým onemocněním.
Skutečnost, že u pacientů s antisyntetázovými nebo s jinými autoprotilátkami pro myozitidu specifickými nebo s myozitidou asociovanými je malignita poměrně vzácná, umožnila Hectoru Chinoyovi (30) výpočet pravděpodobnosti myozitidy asociované s malignitou u pacientů, kteří mají pozitivitu těchto protilátek a zároveň nemají anti-p155/140. Senzitivita a negativní prediktivní hodnota této kombinované strategie se blížila 100 %, čímž vpodstatě vylučuje u daného nemocného přítomnost zhoubného nádoru. Antigen, proti kterému jsou anti-p155/140 zaměřené, byl nedávno identifikován jako TIF1-γ (transcriptional intermediary factor). Průkaz této autoprotilátky tedy může mít velký praktický význam nejen při diagnóze IZM, ale také při odhalování skrytých zhoubných nádorů u pacientů s dermatomyozitidou, kteří nereagují na léčbu. Bohužel průkaz anti-p155/140 je v současné době prováděn pouze v několika výzkumných laboratořích.
Anti-p140
Nedávno byla u 23 % pacientů s JDM (31) popsána další autoprotilátka reagující s proteinem o molekulární hmotnosti 140 kDa, která se liší od anti-p155/140. Tato protilátka, nazvaná anti-p140, byla přítomná výhradně u nemocných s JDM a ne u pacientů s překryvnými syndromy nebo u kontrolních osob. U žádného z nemocných s pozitivitou anti-p140 nebyla zjištěna přítomnost jiné autoprotilátky. Imunodepleční studie ukázaly, že p140 odpovídá NXP-2 (nuclear matrix protein), který je totožný s dříve popsaným antigenem MJ. Pozitivita anti-p140 je významně asociována s kalcinózou podkoží (31).
Anti-CADM-140
Dermatomyozitida, při které mají nemocní typické kožní projevy, ale v průběhu dvou let se neobjeví žádné příznaky svalového postižení se nazývá amyopatická dermatomyozitida. Pokud je přítomné jen subklinické svalové postižení, pak mluvíme o hypomyopatické dermatomyozitidě. Tyto dvě podskupiny označujeme jako klinicky amyopatickou dermatomyozitidu (C-ADM). U 53 % nemocných s C-ADM byla zjištěna přítomnost nové autoprotilátky nazvaná anti-CADM-140 (32). U nemocných s pozitivitou této autoprotilátky bývá přítomno rychle progredující intersticiální plicní postižení (32). Anti-CADM-140 jsou vysoce specifické pro C-ADM, ale zatím byly zjištěny pouze u japonských pacientů s C-ADM a u 1 nemocného s klasickou dermatomyozitidou. Antigen pro CADM-140 byl identifikován jako RNA helikáza kódovaná genem MDA-5 (melanoma differentiation-associated gene 5) (33). Již byla vyvinuta ELISA pro detekci anti-CADM-140 protilátek s velmi dobrou specificitou a, ve srovnání s imunoprecipitací, mírně nižší senzitivitou (33). Pomocí této metody byla zjištěna významně vyšší prevalence anti-CADM-140 protilátek u C-ADM pacientů s rychle progredujícím intersticiálním plicním postižením ve srovnání s nemocnými bez ILD (82 % versus 3 %). Rutinní detekce anti-CADM-140 protilátek může mít klinický význam u nemocných z Japonska a východní Asie, což jsou zeměpisné oblasti, ve kterých byly tyto protilátky zjištěné. Rychle progredující ILD ve spojení s C-ADM není běžná u neasijských pacientů (33).
Anti-SAE
Protilátky namířené proti podjednotkám A a B SUMO-1 (small ubiquitin-like modifier 1) aktivačních enzymů byly původně popsány u 2 pacientů s C-ADM, u nichž se po několika měsících rozvinula proximální myozitida. Stejní autoři následně vyšetřili velký soubor nemocných a zjistili přítomnost této autoprotilátky u 8,4% pacientů s dermatomyozitidou (34). Většina nemocných měla v úvodu onemocnění typické kožní projevy a myozitida se projevila v průměru za 3 měsíce. Více než 75 % pacientů trpělo dysfagií a více než 80 % mělo systémové příznaky jako jsou febrilie, hubnutí a elevace reaktantů akutní fáze. Mírné intersticiální postižení plic bylo zjištěno jen u dvou pacientů (18 %). Tyto autoprotilátky jsou silně asociovány s haplotypem HLA-DRB1 * 04-DQA1 * 03-DQB1 * 03.
