
Nástin genetické architektury primární hyperurikémie a dny
Outline of the genetic architecture of primary hyperuricemia and gout
Gout, arthritis urica, is a metabolic disorder caused by an inflammatory reaction to the deposition of urate crystals into joints and soft tissues. Chronic hyperuricaemia, the cause of the gout, results in an imbalance between endogenous production and excretion of uric acid. The most common mechanism leading to hyperuricaemia is decreased excretion of uric acid. Urate transport is a complex process involving a number of transmembrane proteins that provide reabsorption (mostly URAT1, GLUT9) and secretion (ABCG2) on the apical and basolateral side of the proximal tubules. ABCG2, with a significant proportion, provides transport in the gastrointestinal tract. New knowledge on uric acid excretion has allowed the development of a new strategy in the treatment of hyperuricaemia by blocking urate transporters. Knowledge of the genetic background of uricemia is essential for early identification of the aetiology of the disease, the choice of appropriate treatment, and also the monitoring of compliance by the patient. Detailed examination of purine metabolism and uric acid excretion in specialized laboratories is particularly useful for patients with early onset and / or familial outbreaks of the disease.
Keywords:
Hyperuricaemia, gout, purine metabolism, hypoxanthine-guanine phosphoribosyltransferase deficiency, urate transporters, SLC22A12, SLC2A9, ABCG2.
Authors:
B. Stibůrková
Authors‘ workplace:
Ústav dědičných metabolických poruch, 1. lékařská fakulta, Univerzita Karlova a Všeobecná fakultní nemocnice, Praha
; Revmatologický ústav
Published in:
Čes. Revmatol., 25, 2017, No. 3, p. 116-123.
Category:
Review Article
Overview
Dna, arthritis urica, je metabolické onemocnění způsobené zánětlivou reakcí na ukládání urátových krystalů do kloubů a měkkých tkání. Chronická hyperurikémie, kauzální příčina dny, vzniká nerovnováhou mezi endogenní produkcí a exkrecí kyseliny močové. Nejčastějším mechanismem vedoucím ke vzniku hyperurikémie je snížená exkrece kyseliny močové. Transport urátu je komplexní proces zahrnující řadu transmembránových proteinů zajišťujících reabsorpci (majoritně URAT1, GLUT9) a sekreci (ABCG2) na apikální i bazolaterální straně proximálních tubulů a v případě ABCG2 i s významným podílem transportu v gastrointestinálním traktu. Nové znalosti o exkreci kyseliny močové umožnily vývoj nové strategie v léčbě hyperurikémie mechanismem blokace urátových transportérů. Znalosti genetického pozadí urikémie jsou podstatné pro včasné rozpoznání etiologie onemocnění, volbě vhodné léčby a také k monitorování compliance ze strany pacienta. Detailní vyšetření purinového metabolismu a exkrece kyseliny močové ve specializovaných laboratořích je vhodné zejména u pacientů s časným nástupem a/nebo familiárním výskytem onemocnění.
Klíčová slova:
Hyperurikémie, dna, purinový metabolismus, deficit hypoxantin-guaninfosforibozyltransferázy urátové transportéry, SLC22A12, SLC2A9, ABCG2
Úvod
Dna, arthritis urica, je metabolické onemocnění způsobené zánětlivou reakcí na ukládání urátových krystalů do kloubů a měkkých tkání. Dna je nejčastější zánětlivá artritida, v České republice postihuje přibližně 200 000 pacientů. V rozsáhlém epidemiologickém výzkumu (8 300 000 subjektů) z let 2007–2008 v USA hlásilo téměř 4 % dospělých pacientů diagnózu dny: 5,9 % mužů; 2,0 % žen (1). Výrazně vyšší prevalence dny, více než dvojnásobná, se vyskytuje u vybraných etnických skupin: Maoři, původní obyvatelé Tchajwanu a Pacifiku (2). Prevalence dny narůstá s věkem, nástup onemocnění je typicky mezi čtvrtou až šestou dekádou. V rozsáhlé kohortě dnavých pacientů z Velké Británie (23 857 subjektů) byl průměrný věk nástupu onemocnění 61,9 roku ± 14,5 (3). U žen incidence dny výrazně stoupá po menopauze mechanismem poklesu hladiny estrogenu a jeho urikosurického účinku. Realitou současného vývoje je stoupající prevalence, incidence i časnější nástup onemocnění doložená řadou epidemiologických studií (4–7). Jednou z hlavních příčin se jeví dlouhodobý trend mnohonásobně zvýšené spotřeby fruktózy, který vede k rychlé spotřebě adenosin trifosfátu a jeho degradaci na konečný produkt purinových metabolitů – kyselinu močovou (KM), (8).
KM (2,6,8-trihydroxypurin), z důvodu ztráty aktivity enzymu urát oxidázy v průběhu miocénu, je u lidí a vyšších primátů konečným produktem degradace purinů. Funkční urikáza metabolizuje KM na 5-hydroxyisourát, který je dále degradován v závislosti na druhu organismu na allantoin, ureu nebo amonium. Evoluční odpovědí na tuto enzymovou dysfunkci je efektivní ledvinná reabsorpce (mechanismem glomerulární filtrace, zpětné resorpce, sekrece a následné zpětné resorpce) a více než desetinásobné zvýšení hladiny KM v séru u člověka oproti jiným savcům. Vylučování KM je ze dvou třetin zajišťováno ledvinami, třetinou gastrointestinálním traktem. Clearance KM v ledvinách je obvykle stálá, přibližně 90 % je v proximálních tubulech reabsorbováno, 10 % je vyloučeno močí. Denní produkce a vylučování KM činí přibližně 1000 mg a u dospělého jedince je za běžných podmínek relativně konstantní.
