
Paraneoplastické syndromy
Paraneoplastic syndrom
Paraneoplastic syndromes represent various manifestations of the primarily formed tumor, which, however, can be occult at the time of the appearance of the paraneoplastic symptomatology. They are caused by a wide spectrum of distant effects of tumors, which are not related to the mechanical action of tumor masses or distant metastases. The pathogenesis of paraneoplastic manifestations is not yet quite clear, but these syndromes usually arise as a result of secretions of hormones, peptides and various cytokine cells of the tumor, or as an immune-mediated cross-reaction between malignant and normal tissues. A number of paraneoplastic manifestations precede their own manifestations of malignancy by several months or even years, their good knowledge can contribute to the timely diagnosis of the tumor and thus to the early initiation of treatment. The course of the symptomatology of paraneoplastic syndrome often replicates the actual course of the tumor, when paraneoplastic syndrome is resolved after cure of the malignancy, and the reappearance of its manifestations is a sign of recurrence of malignancy. In some cases, for example, in paraneoplastic syndromes with neurological manifestations, there may be irreversible changes due to cross-reactivity between the tumor and the nervous tissue. Haematological paraneoplastic syndromes are often otherwise clinically asymptomatic, and they are usually a manifestation of advanced malignancy.
Keywords:
paraneoplastic syndrome – cancer-associated myositis – paraneoplastic synovitis – paraneoplastic vasculitis – paraneoplastic bone disease
Authors:
Š. Forejtová
Authors‘ workplace:
Revmatologický ústav Praha
Published in:
Čes. Revmatol., 27, 2019, No. 4, p. 186-202.
Category:
Review Article
Overview
Paraneoplastické syndromy představují rozmanité projevy primárně vzniklého nádoru, který ovšem může být v době objevení se paraneoplastické symptomatologie dosud okultní. Jsou způsobeny širokým spektrem vzdálených účinků nádorů, které nesouvisejí s mechanickým působením nádorových hmot ani vzdálených metastáz. Patogeneze paraneoplastických projevů není dosud zcela jasná, tyto syndromy ale obvykle vznikají v důsledku sekrece hormonů, peptidů a různých cytokinů buňkami nádoru, nebo jako imunitně zprostředkovaná zkřížené reakce mezi maligní a normální tkání. Řada paraneoplastických projevů předchází vlastním projevům malignity o několik měsíců, případně i let, jejich dobrá znalost tak může přispět k včasné diagnostice nádoru a tak i k brzkému zahájení léčby. Průběh symptomatologie paraneoplastického syndromu často kopíruje vlastní průběh nádorového onemocnění, kdy po zaléčení malignity dojde k vymizení paraneoplastického syndromu a znovuobjevení se jeho projevů bývá známkou recidivy malignity. V některých případech, například u paraneoplastických syndromů s neurologickými projevy, může vlivem zkřížené reaktivity mezi nádorem a nervovou tkání dojít ke vzniku ireverzibilních změn. Hematologické paraneoplastické syndromy často bývají naopak jinak klinicky asymptomatické a jsou obvykle projevem pokročilé malignity.
Klíčová slova:
paraneoplastický syndrom – s nádorem asociovaná myopatie – paraneoplastická synovitida – paraneoplastická vaskulitida – paraneoplastické kostní onemocnění
ÚVOD
Již na konci 19. století bylo popsáno, že určité nádory mohou způsobovat rozmanité symptomy, které nesouvisejí s přímou invazí tumoru nebo kompresí tkáně v okolí nádoru (1). V roce 1916 byla poprvé popsána asociace mezi karcinomem a myozitidou (2). Pojem „paraneoplastický syndrom“ byl poprvé použit již ve čtyřicátých letech 20. století (3), nicméně přesná podstata vzniku těchto symptomů zůstává dosud nejasná. Syndromy jsou projevem nádoru, který je v době objevení se symptomů často dosud skrytý, a nejsou způsobeny jeho mechanickým tlakem nebo vzdálenými metastázami. Příznaky obvykle vznikají v důsledku sekrece hormonů, peptidů a různých cytokinů buňkami nádoru, nebo jako imunitně zprostředkovaná zkřížené reakce mezi maligní a normální tkání. V mnoha případech se paraneoplastické syndromy manifestují ještě předtím, než došlo k diagnóze nádoru, a tak jejichvčasné rozeznání může vést k objevení jinak dosud okultního tumoru, což může přispět k včasnému zahájení léčby nádorového onemocnění. Odhaduje se, že paraneoplastické syndromy se projevují až u 8 % pacientů s maligním nádorem (4) a mohou postihovat různé orgány. Nejčastěji se projevují v oblasti nervového, endokrinního, kožního nebo pohybového systému. V řadě případů se jedná o syndromy postihující několik systémů, hovoří se pak o smíšených paraneoplastických syndromech.
REVMATICKÉ PROJEVY SPOJENÉS MALIGNITOU
V rámci revmatických projevů paraneoplastických syndromů mohou být postiženy klouby, svaly, měkké tkáně a cévy, nebo se jedná o smíšené postižení v rámci více orgánů (tab. 1).

Výskyt nejčastějších paraneoplastických syndromů napodobující revmatická onemocnění pak udává tabulka 2.