Anti-200/100 kDa
Nedávno byla u nemocných s převážně nekrotickou myopatií bez přítomnosti zánětu ve svalové biopsii popsána nová autoprotilátka, která precipituje s párem proteinů o molekulové hmotnosti 100 a 200 kDa (35). Nemocní neměli jednotný klinický fenotyp a nebyla u nich přítomna žádná jiná autoprotilátka.
Magnetická rezonance
Magnetická rezonance svalů (MRI) může být velmi užitečná při vyšetřování nemocných s nedostatečnou odpovědí na léčbu, protože umožňuje odlišit přetrvávající aktivitu zánětu od poškození svalů a svalové atrofie (36). Zásadní výhodou vyšetření MRI je schopnost neinvazivně vyšetřit rozsáhlé oblasti svalu najednou a díky tomu ukázat distribuci svalového postižení nebo umožnit opakovaná vyšetření u daného pacienta. MRI může také pomoci identifikovat ložiska aktivního zánětu a tím zvýšit diagnostickou výtěžnost svalové biopsie (37). Svalový edém může být patrný pouze při použití techniky STIR (short tau inversion recovery) nebo na T2-vážených obrazech s potlačením tuku. Senzitivitu MRI vyšetření lze zvýšit použitím gadolinia. Obraz edému svalů na MRI je téměř vždy způsobený zvýšeným obsahem vody ve tkáni, ale není specifický pro zánět a může být přítomen také u nemocných s jinými chorobami nebo poškozením svalů. Svalový edém na MRI bývá přechodně přítomen i po větší fyzické zátěži (38). U nemocných s s-IBM může průkaz typické distribuce svalového postižení na MRI (m. quadriceps femoris, m. gastrocnemius medialis a postižení flexorů předloktí) pomoci při určení správné diagnózy (3).
Svalová biopsie
Svalová biopsie je nejdůležitějším testem užívaným k určení diagnózy IZM a zároveň je nejčastějším zdrojem chyb a omylů. U nemocných s rezistencí na léčbu bychom měli vždy zvážit zopakování svalové biopsie. Histologické změny mohou být ve svalové tkání distribuovány nerovnoměrně, a proto nemusely být na původní biopsii zachyceny, a některé nálezy specifické pro s-IBM se mohou objevit až v průběhu času. Pokud není k dispozici patolog, který má zkušenosti s odečítáním svalových biopsií, nebo pokud nejsou dostupné některé techniky (např. enzymovou histochemii) je vhodné zajistit vyšetření na specializovaném pracovišti. Až ve čtvrtině případů IZM může být svalová biopsie falešně negativní, především v důsledku nerovnoměrného rozložení zánětu ve svalové tkáni (1). Ideálním místem pro svalovou biopsii je středně závažně postižený sval podle výsledku svalového testu a/nebo EMG a MRI. Při odebírání biopsie je třeba se vyhnout místu poraněnému vpichem EMG elektrod, obvykle volíme kontralaterální stranu. Manipulace s odebraným vzorkem a příprava tkáně pro vyšetření mají zásadní význam pro zajištění maximální výtěžnosti biopsie. Odebraný vzorek by měl být zmražen, protože artefakty způsobené parafínovou fixací znemožňují detekci vakuol charakteristických pro s-IBM. Také pro biochemické a imunohistochemické vyšetření tkáně jsou potřeba zmražené vzorky. Přítomnost zánětlivých infiltrátů ve svalové biopsii podporuje diagnózu IZM, ale není specifická – může se vyskytnout i u pacientů s různými dystrofiemi, myastenií gravis nebo s léky indukovanou myopatií (39). U většiny pacientů s IZM dochází k difuzní expresi MHC třídy I, ta není přítomna ve zdravé svalové tkáni. Exprese MHC-I bývá patrná také v oblastech bez zánětu nebo u pacientů, kteří jsou v dlouhodobé remisi. Při vyšetření 224 vzorků svalové biopsie získaných od pacientů s různými svalovými chorobami a od zdravých kontrolních osob, nebyla prokázána exprese MHC-I na povrchu svalových vláken u nemocných s metabolickými nebo kongenitálními myopatiemi ani s neurologickými poruchami (40). Nicméně exprese MHC-I na povrchu svalových vláken může být přítomna i u pacientů s dystrofií a není tedy specifická pro IZM, (40, 41). Invaze CD8 pozitivních T lymfocytů a makrofágů do normálně vypadajících svalových vláken spolu s difuzní expresí MHC-I je přítomna u nemocných s polymyozitidou a s-IBM. Přítomnost červeně-lemovaných vakuol a kongofilních inkluzí amyloidu podporuje diagnózu s-IBM. Elektronová mikroskopie se u nemocných s s-IBM užívá k průkazu charakteristických cytoplazmatických a intranukleárních tubulofilament o velikosti 15–20 nm (39).