KM je považována za jeden z významných antioxidantů v biologických tekutinách a pravděpodobně vzhledem k její významné koncentraci je majoritním nízkomolekulárním antioxidantem v séru. Reaguje bez enzymové katalýzy s volnými radikály za vzniku allantoinu a dalších látek (9). Přestože vliv KM na průběh oxidativního stresu v organismu je široce studované téma, její role v tomto procesu doposud nebyla jednoznačně objasněna (10). V některých epidemiologických studiích je zvýšená hladina KM uváděna ve spojitosti se zvýšeným rizikem výskytu kardiovaskulárních onemocnění, jejichž patogeneze souvisí s přítomností oxidativního stresu (11, 12). Studie zaměřená na antioxidační vlastnosti KM ukázala, že během intenzivní aerobní fyzické zátěže, kdy stoupá produkce aktivních forem kyslíku, byl zaznamenán pokles koncentrace KM močové v organismu a zvýšení koncentrace allantoinu ve svalech (13). Při fyzické zátěži u zdravých jedinců dochází ke krátkodobému zvýšení hladiny KM v séru, což má za následek nárůst jeho antioxidační kapacity (14). Potenciálně protektivní antioxidační efekt KM je popsán v souvislosti se sníženým rizikem rozvoje idiopatické Parkinsonovy choroby u jedinců s trvale zvýšenou hladinou kyseliny močové nad medián (15). Naproti tomu výskyt trvale snížené hladiny KM vzhledem ke kontrolní skupině je asociován se zvýšeným rizikem výskytu neurodegenerativního onemocnění roztroušené sklerózy (16) a schizofrenie (17). Vliv hladiny KM na oxidativní stres a antioxidační kapacitu plazmy ve vztahu k inzulinové rezistenci byl zaznamenán u pacientů s obezitou (13).
Chronická hyperurikémie je kauzální příčinou dny. Elevace sérové KM ovlivňuje i další závažná onemocnění – hypertenzi, kardiovaskulární choroby, inzulinovou rezistenci, metabolický syndrom či postižení ledvin (20–24). Hyperurikémie vzniká nerovnováhou mezi endogenní produkcí a exkrecí kyseliny močové, mechanismus je shrnut na obrázku 1. Populační studie – v závislosti na použité definici a zkoumané populaci – uvádí prevalenci hyperurikémie u mužů 24–29 %, u žen podstatně nižší 2,6–20 %, u dětí je hyperurikémie vzácná. Analýzy se shodují na vysokém podílu hyperurikemických mužů, prevalence žen s nadlimitní hodnotou sérové kyseliny močové je populačně specifická a opakovaně nižší v evropské populaci (25–28). Sérová hladina KM má významnou heritabilitu, dle řady studií v rozmezí 0,38–0,63 (29–32).
V posledním desetiletí díky rozvoji nových molekulárně genetických technologií bylo možné začít s rozsáhlými celogenomovými asociačními studiemi (GWAS) zkoumajícími vztah genetických variant, typicky jednonukleotidových polymorfismů, a vybraných fenotypů. Geny lokalizované v asociovaných oblastech genomu se staly kandidáty pro následné funkční analýzy prokazující jejich případnou kauzalitu. Primární prací týkající se hyperurikémie byla rozsáhlá GWAS studie více než 140 000 evropských subjektů, jejímž výsledkem byla identifikace 18 nových genetických lokusů asociovaných s hladinou sérové KM (33). Následné analýzy konfirmovaly a rozšířily počet genetických lokusů asociovaných s hyperurikémií na více než 30, recentním přírůstkem jsou geny HIST1H2BF-HIST1H4E (role ve formování struktury chromatinu), NIPAL1 (magneziový transportér) a FAM35A (neznámá molekulární funkce) (34). Třetinu z těchto genů tvoří urátové transportéry lokalizované v ledvinách a gastrointestinálním traktu.
Znalosti urátového transportu získané v posledním desetiletí potvrzují dřívější in silico analýzu lidského purinového metabolismu, který ještě před identifikací urátových transportérů stanovil jako pravděpodobně nejvýznamnější příčinu vzniku dny dysfunkce renální exkrece KM. V tomto modelu vede 1% úbytek exkrece k téměř pětinásobnému nárůstu KM, zatímco změny stejného rozsahu v enzymech purinového metabolismu, které způsobují hyperurikémii mechanismem nadprodukce KM (přímým postižením recyklace a syntézy purinů) vedou k nesrovnatelně nižšímu vzestupu sérové KM (35). Většina deficiencí syntézy KM je tedy pravděpodobně nevýznamná a zůstává nedetekovatelná; je diagnostikováno pouze několik významných dysfunkcí zapříčiňujících hyperurikémii jako deficit hypoxantin-guaninfosforibozyltransferázy (36). Naproti tomu i „malé“ defekty v exkreci KM mohou významně ovlivnit sérovou hladinu KM a vést k patologickým hodnotám KM: hyperurikémii i hypourikémii.