Karcinomatózní polyartritida
Tato polyartritida byla poprvé popsána v roce 1984 (5). Na rozdíl od revmatoidní artritidy se častěji vyskytuje u mužů, ve vyšším věku a vyznačuje se náhlým bouřlivým začátkem. Jedná se o ne-erozivní, nedeformující, asymetrickou polyartritidu, často migrující, postihující především velké klouby, a to predilekčně na dolních končetinách, naopak nebývají postiženy ruce a zápěstí. Revmatoidní faktor (RF) je obvykle negativní, nicméně je nutné vzít v potaz, že u jedinců nad 60 let stoupá nespecifická pozitivita RF i ANA, dokonce mohou být pozitivní i anticitrulinové protilátky (6). Karcinomatózní polyartritida je v 1/3 případů asociována s hematologickými a lymfoproliferativními malignitami. Ze solidních tumorů provází nejčastěji adenokarcinom plic a prsou, vyskytuje se ale i u karcinomu střeva a u jiných solidních tumorů. Charakteristickým znakem paraneoplastické polyartritidy je špatná odpověď na léčbu glukokortikoidy i chorobu-modifikujícími léky, naopak artritida dobře reaguje na terapii primárního nádoru. Pokud však karcinom recidivuje, 75 % pacientů nevyvine relaps revmatických syndromů (7).
Hypertrofická osteoartropatie (Pierreův-Marieův-Bambergerův syndrom)
Onemocnění bylo poprvé popsáno v roce 1889 von Bambergerem (8) u nemocných s bronchiektáziemi, klinická asociace hypertrofické osteoartropatie (HOA) s malignitami byla zaznamenána o několik dekád později (9). Častěji postihuje muže a je asociována především s adenokarcinomem plic. Charakteristickým příznakem je typická triáda: paličkovité prsty na rukou i nohou, periostitida a artritida. Vývoj paličkovitých prstů je často asymptomatický, nebo jej provází pálivá bolest konečků prstů. Ke klinickému obrazu dále patří artritida, a to především velkých kloubů, provázená ranní ztuhlostí, dále pálení prstů, někdy dominuje palčivá bolest a proteplení bérců či předloktí nebo periartikulární bolest. Na rentgenu se objevuje akroosteolýza (obr. 1) a narůstání terminální falangy, které může mít až houbovitý charakter. Na dlouhých kostech předloktí a bérců se popisuje periostitida (obr. 2). Onemocnění je někdy asociováno s kožními příznaky typu acanthosis palmaris (tripe palm) (obr. 3), což je hyperkeratóza dlaní s prominencí dermatoglyfických dlaňových čar a sametovým vzhledem kůže.



Hypertrofická osteoartropatie se může vyskytovat i bez přítomnosti nádoru. Až ve 33 % je výskyt HOA dědičný, nebo může být asociován i s jinými nitrohrudními afekcemi (infekce, aneurysma, plicní fibróza), s některými jaterními onemocněními nebo s pravo-levým srdečním zkratem (10).
Pokud je HOA způsobena karcinomem, v případě jeho odstranění dochází k ústupu potíží. V etiologii vzniku příznaků HOA hraje totiž roli nadprodukce některých růstových faktorů, jako je VEGF (vascular endothelial growth factor) a PDGF (platelet derived growth factor), které jsou produkovány nádorovými buňkami. Při léčbě palčivých kostních bolesti může být efektivní léčba bisfosfonáty, především kyselinou zolendronovou (11), jinak základem symptomatické léčby jsou nesteroidní antirevmatika (NSA).
Amyloidová artritida
Vyvíjí se u 0,1–6 % nemocných s mnohočetným myelomem a je dána depozicí AL amyloidových mas v synoviální tkáni (10). Artritida se objevuje v době diagnózy myelomu nebo následně až po jejím stanovení. Postihuje běžně ramena, kdy je popisován tzv. syndrom vycpaného ramene, dále kolena, zápěstí, metakarpofalangeální a proximální interfalangeální klouby. U pacientů s amyloidózou se někdy vyskytují podkožní uzly, které jsou podobné revmatickým uzlům, což také může vést k obtížím v diferenciální diagnostice. Obarvením synoviálního stočeného sedimentu Kongo červení je možné detekovat amyloidová depozita ze synoviálních fragmentů. Léčba artritidy je symptomatická, pomocí NSA, základem je terapie myelomu.
Sekundární dna
Sekundární dna může provázet řadu hematologických malignit, a to jak lymfoproliferativní a myeloproliferativní onemocnění, tak i abnormální tvorbu destiček nebo červené krevní řady. Léčba cytostatiky může přispívat ke vzniku excesivní hyperurikemie, neboť zvýšený rozpad buněk vede ke zvýšenému obratu nukleových kyselin (12).
Pyogenní artritida
V několika případech byla popsána septická artritida způsobená mikrobem Clostridium septicum (13) nebo Streptococcus bovis (14), která může být prvním projevem dosud okultního karcinomu tračníku (15).
Myopatie
Myopatie asociovaná s nádorem tvoří separátní jednotku v klasifikaci zánětlivých myopatií. Malignita se v různých souborech objevuje u zánětlivých myopatií v 6–60 %, přičemž střední hodnota je kolem 15 % (16), silnější asociace je popsána pro dermatomyozitidu ( Standardised Incidence Ratio – SIR = 6,2). Ta se nejčastěji vyskytuje při nádorech plic, vaječníků, žaludku, střeva, slinivky, prsu, prostaty a u non-Hodgkinova lymfomu. Polymyozitida se jako paraneoplastický syndrom projevuje méně často (SIR = 2,0), nejvíce je pak asociována s nádory močového měchýře, plic a s non-Hodgkinovým lymfomem. Myozitida s inkluzními tělísky má podobnou asociaci s malignitou (SIR = 2,4). Pravděpodobnost asociace s malignitou klesá v čase (SIR = 4,4 v prvním roce, 3,4 mezi 1. až 3. rokem trvání myopatie, 2,2 pak mezi 3.až 5. rokem a méně než 1,6 po 5. roce (P pro trend = 0,002) (17). Mírně zvýšené je ale i riziko imunitně zprostředkované nekrotizující myopatie s pozitivitou protilátek proti HMGCR. Paraneoplastická dermatomyozitida má častěji těžší kožní projevy, dysfagii a postižení bránice (15), vyskytuje se více u mužů než u žen. Mezi další rizikové faktory pak patří vyšší věk při diagnóze myozitidy, kožní nekrózy a vaskulitida, neodpovídavost na glukokortikoidy a imunosupresivní léčbu, absence intersticiálního plicního postižení, nízká hladina C4 složky komplementu a přítomnost anti-TIF-1γ a anti-NXP-2 autoprotilátek (16). Ve studii u 40 pacientů s dermatomyozitidou nebo polymyozitidou bylo zjištěno, že vyšší výskyt malignit je u pacientů s následujícími symptomy: absence Raynaudova fenoménu, rychlý vývoj myozitidy, vyšší hodnota sedimenatce (48 vs. 25 mm/h) a vyšší hladinou kreatinkinázy (2840 vs. 1346 U/l). Autoři studie dospěli k názoru, že pacienti s těmito symptomy mají vyšší benefit z extenzivního vyšetřování k vyloučení malignity, a to včetně provedení CT hrudníku, břicha a pánve (18). V současné době se doporučuje u pacientů s rizikovými faktory provádět vyšetření PET/CT, a to opakovaně v pravidelných intervalech, zejména v prvních letech nemoci.