Perifascikulární atrofie svalových vláken je charakteristická pro dermatomyozitidu a může být patrná i bez přítomnosti zánětlivého infiltrátu. Mezi další nálezy, které podporují diagnózu dermatomyozitidy patří snížení počtu kapilár a známky ischemie a zánětlivá infiltrace v perivaskulární distribuci. Etiologie nekrotické myopatie je obvykle paraneoplastická nebo toxická, nicméně nekróza může být přítomna i u pacientů s polymyozitidou spojenou s autoprotilátkami anti-SRP nebo anti-200/100 kDa. V bioptickém nálezu těchto pacientů může chybět zánětlivý infiltrát i difuzní exprese MHC-I, což znesnadňuje určení správné diagnózy (23, 35).
Častou chybou při diagnostice polymyozitidy je svádět nepřítomnost zánětlivého infiltrátu v biopsii na vrub předchozí imunosupresivní léčby. Léčba kratší než 4 týdny bioptický nález významně neovlivňuje a difuzní exprese HLA-I je obvykle přítomna i po dlouhodobé úspěšné léčbě nemocných s IZM (42).
Jsou jiná vysvětlení pro nedostatečný účinek léčby?
Pokud byla diagnóza IZM na základě provedených vyšetření skutečně potvrzena, je třeba zvážit zda lze nedostatečnou účinnost léčby vysvětlit jinak. Někteří nemocní nemají dostatečnou odpověď na léčbu glukokortikoidy, protože léčba nebyla podávána v dostatečně vysokých dávkách nebo po dostatečně dlouhou dobu. Je třeba také zvážit přítomnost steroidní myopatie, která se obvykle projevuje zhoršením svalové slabosti bez vzestupu hodnot CK u nemocných, kteří v úvodu reagovali na léčbu. Je třeba také vyloučit jiná konkomitantní onemocnění. U dospělých pacientů s dermatomyozitidou je významně zvýšené riziko zhoubného onemocnění (43) a proto je zvláště v případě nedostatečné odpovědi na léčbu třeba provést pečlivý skrínink okultních malignit. Myozitida asociovaná s maligním onemocněním obvykle reaguje na chemoterapii podávanou pro léčbu základního onemocnění, ale celková prognóza nemocných závisí především na typu a rozsahu nádoru. Podle publikovaných údajů je obecně odpověď na léčbu lepší u nemocných s dermatomyozitidou ve srovnání s polymyozitidou. Mezi nemocnými s polymyozitidou jsou také významné rozdíly v odpovědi na léčbu spojené s přítomností různých autoprotilátek, přičemž pacienti s pozitivitou anti-SRP bývají nejvíce rezistentní na léčbu. Jak již bylo zmíněno dříve s-IBM obvykle nereaguje na imunosupresivní léčbu. U některých pacientů s polymyozitidou se průběh onemocnění podobá spíše s-IBM, ale v biopsii nejsou přítomné charakteristické změny nutné k určení diagnózy s-IBM ani při opakování svalové biopsie. Tito pacienti by měli být z praktického hlediska považováni za nemocné trpící s-IBM.