Endogenní produkce KM
Produkce KM je dána balancí mezi endogenním příjmem purinů, purinovou de novo syntézou, recyklací a degradací. Hyperurikémie z důvodu nadprodukce KM může být výsledkem zvýšené syntézy purinů de novo nebo urychlené degradace purinových nukleotidů. Mezi dědičné poruchy metabolismu s častým výskytem zvýšené hladiny KM patří glykogenózy, intolerance fruktózy, porucha fruktóza-1,6-bisfosfatázy nebo mitochondriální poruchy energetického metabolismu. Mezi enzymopatie, které způsobují hyperurikémii přímým postižením recyklace a syntézy purinů, patří X-vázané dysfunkce hypoxantin-guaninfosforibozyltransferázy (HPRT1, OMIM 308000) a raritní superaktivita fosforibozylpyrofosfátsyntetázy (PRPS, OMIM 311850).
Deficit HPRT je nejčastější poruchou metabolismu purinů vyskytující se v klinicky rozdílných fenotypech. Parciální deficit HPRT, Kelley-Seegmillerův syndrom, je obvykle asociován pouze s klinickou manifestací nadprodukce purinů a následnou zvýšenou syntézou KM (hyperurikémie/dna, urolitiáza a nefrolitiáza). Může se vyskytovat také variabilní spektrum neurologické manifestace onemocnění, které je typické pro těžkou ztrátu aktivity HPRT (atetoidní nebo choreoatiformní pohyby, hyper-reflexie, torzní dystonie, hypotonie, zpomalení a zástava psychomotorického vývoje). Typickým a nutným klinickým symptomem pro těžký deficit HPRT, Lesh-Nyhanův syndrom, je přítomnost psychiatrických projevů ve formě automutilačního chování (okusování rtů, jazyka, konečků prstů), které se objevuje zhruba od druhého roku života. Děti se rodí klinicky zdravé, jedním z prvních příznaků může být oranžové zabarvení moče a plenek od vysrážených krystalů KM v moči. Diagnóza deficitu HPRT je založena na přítomnosti hyperurikémie, hyperurikosurie a zvýšené hladině hypoxanthinu a xanthinu v moči a/nebo plazmě. Diagnostický algoritmus pokračuje stanovením snížené HPRT aktivity v erytrocytech, případně fibroblastech, a finálně je diagnóza potvrzena molekulárně genetickým nálezem. V současnosti je známo okolo pěti set genetických variant HPRT1 včetně rozsáhlých delecí. Kauzální léčba onemocnění není k dispozici. Nadprodukce KM je ošetřena podáváním inhibitorů xanthin oxidázy/dehydrogenázy, z důvodu možného negativního ovlivnění neurologických symptomů je preferováno podávání nepurinového analogu febuxostatu (37, 38).
Deficit HPRT je spojen s X-vázanou recesivní dědičností. Přenašečky jsou symptomatické v závislosti na podílu inaktivace X chromozómu. V rodinách s tímto onemocněním se tedy můžeme setkat s plně symptomatickými (vzácně), částečně symptomatickými či asymptomatickými nositelkami patogenních variant v genu HPRT1. Diagnostický algoritmus u žen s deficitem HPRT, klinicky se projevujícím jako hyperurikémie/případně dna, může být také narušen nálezem fyziologických hodnot xanthinu a hypoxanthinu i normální aktivitou HPRT v erytrocytech (39). Role molekulárně genetického vyšetření je tedy primární, ačkoli u zhruba 5 % pacientů není nalezena kauzální varianta v genu HPRT1 (40, 41).
Fosforibozylpyrofosfátsyntetáza katalyzuje první krok nukleotidové syntézy, účastní se také biosyntézy pyridinů a pyrimidinů. Superaktivita PRPPs zvyšuje hladinu KM nadměrnou produkcí purinů projevující se hyperurikémií a hyperurikurií. Onemocnění je vysoce klinicky variabilní a bylo diagnostikováno celosvětově u třech desítek pacientů, s nálezem necelé desítky kauzálních alelických variant. Pacienti s mírným fenotypem mají klinický nález purinové nadprodukce (hyperurikémie, urolitiáza, nefrolitiáza, dna) bez neuropatických symptomů. Poruchy růstu, zpomalení psychomotorického vývoje, hypotonie, ataxie, percepční hluchota jsou častými projevy těžšího fenotypu superaktivity PRPPs (42). Diagnostické schéma je obdobné jako u deficitu HPRT. Kauzální léčba není k dispozici, možná je pouze úprava hyperurikémie.
Exkrece kyseliny močové
Posun ve znalostech transportu urátu v poslední dekádě jasně ukázal, že zdaleka nejčastějším mechanismem vedoucím ke vzniku hyperurikémie je snížená exkrece KM. Renální transport urátu zajišťuje řada proteinů ležící na apikální i bazolaterální membráně proximálních tubulů: majoritně urát-aniontový přenašeč URAT1, glukózový transportér GLUT9 a široce exprimovaný transportér ABCG2, který má významnou úlohu v transportu KM i řady xenobiotik v renálním i gastrointestinálním traktu, shrnuto na obrázku 2.