Cílem léčby je zlepšit svalovou sílu, potlačit orgánové postižení a zároveň pokud možno minimalizovat riziko komplikací spojených s terapií. Základem léčby je podávání glukokortikoidů, v úvodu v dávce 1–1,5 mg prednisonu/kg/den. U většiny nemocných si stav vyžádá přidání imunosupresivní léčby. Nejčastěji je používán methotrexát, azathioprin nebo cyklosporin. U pacientů s nedostatečným efektem této léčby je možné podávat cyklofosfamid, takrolimus, mykofenolát mofetil, eventuálně leflunomid. U rezistentních stavů mohou být podávány intravenózní imunoglobuliny, eventuálně může být použita plazmaferéza nebo leukoferéza. V posledních letech se také ukazuje, že k podstatnému zlepšení zdravotního stavu může také vést léčba rituximabem (16).
Lambertův-Eatonův syndrom
Jedná se o vzácné onemocnění projevující se svalovou slabostí, únavností a hyporeflexií, které jsou způsobené nedostatkem uvolňování acetylcholinu v nervových zakončeních. U většiny nemocných jsou detekovatelné autoprotilátky proti vysokonapěťovým kalciovým kanálům. Onemocnění se velmi podobá myasthenia gravis (také se někdy nazývá Lambertova-Eatonova myastenie), více jsou při něm postiženy dolní končetiny. Současně může být přítomna i porucha sekrece slin, snížené pocení, posturální hypotenze a impotence. Onemocnění je až v 60 % asociováno s malobuněčným karcinomem plic, ve většině případů se příznaky syndromu objeví ještě před stanovením diagnózy karcinomu. Eatonův-Lambertův syndrom je sdružen s EMG obrazem snížení amplitudy sumačního potenciálu, při vysokofrekvenční stimulaci dochází na rozdíl od myastenie k nárůstu amplitudy akčního potenciálu.
Oproti myastenii jsou při léčbě Lambertova--Eatonova syndromu méně účinné inhibitory cholinesterázy. Účinnější je 3,4-diamidopyridin, glukokortikoidy a imunosupresiva. V případě průkazu nádoru je patrný ústup klinických příznaků po jeho operativním odstranění s tím, že se výrazně zlepší i celková prognóza. Mezi další léčebné možnosti patří plazmaferéza a intravenózní imunoglobuliny (19).
Paraneoplastické kostní onemocnění
Paraneoplastické kostní onemocnění zahrnuje hypertrofickou osteoartropatii, která se ovšem mimo jiné vyskytuje i na kloubech ve smyslu artritidy (viz výše), dále tumorem-indukovanou osteomalacii a hyperkalcemii při malignitě. Samostatnou jednotku představuje syndrom Schnitzlerové.
Tumorem-indukovaná osteomalacie (TIO) je takéjinak nazývána onkogenní hypofosfatemická osteomalacie. Je to vzácné onemocnění, kdy abnormální renální ztráty fosfátů vedou k těžké hypofosfatemii. Velmi nízká hladina kalcitriolu vzniklá jako následek těžké poruchy metabolismu vitaminu D v ledvinách vede ke vzniku osteomalacie. Většina TIO je provázena zvýšením FGF-23 (fibroblast growth factor 23) a/nebo zvýšenou hladinou tzv. frizzled-4-proteinu, které jsou produkovány tumorem (20). Léčba spočívá, pokud je to možné, v chirurgickém odstranění tumoru a dále v suplementaci fosfáty a kalcitriolem.
Hyperkalcemie při malignitě vzniká v 80 % jako důsledek ektopické sekrece PTHrp (parathyroid hormone-related peptide). Podrobněji je popsána v části o paraneoplastických endokrinních syndromech.
Syndrom Schnitzlerové je vzácné onemocnění, které je charakterizováno chronickou, nesvědivou urtikou, asociovanou s opakovanými horečkami, bolestmi kostí, svalů a kloubů, někdy i s artritidami. Je provázen leukocytózou a vysokými hodnotami reaktantů akutní fáze. Charakteristické jsou i kostní změny (kombinace hyperostózy a osteolýzy). Typický je výskyt monoklonální komponenty v IgM, vzácněji v IgG. Přibližně 15 % pacientů má riziko vývoje malignity spojené s monoklonální komponentou. V terapii se užívají především blokátory IL-1, které však tlumí pouze zánětlivou symptomatologii, nemají vliv na provázející monoklonální gamapatii.
Raynaudův syndrom
Také Raynaudův syndrom byl popsán jako jeden z prvních projevů malignity, např. u karcinomu jícnu, žaludku, střeva, slinivky, ledvin, prsu, plic, ovarií, dělohy, prostaty a u hematologických malignit, především u lymfoproliferativních typů nádorů. Většinou se objevuje společně s nádorem nebo těsně před stanovením diagnózy, v některých případech může být přítomen až 2 roky před nádorem.