Možnosti léčby u pacientů, kteří nereagují na konvenční imunosupresivní léčbu
Farmakoterapie
U nemocných se skutečně refrakterní polymyozitidou nebo dermatomyozitidou může být zvážena léčba kombinací metotrexátu a azatioprinu (44). Další alternativou farmakologické léčby je podání vysokých dávek intravenózních imunoglobulinů (IVIG), které měly v jedné placebem kontrolované studii dobré účinky u rezistentních nemocných s dermatomyozitidou (45). Tento výsledek však nebyl potvrzen v dalším otevřeném klinickém hodnocení u pacientů s dermatomyozitidou, polymyozitidou a s-IBM (46). Další léčiva, která mohou být účinná v refrakterních případech jsou cyklosporin a mykofenolát mofetil (47). Klinické zkušenosti ukazují, že u některých pacientů může být účinná kombinace cyklosporinu s metotrexátem. Cyklofosfamid je obvykle vyhrazen pro léčbu přidruženého intersticiálního plicního postižení, údaje týkající se účinků na postižení svalů jsou nejednotné (48). Takrolimus byl s úspěchem použit při léčbě pacientů s antisyntetázovým syndromem (49).
Z nových biologických léčiv se zatím jako nejvíce nadějná jeví léčba monoklonální protilátkou proti CD-20 lymfocytům – rituximabem, která byl účinný ve skupině nemocných s rezistentní dermatomyozitidou a polymyozitidou (50). Později publikované výsledky ale byly méně povzbudivé. Kombinace rituximabu s výměnnou plazmaferézou byla účinná při léčbě 2 pacientů s anti-SRP pozitivní myozitidou (51). Nedávno byla dokončena randomizovaná placebem kontrolovaná studie zaměřená na účinnost rituximabu u IZM, jejíž výsledek by měl pomoci při objasnění terapeutického potenciálu tohoto léčiva. Zprávy o účinnosti blokády TNF při léčbě rezistentních případů IZM jsou rozporuplné, výsledky jsou většinou negativní. Léčba anakinrou - blokátorem IL-1 byla účinná u jednoho pacienta s pozitivitou anti-Jo-1 protilátek a s intersticiálním plicním postižením, ovšem tento výsledek je třeba potvrdit na větším počtu nemocných (52). I nadále je třeba hledat účinné možnosti léčby nemocných, kteří jsou rezistentní na léčbu glukokortikoidy v kombinaci s metotrexátem a/nebo azatioprinem.
Rehabilitace
IZM se projevují svalovou slabostí a svalovou únavou. Etiologie svalové slabosti je multifaktoriální a není zatím zcela objasněna. Jedním z možných mechanismů je ztráta svalové hmoty, která může být vyvolána poškozením svalových vláken v důsledku autoimunitního procesu, fyzickou inaktivitou nebo může být nežádoucím účinkem léčby glukokortikoidy. Cvičení pomáhá zmírnit negativní dopady nemoci a léčby a zároveň umožňuje obnovit svalovou hmotu. Rehabilitace může být zahájena zároveň s imunosupresivní léčbou a měla by probíhat pod dohledem erudovaných fyzioterapeutů (53).
Léčba s-IBM
Pacienti s s-IBM mohou sice přechodně reagovat na imunosupresivní léčbu, ale celkově jsou možnosti léčby tohoto onemocnění neuspokojivé. Nedávno byly publikovány výsledky klinického hodnocení, ve kterém se podařilo zpomalit progresi s-IBM jednou sérii infuzí monoklonální protilátky proti CD-52 alemtuzumabu (54). Předpokladem rozvoje nových terapeutických možností s-IBM je především lepší pochopení etiopatogenze tohoto onemocnění (3).