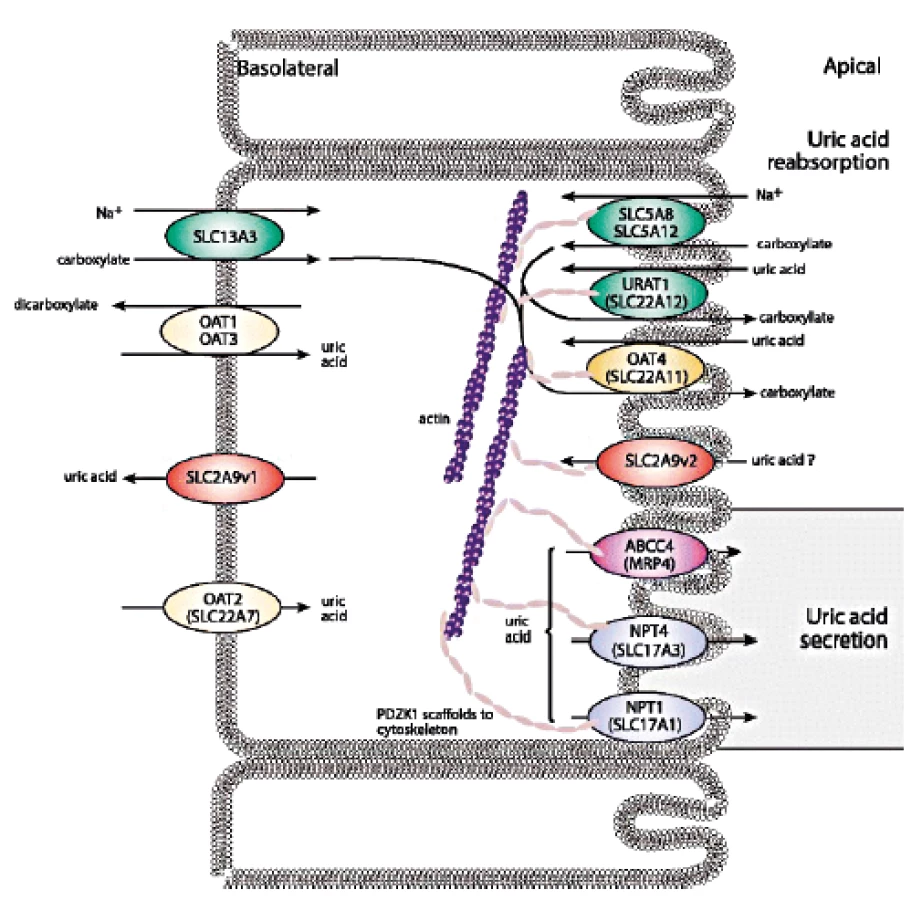
Produkt genu SLC22A12, protein URAT1, je vysoce specificky exprimovaný pouze v apikální membráně buněk proximálních tubulů (43). URAT1 se podílí na reabsorpci urátu, jeho poruchy způsobují dědičnou renální hypourikémii typu 1 (RHUC1, OMIM #220150) mechanismem endoteliální dysfunkce (44), která byla celosvětově popsána u přibližně 150 pacientů především asijského původu. RHUC je heterogenní dědičné onemocnění způsobené poškozením tubulárního transportu KM, nedostačující reabsorpcí a/nebo urychlenou sekrecí. Biochemickými markery jsou hypourikémie a zvýšená exkreční frakce KM. Klinicky se onemocnění projevuje urolitiázou, nefrolitiázou a u části pacientů akutním renálním selháním (často opakované po předchozí fyzické námaze). Vysoká incidence japonských a korejských pacientů je odrazem alelické frekvence 2,3–2,37% predominantní dysfunkční varianty rs121907892 (p.W258X), (45). Nicméně světově nejvyšší frekvence predominantních dysfunkčních variant byla recentně identifikována u romské populace (1016 jedinců z regionů České Republiky, Slovenska a Španělska): varianta rs200104135 (p.T467M) se vyskytovala s frekvencí 5,56 %, deleční varianta a p.L415_G417 s frekvencí 1,92 % (46). Vlivem genetického driftu se tedy u vybraných populací výrazně zvyšuje prevalence tohoto jinak raritního dědičného onemocnění.
V roce 2007–2009 byl nezávislými studiemi objeven vztah mezi genetickými variantami genu SLC2A9, sérovou hladinou KM a výskytem dny (47–50). Gen SLC2A9 kóduje dvě isoformy proteinu: GLUT9a (pravděpodobně bazolaterální membrána proximálních ledvinných tubulů, játra, placenta, leukocyty), GLUT9b (pravděpodobně apikální membrána proximálních tubulů, placenta). Dysfunkční varianty v SLC2A9 způsobují dědičnou renální hypourikémii typu 2 (RHUC2, OMIM #612076), která byla popsána u desítky pacientů a mezi klinické příznaky patří i totální ztráta reabsorpce KM (hladiny sérové KM < 10 µmol/l, exkreční frakce KM okolo 100 %). Finální diagnostika RHUC typu 1 a 2 je molekulárně genetická, s případným doplněním funkční studií. Léčba onemocnění není k dispozici, profylaxe pomocí allopurinolu byla popsána u pacienta s rekurentními ataky akutního renálního selhání (51).
Reabsorpci KM dále zajišťují apikální membránové transportéry OAT4 (gen SLC22A11) a OAT 10 (gen SLC22A13). OAT4 je exprimován tkáňově specificky v ledvinách a placentě (52), OAT 10 navíc v srdci, mozku a střevě (53).
Sekreční podíl transportu KM zajišťuje skupina proteinů v apikální membráně proximálních tubulů – zejména produkt genu ATP-binding cassette, subfamily G, member 2 (ABCG2/BCRP) ABCG2. ABCG2 je exprimován v plazmatických membránách různých tkání (placenta, ledviny, mozek, střevo aj.) a zajišťuje transport řady substrátů včetně xenobiotik (54, 55). Výrazně ovlivňuje sérovou hladinu KM, se statisticky významným rozdílem daným pohlavím a populací. Běžně se vyskytující alelická varianta rs2231142 (p.Q141K), s frekvencí v evropské populaci 9 %, způsobuje zhruba poloviční redukci urátového transportu a byla nalezena u 10 % pacientů s primární dnou (56–59). V regresním modelu (kontrolní kohorta 589 českých subjektů z běžné populace) varianta rs2231142 (p.Q141K) v heterozygotní/homozygotní formě výrazně zvýšila sérovou hladinu KM o 39/83 µmol/l u mužů, o 8/15 µmol/l u žen (60).