Paraneoplastická vaskulitida
Paraneoplastické vaskulitidy představují přibližně 2–5 % všech vaskulitických syndromů (21, 22). Leukocytoklastické vaskulitidy (LCV) reprezentují 50–60 % všech paraneoplastických vaskulitid a provází jak hematologické malignity, tak i solidní tumory, z nichž nejčastěji jde o karcinom močového měchýře a plic (21). Jednou z forem LCV je Heno-chova-Schönleinova purpura, která tvoří až 15 %paraneoplastických vaskulitid. Paraneoplastická forma Henochovy-Schönleinovy purpury se vyskytuje především u starších mužů s karcinomem plic či urogenitálního nebo gastrointestinálního traktu, u této formy je běžné i renální postižení (23). Také polyarteritis nodosa představuje až 15% paraneoplastických vaskulitid. Postihuje především malé a středně velké tepny kůže, periferních nervů a gastrointestinálního traktu. Paraneoplastická polyarteritis nodosa bývá asociována především s leukemií z vlasatých buněk. V etiopatogenezi zřejmě hraje roli zkřížená reaktivita protilátek proti vlasatým buňkám s endoteliálními buňkami cév (24). Z dalších vaskulitid může mít paraneoplastickou etiologii temporální arteritida, byly popsány i jednotlivé případy paraneoplastické granulomatózy s polyangiitidou, mikroskopické polyangiitidy i syndromu Churga-Straussové.
Paraneoplastická vaskulitida může reagovat na glukokortikoidní a imunosupresivní léčbu, účinné bývá i odstranění tumoru, naopak s recidivou malignity se vaskulitida často znovu vrací.
Erytromelalgie
Erytromelagie je poměrně vzácné onemocnění, které je charakterizováno intenzivní pálivou bolestí, erytémem kůže a zvýšenou teplotou nohou, méně často rukou (obr. 4). Příznaky se zhoršují v teple, naopak zlepšení nastává v chladu a při elevaci končetin. Erytromelalgie u dospělých může být idiopatická, 18 % případů se ovšem vyskytuje u polycytemia vera nebo esenciální trombocytózy. Symptomy erytromelalgie mohou předcházet vývoji trombocytózy až 2,5 roku. Paraneoplastická erytromelalgie může provázet i jiné malignity, jako je chronická myeloidní leukemie, myelo-fibróza, karcinom prsu, prostaty, ovaria nebo střeva. V léčbě používáme kyselinu acetylsalicylovou, která většinou přináší pouze částečnou úlevu (15, 25). Dále jsou užívány blokátory kalciového kanálu, různá antidepresiva, antihistaminika, zkouší se i gabapentin nebo pregabalin, případně transdermální lidokain (26).

Palmární fascitida se symetrickou artritidou
Jedná se o poměrně raritní onemocnění, které se nejvíce vyskytuje u žen s ovariálním adenokarinomem, může se ovšem objevit i u jiných tumorů. Onemocnění bylo popsáno v roce 1966 jako syndrom rameno-ruka (27). V roce 1982 pak byla separátně popsána u šesti posmenopauzálních žen s ovariálním tumorem nová jednotka s názvem palmární fascitida se symetrickou polyartritidou (obr. 5), kdy u uvedených případů symptomy na pohybovém ústrojí předcházely diagnóze adenokarcinomu ovaria o 5–25 měsíců (28). Onemocnění se projevuje difuzním otokem končetin s fibrotizujícím charakterem (dif. dg. je nutné zvažovat sklerodermii) a může vést k rychle progredujícím flekčním kontrakturám rukou (dif. dg. podobná Dupuytrenově kontraktuře). Fascitidou mohou být ovšem postiženy i nohy a dále symetricky i řada větších kloubů, jako jsou zápěstí, lokte, ramena a kolena. V léčbě používáme nesteroidní antirevmatika, glukokortikoidy a fyzikální léčbu, bohužel ve většině případů je efekt této terapie nedostatečný.

Eozinofilní fascitida
Jedná se o vzácnější onemocnění charakterizované symetrickým postižením končetin a trupu se zarudnutím a otokem kůže. Typicky je popisována tzv. „pomerančová kůže“ a současné kolagenní ztluštění podkožní fascie. Eozinofilie je přítomna již na počátku onemocnění. Posti-žení se typicky vyhýbá kůži rukou i nohou. Eozinofilní fascitida může být idiopatická, nebo je asociována s některými malignitami, především hematologickými, například s aplastickou anemií, myelodysplastickým syndromem nebo myeloproliferativním onemocněním, lymfomem, mnohočetným myelomem, případně s lymfatickou nebo eozinofilní leukemií (29).
Základem léčby eozinofilní fascitidy je podávání glukokortikoidů, dlouhodobě podáváme prednison v dávce 20 mg nebo ekvivalent methylprednisolonu. U pacientů refrakterních k léčbě glukokortikoidy je indikovaná léčba methotrexátem. Důležitou součástí léčby je samozřejmě rehabilitace a fyzikální léčba. U paraneoplastické etiologie je základem léčby samozřejmě terapie primárního tumoru (30).
Polyartritida s panikulitidou
Lobulární panikulitida s rozsáhlou nekrózou tuku společně s artritidou a/nebo s kostní nekrózou je popisována u onemocnění slinivky. Známý je například PPP syndrom: pankreatitida, panikulitida a polyartritida (31) nebo polyartritida s panikulitidou u nádorů slinivky (32, 33). Nejčastěji jsou postiženy kotníky, kolena, zápěstí a metakarpofalangeální klouby (obr. 6). Na rentgenu bývá typicky popisováno zúžení kloubní štěrbiny, osteolytické léze, patologické fraktury a osteonekróza. Potíže neodpovídají na léčbu nesteroidními antirevmatiky ani glukokortikoidy.