Závěr
I opakovaná svalová biopsie u našeho nemocného potvrdila nekrotickou myopatii bez přítomnosti zánětu. Doplňující imunologické vyšetření provedené ve specializované zahraniční laboratoři prokázalo přítomnost anti-SRP autoprotilátek. Na základě tohoto výsledku a typických klinických projevů (dysfagie, těžký průběh onemocnění, rezistence na léčbu) byla u nemocného prokázána anti-SRP pozitivní polymyozitida a nemocnému byl podán rituximab. Po této léčbě došlo k výraznému zlepšení svalové síly, nemocný přestal být upoután na invalidní vozík a dokáže již chodit jen s holí. Hodnoty svalových enzymů poklesly do normy a kontrolní, celkově třetí v pořadí, svalová biopsie prokázala zřejmý ústup nekrotizující složky a absenci exprese HLA-I na povrchu svalových vláken.
MUDr. Heřman Mann
Revmatologický ústav
Na Slupi 4
128 50 Praha 2
e-mail: mann@revma.cz
Sources
1. Christopher-Stine L, Plotz PH. Adult inflammatory myopathies. Best Pract Res Clin Rheumatol 2004;18 : 331-344.
2. Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med 1975;292 : 344-347.
3. Needham M, Mastaglia FL. Inclusion body myositis: current pathogenetic concepts and diagnostic and therapeutic approaches. Lancet Neurol 2007;6 : 620-631.
4. Alexanderson H, Lundberg IE. The role of exercise in the rehabilitation of idiopathic inflammatory myopathies. Curr Opin Rheumatol 2005;17 : 164-171.
5. Miller FW, Rider LG, Chung YL, et al. Proposed preliminary core set measures for disease outcome assessment in adult and juvenile idiopathic inflammatory myopathies. Rheumatology (Oxford) 2001;40 : 1262-1273.
6. Dugan EM, Huber AM, Miller FW, et al. Photoessay of the cutaneous manifestations of the idiopathic inflammatory myopathies. Dermatol Online J 2009;15 : 1.
7. Gerami P, Schope JM, McDonald L, et al. A systematic review of adult-onset clinically amyopathic dermatomyositis (dermatomyositis sine myositis): a missing link within the spectrum of the idiopathic inflammatory myopathies. J Am Acad Dermatol 2006;54 : 597-613.
8. Hoffman EP, Rao D, Pachman LM. Clarifying the boundaries between the inflammatory and dystrophic myopathies: insights from molecular diagnostics and microarrays. Rheum Dis Clin North Am 2002;28 : 743-757.
9. Chahin N, Engel AG. Correlation of muscle biopsy, clinical course, and outcome in PM and sporadic IBM. Neurology 2008;70 : 418-424.
10. Walsh RJ, Amato AA. Toxic myopathies. Neurol Clin 2005;23 : 397-428.
11. Seidler A, Gottlieb A. Dermatomyositis induced by drug therapy. J Am Acad Dermatol 2008;59 : 872-880.
12. Yazici Y, Kagen LJ. Clinical presentation of the idiopathic inflammatory myopathies. Rheum Dis Clin North Am 2002;28 : 823-832.
13. Wortmann RL, DiMauro S. Differentiating idiopathic inflammatory myopathies from metabolic myopathies. Rheum Dis Clin North Am 2002;28 : 759-778.
14. Lundberg IE. The heart in dermatomyositis and polymyositis. Rheumatology (Oxford) 2006;45 : 18-21.
15. Rider LG. Outcome assessment in the adult and juvenile idiopathic inflammatory myopathies. Rheum Dis Clin North Am 2002;28 : 935-977.
16. Marguerie C, Bunn CC, Beynon HL,, et al. Polymyositis, pulmonary fibrosis and autoantibodies to aminoacyl-tRNA synthetase enzymes. Q J Med 1990;77 : 1019-1038.
17. Brouwer R, Hengstman GJ, Vree Egberts W, et al. Autoantibody profiles in the sera of European patients with myositis. Ann Rheum Dis 2001;60 : 116-123.
18. Betteridge Z, Gunawardena H, North J, et al. Anti-synthetase syndrome: a new autoantibody to phenylalanyl transfer RNA synthetase (anti-Zo) associated with polymyositis and interstitial pneumonia. Rheumatology (Oxford) 2007;46 : 1005-1008.