Na transportu KM se podílí také protein hNPT4 (human sodium phosphate transporter 4), produkt genu SLC17A3, exprimovaný kromě ledvin i v játrech. Kauzální alelické varianty v SLC17A3 způsobující snížení urátové sekrece byly popsány u pacientů s hyperurikémií (61). Dalším urátovým transportérem je vysoce polymorfní MRP4 protein, člen MRP/ABCC subfamily ATP-binding cassette transporters, nacházející se na plazmatické membráně řady odlišných tkání s bariérovou funkcí (ledviny, játra, střevo, placenta, mozek), (62–64). U urátového transportéru NPT1 (gen SLC17A1) byla analýzou polymorfismů nalezena signifikantní asociace se sníženou hladinou KM (65–67). Sekreci urátu na bazolaterální membráně zajišťuje kromě majoritního GLUT9 také OAT1 (gen SLC22A6) a OAT3 (gen SLC22A8). Oba proteiny jsou exprimovány predominantně v ledvinách, se sníženou specifitou v dalších tkáních, kde přenášejí široké spektrum substrátů včetně řady léčiv (68–70).
Transport urátu je tedy komplexní proces zahrnující řadu transmembránových proteinů zajišťujících reabsorpci a sekreci na apikální i bazolaterální straně membrán proximálních tubulů a v případě ABCG2 i s významným podílem transportu urátu v gastrointestinálním traktu. Ačkoli jejich vzájemné ovlivnění není dostatečně známo, studie ukazují na model „urátového transportozómu“ se značným vzájemným ovlivněním aktivity přenosu (71).
Populační atributivní riziko (PAR) transportéru ABCG2 pro hyperurikémii (29,2 %) převyšuje ostatní rizikové faktory spojené se zvýšením KM jako je věk (≥60 let věku; PAR% = 5,74 %), BMI index (BMI ≥ 25,0; PAR% = 18,7 %) nebo zvýšená konzumace alkoholu (>196 gramů čistého alkoholu/týden u mužů, >98 u žen; PAR% = 15,4 %), (72). Recentní studie navíc ukázaly signifikantně zvýšené riziko pro nedosažení hodnot normourikémie u pacientů nesoucích minoritní dysfunkční alelu p.Q141K (rs2231142) v genu ABCG2 (73).
Autosomálně dominantní tubulointersticiální ledvinné onemocnění
Mezi mendeliánská onemocnění s předním klinickým nálezem hyperurikémie a dny patří autosomálně dominantní tubulointersticiální ledvinné onemocnění. Časná hyperurikémie/dna spolu s pomalu progredujícím chronickým tubulointersticiálním onemocněním ledvin, z důvodu redukované exkreční frakce kyseliny močové, je typickým klinickým markerem u tohoto dědičného onemocnění způsobeného patogenními variantami v uromodulinu (ADTKD-UMOD). Tato recentně definovaná jednotka (http://www.genereviews.org) zahrnuje dříve definovaná onemocnění familiární hyperurikemickou nefropatii typ 1 (FJHN1, OMIM 162000), medulární cystické onemocnění ledvin typ 2 (MCKD2, OMIM 603860) a uromodulin asociované ledvinné onemocnění. Diagnóza je založena na nálezu hyperurikémie a snížené exkreční frakci KM, následuje průkaz snížené exkrece uromodulinu v moči a finální genetická konfirmace. V časném věku mohou být hladiny sérového kreatininu fyziologické, později nastupuje snížená schopnost glomerulární filtrace. Kauzální léčba není k dispozici, možná je pouze úprava hyperurikémie.
Závěr
Sérová koncentrace KM je komplexním fenotypem, do kterého se promítá kombinace vnějších vlivů a genetického pozadí. Klinická manifestace dysfunkce v endogenní produkci KM může koexistovat s genetickými variantami ovlivňujícími její transport – jak jsme prokázali u pacienta s dlouholetou tofózní dnou s neodpovídající odpovědí na léčbu (nadprodukce KM způsobená deficitem HPRT spojená se sníženou renální exkrecí KM způsobenou dysfunkční variantou transportéru ABCG2), (74).
Pochopení molekulární podstaty a komplexního vztahu mezi genotypem a fenotypem hyperurikémie/dny vede k současnému přenosu těchto poznatků do klinické praxe. Nové znalosti o exkreci KM umožnily vývoj nové strategie v léčbě hyperurikémie mechanismem blokace urátových transportérů. Jedná se zejména o aktivní metabolit Lesinurad (RDEA594), který selektivně inhibuje transportér URAT1 (75).
Diferenciálně diagnostická rozvaha nad pacientem s primární hyperurikémií/dnou je poměrně široká. Detailní vyšetření purinového metabolismu a exkrece kyseliny močové (zahrnující biochemická, případně enzymová a molekulárně genetická vyšetření) ve specializovaných laboratořích je vhodné zejména u pacientů s časným nástupem a/nebo familiárním výskytem onemocnění. Znalosti genetického pozadí urikémie jsou nezbytné pro včasné rozpoznání etiologie onemocnění, volbě vhodné léčby a také k monitorování compliance ze strany pacienta.