Polymyalgia revmatika
Paraneoplastická polymyalgia revmatika (PMR) je nejčastěji asociována s myelodysplastickým syndromem (34). Projevuje se typicky bolestí a ztuhlostí krku, pletenců ramenních a kyčelních kloubů. Padesát procent pacientů současně vyvine periferní synovitidu. Atypická PMR byla popsána u pacientů s metastatickým rozsevem nádoru. Tito pacienti mají časný výskyt symptomů, asymetrickou lokalizaci, hodnota sedimentace erytrocytů je často < 40 nebo > 100 mm/hodinu. Vyskytuje se nejvíce u leukemií a lymfomů, myelodysplastického syndromu, karcinomu tlustého střeva, plic, ledvin, prostaty nebo prsu. U těchto jedinců je typická špatná odpověď na glukokortikoidy a prolongovaný výskyt symptomů.
Systémová sklerodermie
Malignita také může iniciovat imunitní odpověď, která je specifická pro sklerodermii. Vyvolá tak tzv. sklerodermii synchronní s nádorem, která patří do subsetu systémové sklerodermie. Nádor se může objevit až do 2 let od diagnózy sklerodermie. Nejčastěji asociovanou malignitou je karcinom plic, ale je popsána souvislost i s jinými solidními nádory, jako je tumor ovaria, prsu, prostaty atd. Obezřetní musíme být především v případě, že je při imunologickém nálezu přítomná pozitivita RNA-III polymerázy. Naopak podle výsledků jedné studie vyplývá, že plicní fibróza a přítomnost protilátek proti topoizomeráze nenavyšují riziko vzniku plicního nádoru. Kouření zato zvyšuje riziko vývoje tumoru plic u sklerodermie sedmkrát v porovnání s nekuřáky (P = 0,008) (P35) Některé další skleroderma-like dermatózy, jako je skleromyxedém nebo skleroedém, byly popsány v souvislosti s paraproteinemií.
Lupus-like syndrom
Subakutní kožní lupus erythematodes (SCLE) je fotosenzitivní, nejizvící se kožní vyrážka, která je až v 50 % asociována s výskytem systémového lupusu. Většinou se jedná o idiopatické onemocnění, někdy se může jednat i o léky indukovanou formu. Osmdesát procent pacientů má pozitivní protilátky anti-Ro. Byly popsány i případy paraneoplastického výskytu lupus-like syndromu, například u malobuněčného karcinomu plic, kdy se u pacienta vyskytuje SCLE, současně bývají pozitivní i antinukleární protilátky (36). Dále byl výskyt zaznamenám u thymomu, Hodgkinova onemocnění, nádoru prsu a vaječníků.
Sweetův syndrom (akutní febrilní neutrofilní dermatóza)
Sweetův syndrom se projevuje teplotami, neutrofilií, artralgiemi, myalgiemi a malátností. Pro onemocnění je dále charakteristický výskyt zarudlých, bolestivých kožních lézí, které vznikají jako následek infiltrace neutrofily, přičemž tato infiltrace není typicky vázána na cévy. Kožní léze se obvykle vyskytují v obličeji, na krku, na dorzu rukou a na končetinách (obr. 7). Léze jsou palpačně bolestivé, ale nesvědivé. Sweetův syndrom se nejčastěji vyskytuje při myeloidní leukemii a myelodysplastickém syndromu, i když popsán byl i výskyt u solidních tumorů a lymfomů (37). Sweetův syndrom může být asociován i s panikulitidou, eventuálně erythema nodosum. V léčbě se používají glukokortikoidy, při jejich nedostatečném efektu pak imunosupresiva, u nepara-neoplastických forem pak eventuálně i biologická léčba (infliximab).

Komplexní regionální syndrom
Dříve byl nazýván algodystrofický syndrom, dnes jej spíše označujeme jako komplexní regionální syndrom (KRS) nebo syndrom reflexní sympatické dystrofie. Obvykle vzniká jako abnormální reakce na úraz, operaci či jiné bolestivé podněty z periferie, vzácně je asociovaný s karcinomem plic, prsu, ovarií, gastrointestinálního traktu nebo myeloidní leukemií.
Jako paraneoplastický syndrom je nejčastěji vyvolán Pancoastovým tumorem plic, který infiltruje ganglion stellatum nebo brachiální plexus, stejně tak může být asociován karcinomem plic s postižením axily.
Onemocnění prochází několika fázemi. Akutní fáze je dána sníženou činností sympatiku, kdy vzniká zvýšené prokrvení a následně zvýšená teplota, potivost a lesk kůže, provází jej i urychlený růst ochlupení a nehtů. Nápadný je místní edém, zarudnutí a snížený rozsah pohybu (obr. 8).Dystrofická fáze je naopak dána zvýšenou činností sympatiku: dochází při ní ke sníženému prokrvení a snížené teplotě kůže, zpomalení růstu ochlupení, k větší lomivosti nehtů a edému končetiny. Na rtg je typická skvrnitá poróza. V konečné fázi onemocnění vzniká atrofická fáze. Projevuje se prohloubením ireverzibilních tkáňových změn v postižených svalech, vazivu i kostech a nakonec vede k poruše konfigurace a postavení kloubů. Finálně může onemocnění dospět dokonce až ke vzniku osteonekrózy v postižené oblasti.