19. Hashish L, Trieu EP, Sadanandan P, et al. Identification of autoantibodies to tyrosyl-tRNA synthetase in dermatomyositis with features consistent with antisynthetase syndrome (abstract). Arthritis Rheum 2005;52:S312.
20. Tillie-Leblond I, Wislez M, Valeyre D, et al. Interstitial lung disease and anti-Jo-1 antibodies: difference between acute and gradual onset. Thorax 2008;63 : 53-59.
21. Stone KB, Oddis CV, Fertig N, et al. Anti-Jo-1 antibody levels correlate with disease activity in idiopathic inflammatory myopathy. Arthritis Rheum 2007;56 : 3125-3131.
22. Kao AH, Lacomis D, Lucas M, et al. Anti-signal recognition particle autoantibody in patients with and patients without idiopathic inflammatory myopathy. Arthritis Rheum 2004;50 : 209-215.
23. Miller T, Al-Lozi MT, Lopate G, et al. Myopathy with antibodies to the signal recognition particle: clinical and pathological features. J Neurol Neurosurg Psychiatry 2002;73 : 420-428.
24. Hengstman GJ, Vree Egberts WT, Seelig HP, et al. Clinical characteristics of patients with myositis and autoantibodies to different fragments of the Mi-2 beta antigen. Ann Rheum Dis 2006;65 : 242-245 .
25. Marguerie C, Bunn CC, Copier J, et al. The clinical and immunogenetic features of patients with autoantibodies to the nucleolar antigen PM-Scl. Medicine (Baltimore) 1992;71 : 327-336 .
26. Cavazzana I, Ceribelli A, Quinzanini M, et al. Prevalence and clinical associations of anti-Ku antibodies in systemic autoimmune diseases. Lupus 2008;17 : 727-732.
27. Targoff IN, Mamyrova G, Trieu EP, et al. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum 2006;54 : 3682-3689.
28. Kaji K, Fujimoto M, Hasegawa M, et al. Identification of a novel autoantibody reactive with 155 and 140 kDa nuclear proteins in patients with dermatomyositis: an association with malignancy. Rheumatology (Oxford) 2007;46 : 25-28.
29. Gunawardena H, Wedderburn LR, North J, et al. Juvenile Dermatomyositis Research Group UK. Clinical associations of autoantibodies to a p155/140 kDa doublet protein in juvenile dermatomyositis. Rheumatology (Oxford) 2008;47 : 324-328.
30. Chinoy H, Fertig N, Oddis CV, et al. The diagnostic utility of myositis autoantibody testing for predicting the risk of cancer-associated myositis. Ann Rheum Dis 2007;66 : 1345-1349.
31. Gunawardena H, Wedderburn LR, Chinoy H, et al. Juvenile Dermatomyositis Research Group, UK and Ireland. Autoantibodies to a 140-kd protein in juvenile dermatomyositis are associated with calcinosis. Arthritis Rheum 2009;60 : 1807-1814.
32. Sato S, Hirakata M, Kuwana M, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum 2005;52 : 1571-1576.
33. Sato S, Hoshino K, Satoh T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: Association with rapidly progressive interstitial lung disease. Arthritis Rheum 2009;60 : 2193-2200.
34. Betteridge ZE, Gunawardena H, Chinoy H, et al. Clinical and human leucocyte antigen class II haplotype associations of autoantibodies to small ubiquitin-like modifier enzyme, a dermatomyositis-specific autoantigen target, in UK Caucasian adult-onset myositis. Ann Rheum Dis 2009;68 : 1621-1625.
35. Christopher-Stine L, Hong G, Casciola-Rosen L, et al. A Novel Autoantibody against 200/100 Kd Proteins Is Associated with Necrotizing Myopathy (abstract). Arthritis Rheum 2009;60:S430.
36. Lovitt S, Moore SL, Marden FA. The use of MRI in the evaluation of myopathy. Clin Neurophysiol 2006;117 : 486-495.
37. Tomasova-Studynkova J, Charvat F, Jarosova K, Vencovsky J. The role of MRI in the assessment of polymyositis and dermatomyositis. Rheumatology 2007;46 : 1174-1179.
38. Curiel RV, Jones R, Brindle K. Magnetic resonance imaging of the idiopathic inflammatory myopathies: structural and clinical aspects. Ann NY Acad Sci 2009;1154 : 101–114.