Tato práce byla podpořena projektem Ministerstva zdravotnictví České republiky pro konceptuální rozvoj výzkumné organizace VFN64165 a 00023728 (Revmatologický ústav).
adresa pro korespondenci:
Ing. et Mgr. Blanka Stibůrková, Ph.D.
Revmatologický ústav
Revmatologická klinika 1. LF UK
Na Slupi 4
128 50 Praha 2
e-mail: stiburkova@revma.cz
Sources
1. Zhu Y1, Pandya BJ, Choi HK, et al. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007–2008. Arthritis Rheum. 2011 Oct; 63(10): 3136–41.
2. Winnard D, Wright C, Taylor WJ, et al. National prevalence of gout derived from administrative health data in Aotearoa New Zealand. Rheumatology (Oxford). 2012 May; 51(5): 901–9.
3. Rothenbacher D, Primatesta P, Ferreira A, et al. Frequency and risk factors of gout flares in a large population–based cohort of incident gout. Rheumatology (Oxford). 2011; 50(5): 973–81.
4. Kuo CF, Grainge MJ, Mallen C, et al. Rising burden of gout in the UK but continuing suboptimal management: a nationwide population study. Ann Rheum Dis. 2015; 74 : 661–67.
5. Saag K, Choi H. Epidemiology, risk factors, and lifestyle modifications for gout. Arthritis Res Ther. 2006; 8(Suppl 1): S2.
6. Arromdee E, Michet CJ, Crowson CS, et al. Epidemiology of gout: is the incidence rising? J Rheumatol. 2002; 29 : 2403–2406.
7. López López CO, Lugo EF, Alvarez–Hernández E, et al. Severe tophaceous gout and disability: changes in the past 15 years. Clin Rheumatol. 2017; 36(1): 199–204.
8. Johnson RJ, Segal MS, Sautin Y, et al. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am J Clin Nutr. 2007; 86(4): 899–906.
9. Kim KM, Henderson GN, Frye RF, et al. Simultaneous determination of uric acid metabolites allantoin, 6-aminouracil, and triuret in human urine using liquid chromatography–mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2009; 1; 877(1–2): 65–70.
10. Glantzounis GK, Tsimoyiannis EC, Kappas AM, et al. Uric acid and oxidative stress. Curr Pharm Des. 2005; 11(32): 4145–51.
11. Bickel C, Rupprecht HJ, Blankenberg S, et al. Serum uric acid as an independent predictor of mortality in patients with angiographically proven coronary artery disease. Am J Cardiol. 2002; 89(1): 12–7.
12. Viazzi F, Parodi D, Leoncini G, et al. Serum uric acid and target organ damage in primary hypertension. Hypertension. 2005; 45(5): 991–6.
13. Palmer TM, Nordestgaard BG, Benn M, et al. Association of plasma uric acid with ischaemic heart disease and blood pressure: mendelian randomisation analysis of two large cohorts. BMJ. 2013; 347: f4262.
14. Fabbrini E, Serafini M, Colic Baric I, et al. Effect of plasma uric acid on antioxidant capacity, oxidative stress, and insulin sensitivity in obese subjects. Diabetes. 2014; 63(3): 976–81.
15. Mikami T, Kita K, Tomita S, et al. Is allantoin in serum and urine a useful indicator of exercise–induced oxidative stress in humans? Free Radic Res. 2000; 32(3): 235–44.
16. Waring WS, Convery A, Mishra V, et al. Uric acid reduces exercise–induced oxidative stress in healthy adults. Clin Sci (Lond). 2003; 105(4): 425–30.
17. Davis JW1, Grandinetti A, Waslien CI, et al. Observations on serum uric acid levels and the risk of idiopathic Parkinson’s disease. Am J Epidemiol. 1996; 144(5): 480–4.
18. Hooper DC1, Spitsin S, Kean RB, et al. Uric acid, a natural scavenger of peroxynitrite, in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci U S A. 1998; 95(2): 675–80.
19. Yao JK, Reddy R, van Kammen DP, et al. Reduced level of plasma antioxidant uric acid in schizophrenia. Psychiatry Res. 1998; 80(1): 29–39.
20. Hayden MR, Tyagi SC. Uric acid: A new look at an old risk marker for cardiovascular disease, metabolic syndrome, and type 2 diabetes mellitus: The urate redox shuttle. Nutr Metab (Lond). 2004; 1(1): 10.
21. Ebrahimpour P, Fakhrzadeh H, Heshmat R, et al. Serum uric acid levels and risk of metabolic syndrome in healthy adults. Endocr Pract. 2008; 14(3): 298–304.
22. Heinig M, Johnson RJ. Role of uric acid in hypertension, renal disease, and metabolic syndrome. Cleve Clin J Med. 2006; 73(12): 1059–64.
23. Sui X, Church TS, Meriwether RA, et al. Uric acid and the development of metabolic syndrome in women and men. Metabolism. 2008; 57(6): 845–52.
24. Tamba S, Nishizawa H, Funahashi T, et al. Relationship between the serum uric acid level, visceral fat accumulation and serum adiponectin concentration in Japanese men. Intern Med. 2008; 47(13): 1175–80.
25. Chen S, Du H, Wang Y, et al. The epidemiology study of hyperuricemia and gout in a community population of Huangpu District in Shanghai. Chin Med J (Engl).1998; 111(3): 228–30.