V léčbě KRS bylo navrženo celé spektrum léčebných postupů zaměřených zejména na potlačení bolesti a zlepšení hybnosti. Základem léčby je určení správné medikace, kdy se pomocí léků snažíme o potlačení bolesti, obnovení spánku, úpravu mikrocirkulace a o ovlivnění vegetativních změn. Vzhledem ke kostní resorpci bývají používány bisfosfonáty. Důležité jsou postupy fyzikální a rehabilitační léčby podle stadia a vývoje. V akutním stadiu je nutné chlazení oteklé části těla, vhodná je magnetoterapie a cvičení končetinou, a to ovšem pouze do bolesti. Cvičení nikdy nemá přesáhnout přes bolest, neboť ta může stav naopak zhoršit. Dle okolností jsou někdy vyžadovány speciální invazivní postupy ve spolupráci s anesteziologem či odborníkem na léčbu bolesti, kteří aplikují bloky nervů postižené končetiny, což vede ke snížení bolestivosti s možností další rehabilitační terapie. Velmi důležitá je spolupráce s psychologem či psychiatrem. Pokud se KRS dostane do třetího stadia, jsou změny nevratné a mohou vést až k invalidizaci pacienta (38).
Remitující séronegativní symetrická synovitida s plastickým edémem (RS3PE)
Remitující séronegativní symetrická synovitida s edémem je charakterizována symetrickou synovitidou s otokem, který postihuje hlavně ruce a zápěstí starších osob. V 50 % provází maligní onemocnění, především lymfomy, myelodysplastické syndromy, adenokarcinomy a jiné solidní tumory. Pokud onemocnění hůře odpovídá na léčbu glukokortikoidy, je nutné na paraneoplastickou etiologii vždy pomýšlet. V patogenezi vzniku RS3PE hraje klíčovou roli VEGF. Nadprodukce tohoto faktoru například nádorovými buňkami vede k hypervaskularizaci synoviální tkáně s následným vznikem artritidy, zvýšení vaskulární permeability přispívá ke vzniku otoku.
NSA nejsou zpravidla příliš účinná. V léčbě používáme nízkou dávku glukokortikoidů, délka léčby se udává průměrně 18 měsíců. Dále mohou být podávána antimalarika, nutnost podávat imunosupresiva je spíše výjimečná (39).
Stiff-man syndrom
Někdy také bývá nazýván Canale-Smith-stiff-person syndrom (SPS). Je to vzácné onemocnění projevující se progresivní svalovou rigiditou a spazmem, kdy postiženo je především axiální svalstvo. Výskyt SPS bývá asociován s výskytem jiných autoimunitních onemocnění, jako je diabetes mellitus I. typu, tyreoiditida, vitiligo nebo perniciózní anemie. U pacientů s autoimunitními chorobami je SPS často asociován s přítomností protilátek proti GAD (dekarboxyláza kyseliny glutamové). Paraneoplastický SPS byl popsán u pacientů s karcinomem prsu a u malobuněčného karcinomu plic. Pacienti zde nemívají pozitivitu protilátek proti GAD, naopak jsou u nich často přítomny protilátky proti amphiphysinu (především u starších žen s karcinomem prsu), kdy dominuje hlavně postižení cervikální oblasti. Příznaky bývají ovlivnitelné podáváním benzodiazepinů, případně glukokortikoidů. U pacientů obvykle dochází k dramatickému zlepšení po úspěšné léčbě primární malignity (15).
Multicentrická retikulohistiocytóza
Multicentrická retikulohistiocytóza (MRH) je vzácná multisystémová granulomatózní histiocytóza z non-Langerhansových buněk. Projevuje se vznikem kožních papul a uzlů asociovaných s těžkou artritidou, která může progredovat až do mutilujících forem s destrukcí kloubů. Kůže je infiltrována CD68-pozitivními histiocyty a mnohojadernými obrovskými buňkami s eozinofilní cytoplazmou. Tyto infiltrace tvoří načervenalé kožní papuly, charakteristický je také Köbnerův fenomén. Typické je shlukování uzlíků kolem nehtů, což připomíná šňůry perel (obr. 9). Infiltrace se kromě kůže mohou vyskytovat i ve šlachových pochvách, synoviálních tkáních a v kostech, méně často jsou infiltrována játra, slinné žlázy, ledviny, lymfatické uzliny, srdce a plíce. Typický je také výskyt xantelesmat kolem očí (40). Onemocnění zpravidla začíná ve středním věku, častěji jsou postiženy ženy. V asociaci s MRH byl také popsán pozitivní kožní tuberkulinový test, systémová vaskulitida a v 15–30 % případů malignita, především karcinom prsu, žaludku a hematologické malignity (41, 42). Onemocnění je zpravidla dosti rezistentní k léčbě glukokortikoidy i imunosupresivními nebo chorobu modifikujícími léky.

POEMS syndrom
POEMS syndrom (P = polyneuropatie, O = organomegalie, E = endokrinopthie, M = monoklonální gamapatie a S = skin = kožní změny) je poměrně vzácná klinická jednotka patřící do skupiny plazmatických dyskrazií (43). Pacienti také mívají často zvýšený extravaskulární objem tekutin, sklerotické kostní léze, trombocytózu a zvětšení uzlin, které bývá podmíněno benigní angiofolikulární hyperplazií s příměsí plazmocytů. Diagnóza bývá potvrzena nálezem zvýšené hladiny vaskulárního endoteliálního růstového faktoru (44) a často se překrývá se symptomy multicentrické Castelmanovy choroby.
Na rozdíl od mnohočetného myelomu se vyskytuje zejména u mladších jedinců a má poněkud méně agresivní průběh. Přesto je prognóza onemocnění nepříznivá, s udávaným mediánem celkového přežití 33–165 měsíců. Nemocní často umírají na projevy srdečního či renálního selhání, s refrakterními edémy, nebo na infekční komplikace (45). Léčba POEMS syndromu je dána především tím, že se jedná o plazmocelulární dyskrazii, a spadá tedy plně do kompetence hematoonkologa. Jistě je možné zmírnit přidružené příznaky symptomatickou léčbou, rozhodující je ale stále celková chemoterapie s radioterapií osteosklerotických lézí. U mladších jedinců je pak vhodné zařazení autologní transplantace kostní dřeně (46).