39. *Dalakas MC. Muscle biopsy findings in inflammatory myopathies. Rheum Dis Clin North Am 2002;28 : 779-798.
40. van der Pas J, Hengstman GJ, ter Laak HJ, et al. Diagnostic value of MHC class I staining in idiopathic inflammatory myopathies. J Neurol Neurosurg Psychiatry 2004;75 : 136-139.
41. Jain A, Sharma MC, Sarkar C, et al. Major histocompatibility complex class I and II detection as a diagnostic tool in idiopathic inflammatory myopathies. Arch Pathol Lab Med 2007;131 : 1070-1076.
42. Lundberg I, Kratz AK, Alexanderson H, Patarroyo M. Decreased expression of interleukin-1alpha, interleukin-1beta, and cell adhesion molecules in muscle tissue following corticosteroid treatment in patients with polymyositis and dermatomyositis. Arthritis Rheum 2000;43 : 336-348.
43. Madan V, Chinoy H, Griffiths CE, Cooper RG. Defining cancer risk in dermatomyositis. Clin Exp Dermatol 2009;34 : 451-455.
44. Villalba L, Hicks JE, Adams EM, et al. Treatment of refractory myositis: a randomized crossover study of two new cytotoxic regimens. Arthritis Rheum 1998;41 : 392-399.
45. Dalakas MC, Illa I, Dambrosia JM, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med 1993;329 : 1993-2000.
46. Barbasso Helmers S, Dastmalchi M, Alexanderson H, et al. Limited effects of high-dose intravenous immunoglobulin (IVIG) treatment on molecular expression in muscle tissue of patients with inflammatory myopathies. Ann Rheum Dis 2007;66 : 1276-1283.
47. Pisoni CN, Cuadrado MJ, Khamashta MA, et al. Mycophenolate mofetil treatment in resistant myositis. Rheumatology (Oxford) 2007;46 : 516-518.
48. Yamasaki Y, Yamada H, Yamasaki M, et al. Intravenous cyclophosphamide therapy for progressive interstitial pneumonia in patients with polymyositis/dermatomyositis. Rheumatology (Oxford) 2007;46 : 124-130.
49. Wilkes MR, Sereika SM, Fertig N, et al. Treatment of antisynthetase-associated interstitial lung disease with tacrolimus. Arthritis Rheum 2005;52 : 2439-2446.
50. Noss EH, Hausner-Sypek DL, Weinblatt ME. Rituximab as therapy for refractory polymyositis and dermatomyositis. J Rheumatol 2006;33 : 1021-1026.
51. Arlet JB, Dimitri D, Pagnoux C, et al. Marked efficacy of a therapeutic strategy associating prednisone and plasma exchange followed by rituximab in two patients with refractory myopathy associated with antibodies to the signal recognition particle (SRP). Neuromuscul Disord 2006;16 : 334-336.
52. Furlan A, Botsios C, Ruffatti A, et al. Antisynthetase syndrome with refractory polyarthritis and fever successfully treated with the IL-1 receptor antagonist, anakinra: A case report. Joint Bone Spine 2008;75 : 366-367.
53. Alexanderson H, Stenström CH, Jenner G, Lunberg I. The safety of a resistive home exercise program in patients with recent onset active polymyositis or dermatomyositis. Scand J Rheumatol 2000;29 : 295-301.
54. Dalakas, Rakocevic G, Schmidt J, et al. Effect of Alemtuzumab (CAMPATH 1-H) in patients with inclusion-body myositis. Brain 2009 : 132;1536–1544.
Labels
Dermatology & STDs Paediatric rheumatology RheumatologyArticle was published in
Czech Rheumatology

2010 Issue 2
Most read in this issue
- Myozitida rezistentní na léčbu
- Posuzování funkční schopnosti u pacientů s revmatoidní artritidou; validace české verze Stanfordského dotazníku Health Assessment Questionnaire (HAQ)
- Jaccoudova artropatie u systémového lupus erytematodes
- Kognitivní dysfunkce u české populace nemocných se systémovým lupus erytematodes