26. Kuntz D, Chretien JM, Ryckewaert A, et al. [Distribution and correlations of serum uric–acid in two French adult populations: 13,885 men and 6,861 women (author’s transl)]. Sem Hop. 1979; 55(5–6): 241–8.
27. Lin K, Lin HY, Chou P. Community based epidemiological study on hyperuricemia and gout in Kin–Hu, Kinmen. J Rheumatol. 2000; 27(4): 1045–50.
28. Chang HY, Pan WH, Yeh WT et al. Hyperuricemia and gout in Taiwan: results from the Nutritional and Health Survey in Taiwan (1993–96). J Rheumatol. 2001; 28(7): 1640–6.
29. Tang W, Miller MB, Rich SS, et al. Linkage analysis of a composite factor for the multiple metabolic syndrome: the National Heart, Lung, and Blood Institute Family Heart Study. Diabetes. 2003; 52(11): 2840–7.
30. Wilk JB, Djousse L, Borecki I, et al. Segregation analysis of serum uric acid in the NHLBI Family Heart Study. Hum Genet. 2000; 106(3): 355–9.
31. Nath SD, Voruganti VS, Arar NH, et al. Genome scan for determinants of serum uric acid variability. J Am Soc Nephrol. 2007; 18(12): 3156–63.
32. Yang Q, Guo CY, Cupples LA, et al. Genome–wide search for genes affecting serum uric acid levels: the Framingham Heart Study. Metabolism. 2005; 54(11): 1435–41.
33. Köttgen A, Albrecht E, Teumer A, et al. Genome–wide association analyses identify 18 new loci associated with serum urate concentrations. Nat Genet. 2013; 45(2): 145–54.
34. Nakayama A, Nakaoka H, Yamamoto K, et al. GWAS of clinically defined gout and subtypes identifies multiple susceptibility loci that include urate transporter genes. Ann Rheum Dis. 2016; doi: 10.1136/annrheumdis–2016–209632. [Epub ahead of print]
35. Curto R, Voit EO, Sorribas A. Mathematical models of purine metabolism in man. Math Biosci. 1998; 151(1): 1–49.
36. Curto R, Voit EO, Cascante M. Analysis of abnormalities in purine metabolism leading to gout and to neurological dysfunctions in man. Biochem J. 1998; 329( Pt 3): 477–87.
37. Simmonds HA, Reiter S, Davies PM, et al. Orotidine accumulation in human erythrocytes during allopurinol therapy: association with high urinary oxypurinol-7-riboside concentrations in renal failure and in the Lesch-Nyhan syndrome. Clin Sci (Lond) 1991; 80 : 191–7.
38. Duley JA, Christodoulou J, de Brouwer AP. The PRPP synthetase spectrum: what does it demonstrate about nucleotide syndromes? Nucleosides Nucleotides Nucleic Acids
2011; 30 : 1129–39.
39. Kostalova E, Pavelka K, Vlaskova H, et al. Hyperuricemia and gout due to deficiency of hypoxanthine–guanine phosphoribosyltransferase in female carriers: New insight to differential diagnosis. Clin Chim Acta. 2015; 440 : 214–7.
40. Dawson PA, Gordon RB, Keough DT, et al. Normal HPRT coding region in a male with gout due to HPRT deficiency. Mol Genet Metab. 2005; 85 : 78–80.
41. Nguyen KV, Naviaux RK, Paik KK, et al. Lesch–Nyhan syndrome: mRNA expression of HPRT in patients with enzyme proven deficiency of HPRT and normal HPRT coding region of the DNA. Mol Genet Metab. 2012; 106 : 498–501.
42. Rahul Mittal, 1 Kunal Patel, 1 Jeenu Mittal, et al. Association of PRPS1 Mutations with Disease Phenotypes. Dis Markers. 2015; 2015 : 127013.
43. Enomoto A, Kimura H, Chairoungdua A, et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002; 417(6887): 447–52.
44. Sugihara S, Hisatome I, Kuwabara M. Depletion of Uric Acid Due to SLC22A12 (URAT1) Loss–of–Function Mutation Causes Endothelial Dysfunction in Hypouricemia. Circ J. 2015; 79(5): 1125–32.
45. Ichida K, Hosoyamada M, Kamatani N, et al. Age and origin of the G774A mutation in SLC22A12 causing renal hypouricemia in Japanese. Clin Genet. 2008; 74(3): 243–51.
46. Stiburkova B, Gabrikova D, Čepek P, et al. Prevalence of URAT1 allelic variants in the Roma population. Nucleosides Nucleotides Nucleic Acids. 2016 Dec;35(10-12): 529–535.
47. Doring A, Gieger C, Mehta D, et al. SLC2A9 influences uric acid concentrations with pronounced sex–specific effects. Nat Genet. 2008; 40(4): 430–6.
48. Vitart V, Rudan I, Hayward C, et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat Genet. 2008; 40(4): 437–42.
49. Hollis–Moffatt JE, Xu X, Dalbeth N, et al. Role of the urate transporter SLC2A9 gene in susceptibility to gout in New Zealand Māori, Pacific Island, and Caucasian case–control sample sets. Arthritis Rheum. 2009; 60(11): 3485–92.
50. Hollis–Moffatt JE, Gow PJ, Harrison AA, et al. The SLC2A9 nonsynonymous Arg265His variant and gout: evidence for a population – specific effect on severity. Arthritis Res Ther. 2011; 13(3): R85.