Antifosfolipidové protilátky
Vyskytují se častěji u pacientů jak s různými solidními nádory, tak u lymfoproliferativních onemocnění. Dosud není jednotný názor, zda přítomnost antifosfolipidových protilátek u maligních onemocnění zvyšuje riziko vzniku tromboembolické nemoci.
PARANEOPLASTICKÉ ENDOKRINNÍSYNDROMY
Paraneoplastické endokrinní syndromy vznikají obvykle jako následek produkce hormonů nebo různých peptidů nádorovými buňkami. Zvýšená tvorba těchto působků může vést ke vzniku různých metabolických odchylek. Úspěšná léčba primárního nádoru často zlepšuje sekundárně vzniklý endokrinní syndrom (47), nicméně tíže postižení v rámci tohoto syndromu nekoreluje přímo se stadiem nádoru nebo jeho prognózou (48). Přehled paraneoplastických endokrinních syndromů udává tabulka 3.

Syndrom nadměrné produkce antidiuretického hormonu (Schwartzův-Bartterův syndrom)
SIADH (syndrom nadměrné produkce antidiuretického hormonu) postihuje přibližně 1–2 % pacientů s karcinomem. Projevy syndromu vyplývají přímo z nadměrné produkce antidiuretického hormonu (ADH), který zvyšuje reabsopci vody nebo atriálního natriuretického peptidu, který má natriuretické a antidiuretické vlastnosti. Pacienti se SIADH typicky trpí hypoosmotickou euvolemickou hyponatremií, tedy nemají ortostatické problémy ani otoky, naopak mají typicky normální centrální žilní tlak. Hladiny urey i kyseliny močové jsou nízké. V kontrastu s hyponatremií při nedostatečném příjmu sodíku mají pacienti se SIADH zvýšenou hladinu sodíku v moči, která je pak hyperosmolární.
Symptomy mohou značně kolísat v závislosti na tíži a rychlosti vzniku hyponatremie. Mírné symptomy zahrnují bolesti hlavy, slabost a poruchy paměti. V těžších případech se objevuje letargie, zmatenost, poruchy dýchání a kóma (49, 50). V léčbě SIADH je účinná restrikce podávaných tekutin (< 1000 ml/den) a podpora adekvátního příjmu soli a proteinů. Z farmakologické léčby používáme demeklocyklin nebo antagonisty receptoru pro vasopresin. Základem je ale samozřejmě léčba primárního tumoru.
Hyperkalcemie
Hyperkalcemie asociovaná s malignitou se objevuje až u 10 % pacientů s pokročilým maligním onemocněním a obecně je pak spojována se špatnou prognózou. Nejčastěji se vyskytuje při dlaždicobuněčných karcinomech, při karcinomu ledvin, plic, u mnohočetného myelomu, u lymfomů včetně HTLV (lidský T-lymfotropní virus) asociovaného lymfomu, u karcinomu vaječníku, endometria atd. Hyperkalcemie vychází z nadměrné sekrece parathormonu nebo PTHrP (parathormon related peptid), který vede ke zvýšené kostní resorpci, snížené renální reabsorpci fosfátů a zvýšené retenci kalcia v distálním tubulu v ledvině. Další příčinou hyperkalcemie při malignitě mohou být osteolytické metastázy uvolňující různé cytokiny a dále zvýšená hladina 1,25-dihydroxyvitaminu D, který je produkován tumorózními buňkami (51).
Ke klinickým příznakům patří nauzea, zvracení, latergie, renální selhání a kóma. U mnohočetného myelomu dochází k hyperkalcemii zejména ve spojení s vysokou celkovou bílkovinou a hyperviskozitou. Při ložiscích mnohočetného myelomu v páteři, v dlouhých kostech nebo v lebce se onemocnění projevuje bolestmi v postižených oblastech.
Při léčbě paraneoplastické hyparkalcemie je samozřejmě základem léčba primárního nádoru. Dále podáváme normosalické tekutiny, které zvyšují glomerulární filtraci a inhibují renální reabsopci kalcia. Užíváme i kličková diuretika, která rovněž snižují reabsorpci kalcia v ledvinách. Také podání intravenózních bisfosfonátů (pamidronát nebo zoledronát) obvykle vede k rychlému poklesu hladiny vápníku v krvi. V krajním případě možné při léčbě užít i hemodialýzu (52, 53).
Hypoglykemie
S tumorem asociovaná hypoglykemie se vyskytuje poměrně raritně. Může vycházet z buněk Langerhansových ostrůvků nebo z nádorů vznikajících mimo pankreas, kdy syndrom je pak nazýván NICTH (Non-islet Cell Tumor Hypoglycemia) a projevuje se epizodickými nebo trvalými stavy hypoglykemie (54). Postihuje především starší pacienty s pokročilým karcinomem, nicméně v některých případech může hypoglykemie předcházet i dosud neobjevenému karcinomu.
NICTH syndrom je způsoben produkcí IGF-2 (insulin-like growth factor 2), který má za následek stimulaci inzulinových receptorů a zvýšenou utilizaci glukózy. Syndrom je provázen nízkou hladinou inzulinu i C-peptidu, nízkou hladinou IGF-l a normální nebo zvýšenou hladinou IGF-2. Léčba syndromu spočívá především v léčbě bazálního tumoru. Při léčbě dlouhotrvajících hypoglykemií užíváme glukokortikoidy, růstové hormony, diazoxid, oktreotid a glukagon (55).