51. Bhasin B, Stiburkova B, De Castro–Pretelt M, et al. Hereditary renal hypouricemia: a new role for allopurinol? Am J Med. 2014; 127(1): e3–4.
52. Ekaratanawong S, Anzai N, Jutabha P, et al. Human organic anion transporter 4 is a renal apical organic anion/dicarboxylate exchanger in the proximal tubules. J Pharmacol Sci. 2004; 94(3): 297–304.
53. Bahn A, Hagos Y, Reuter S, et al. Identification of a new urate and high affinity nicotinate transporter, hOAT10 (SLC22A13). J Biol Chem. 2008; 283(24): 16332–41.
54. Allikmets R, Schriml LM, Hutchinson A, et al. A human placenta–specific ATP–binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res. 1998; 58(23): 5337–9.
55. Mao Q, Unadkat JD. Role of the breast cancer resistance protein (ABCG2) in drug transport. AAPS J. 2005; 7(1): 118–33.
56. Kolz M, Johnson T, Sanna S, et al. Meta-analysis of 28,141 individuals identifies common variants within five new loci that influence uric acid concentrations. PLoS Genet. 2009; 5(6): 1000504.
57. Matsuo H, Takada T, Ichida K, et al. Common defects of ABCG2, a high–capacity urate exporter, cause gout: a function-based genetic analysis in a Japanese population. Sci Transl Med. 2009; 1(5): 5ra11.
58. Woodward OM, Köttgen A, Coresh J, et al. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc Natl Acad Sci U S A. 2009; 106(25): 10338–42.
59. Ichida K, Matsuo H, Takada T, et al. Decreased extra–renal urate excretion is a common cause of hyperuricemia. Nat Commun. 2012; 3 : 764.
60. Stibůrková B, Pavlíková M, Sokolová J, et al. Metabolic syndrome, alcohol consumption and genetic factors are associated with serum uric acid concentration. PLoS One. 2014; 9(5): e97646.
61. Jutabha P, Anzai N, Kitamura K, et al. Human sodium phosphate transporter 4 (hNPT4/SLC17A3) as a common renal secretory pathway for drugs and urate. J Biol Chem. 2010; 285(45): 35123–32.
62. Russel FG, Koenderink JB, Masereeuw R. Multidrug resistance protein 4 (MRP4/ABCC4): a versatile efflux transporter for drugs and signalling molecules. Trends Pharmacol Sci. 2008; 29(4): 200–7.
63. Hoque MT, Conseil G, Cole SP. Involvement of NHERF1 in apical membrane localization of MRP4 in polarized kidney cells. Biochem Biophys Res Commun. 2009; 379(1): 60–4.
64. Eraly SA, Vallon V, Rieg T, et al. Multiple organic anion transporters contribute to net renal excretion of uric acid. Physiol Genomics. 2008; 33(2): 180–92.
65. Chong SS, Kristjansson K, Zoghbi HY, et al. Molecular cloning of the cDNA encoding a human renal sodium phosphate transport protein and its assignment to chromosome 6p21.3–p23. Genomics. 1993; 18(2): 355–9.
66. Uchino H, Tamai I, Yamashita K, et al. p-aminohippuric acid transport at renal apical membrane mediated by human inorganic phosphate transporter NPT1. Biochem Biophys Res Commun. 2000; 270(1): 254–9.
67. Urano W, Taniguchi A, Anzai N, et al. Sodium–dependent phosphate cotransporter type 1 sequence polymorphisms in male patients with gout. Ann Rheum Dis. 2010; 69(6): 1232–4.
68. Sekine T, Watanabe N, Hosoyamada M, et al. Expression cloning and characterization of a novel multispecific organic anion transporter. J Biol Chem. 1997; 272(30): 18526–9.
69. Cha SH, Sekine T, Fukushima JI, et al. Identification and characterization of human organic anion transporter 3 expressing predominantly in the kidney. Mol Pharmacol. 2001; 59(5): 1277–86.
70. Xu G, Chen X, Wu D, et al. Development of high–specificity antibodies against renal urate transporters using genetic immunization. J Biochem Mol Biol. 2006; 39(6): 696–702.
71. Anzai N, Kanai Y, Endou H. New insights into renal transport of urate. Curr Opin Rheumatol. 2007; 19(2) 151–57.
72. Nakayama A, Matsuo H, Nakaoka H, et al. Common dysfunctional variants of ABCG2 have stronger impact on hyperuricemia progression than typical environmental risk factors. Scientific Reports. 2014; 4 : 5227.
73. Roberts RL, Wallace MC, Phipps–Green AJ, et al. ABCG2 loss–of–function polymorphism predicts poor response to allopurinol in patients with gout. Pharmacogenomics J. 2017; 17(2): 201–203.
74. Petru L, Pavelcova K, Sebesta I, et al. Genetic background of uric acid metabolism in a patient with severe chronic tophaceous gout. Clin Chim Acta. 2016; 460 : 46–9.
75. Perez–Ruiz F, Sundy JS, Miner JN, et al. Lesinurad in combination with allopurinol: results of a phase 2, randomised, double-blind study in patients with gout with an inadequate response to allopurinol. Ann Rheum Dis. 2016; 75(6): 1074–80.
Labels
Dermatology & STDs Paediatric rheumatology RheumatologyArticle was published in
Czech Rheumatology

2017 Issue 3
Most read in this issue
- Současné možnosti terapie entezitid
- Nástin genetické architektury primární hyperurikémie a dny
- Kardiovaskulární komorbidity a ateroskleróza u systémového lupus erythematodes