Cuschingův syndrom
Přibližně 5–10 % případů Cuschingova syndromu je paraneoplastické etiologie (56). Padesát až 60 % z toho představuje syndrom vzniklý při neuroendokrinním tumoru plic (malobunečný karcinom a bronchiální karcinoid). Cushingův syndrom se často projeví ještě před stanovením diagnózy nádoru. Paraneoplastický Cushingův syndrom vychází ze sekrece ACTH (adrenokortikotropní hormon) nebo faktoru uvolňující kortikotropin. Klinicky se projevuje hypertenzí, hypokalemií, svalovou slabostí, generalizovanými otoky a přírůstkem na váze s tvorbou strií. Centripetální rozložení tuku, které je pro Cushingův syndrom typické, u paraneoplastického cushingoidního syndromu nebývá příliš vyjádřeno. Neschopnost odpovědět na supresivní dexamethasonový test pomáhá odlišit ektopickou produkci ACTH od produkce hypofýzou. Základem léčby je samozřejmě léčba primárního nádoru, kromě toho používáme látky, které přímo snižují produkci steroidů, např. ketokonazol, mitotan, metyrapon a aminoglutethimid (57).
PARANEOPLASTICKÉ NEUROLOGICKÉSYNDROMY
Paraneoplastické neurologické syndromy (PNS) vznikají jako důsledek zkřížené reakce mezi tumorovými buňkami a jednotlivými komponentami nervového systému. U pacientů dochází k produkci protilátek namířených proti tumoru, které jsou známé jako onkoneurální protilátky. Vzhledem k podobnosti antigenů tyto onkoneurální protilátky a asociované onkoneurální T-lymfocyty napadají jednotlivé komponenty nervového systému. Na rozdíl od paraneoplastických endokrinních syndromů, PNS se v 80 % objevují ještě před manifestací primárního nádoru a vzhledem k tomu, že onkoneurální protilátky mohou způsobit ireverzibilní postižení, úspěšná léčba nádorů nemusí vést ke zlepšení neurologických symptomů. Hlavní způsob léčby je tedy podávání imunosupresivní léčby, úspěch terapie je však značně variabilní. PNS se vyskytují velmi vzácně, postihují pouhé 1 % jedinců s malignitami. Nejčastěji se objevují při malobuněčném karcinomu plic, u pacientů s lymfomem, myelomem nebo neuroblastomem. Nádorové buňky produkující neuroendokrinní proteiny vycházejí z neuronálních buněk, například v teratomu, nebo z buněk imunoregulatorních orgánů (thymom) (58).
V závislosti na tom, která komponenta nervového systému je postižena, se PNS mohou manifestovat jako kognitivní či personální změny, ataxie, deficit hlavových nervů, svalová slabost nebo porucha čití. PNS mohou postihovat centrální nervový systém (limbická encefalitida, paraneoplastická cerebelární degenerace), neuromuskulární junkci (myasthenia gravis a Lambert-Eatonova myastenie) nebo periferní nervový systém (autonomní neuropatie a senzorická subakutní neuropatie). Na druhou stranu je nutné dodat, že u více než 70 % pacientů s limbickou encefalitidou nebo subakutní senzorickou neuropatií není asociace s žádnou malignitou. V diferenciální diagnóze musíme zvažovat infekci, toxické a metabolické vlivy, metastázy do mozku, onemocnění mening či kompresi míchy, případně nervových kořenů. Přehled PNS uvádí tabulka 4.

Při diagnostice PNS užíváme zobrazovací metody, sérologické vyšetření, elektroencefalografii, elektromyografii a vyšetření cerebrospinálního moku. Třicet procent všech pacientů s PNS nemají detekovatelné žádné protilátky v séru ani v moku. Na druhou stranu některé onkoneurální protilátky mohou být přítomny i u zdravých jedinců. Proto byla vytvořena nová kritéria pro diagnózu PNS: výskyt nádoru, definice klasických symptomů a detekce onkoneurálních protilátek. PNS pak bývají rozdělované jako „definitivní“ PNS a „možný“ PNS.
Onkoneurální protilátky klasifikujeme do tří různých kategorií. V prvním případě jsou to molekulárně velmi dobře charakterizované protilátky se silnou asociací s nádorem (anti-amphysin, anti-CV2, anti-Hu [ANNA-1], anti-Ma2, anti-rekoverin, anti-Ri [ANNA-2], anti-Yo), ve druhém případě jsou to protilátky s parciální asociací (ANNA-3, anti-mGluR1, anti-Tr, anti-Zic4, PCA-2) a ve třetím případě se protilátky vyskytují v obou případech, a to jako asociované i neasociované s nádorem (anti-acetylcholinR, anti nikotin AchR, ani-VGCC, anti-VGKC). Pro většinu PNS přesný mechanismus působení protilátek zůstává nejasný (59).
Jak již bylo zmíněno, imunosupresivní léky hrají klíčovou roli v léčbě PNS. Užívají se glukokortikoidy, azathioprin, cyklofosfamid, rituximab, intravenózní imunoglobuliny i plazmaferéza.
Při prognóze onemocněním PNS hraje roli řada faktorů. Na jednu stranu manifestace PNS může pomoci k diagnóze dosud okultního primárního nádoru, na druhou stranu sama symptomatologie PNS může vést až k úmrtí pacienta, neboť PNS může zapříčinit ireverzibilní změny v oblasti centrálního i periferního nervového systému.
PARANEOPLASTICKÉ SYNDROMY S PŘEVAŽUJÍCÍ KOŽNÍ MANIFESTACÍ
Mnoho z dále popsaných syndromů se objevuje běžně bez asociované malignity. Nicméně pokud se některý z nich nově objeví, měl by být pacient vyšetřen k vyloučení paraneoplastické etiologie, přičemž samozřejmě bereme v úvahu věk a rizikové faktory u pacienta. Léčba paraneoplastických kožních syndromů spočívá v léčbě základního maligního onemocnění, současně užíváme stejnou terapii, jaká se aplikuje u stejných kožně-kloubních projevů neparaneoplastické etiologie (60, 61). Klinické příznaky, laboratorní a ostatní diagnostické nálezy uvádí tabulka 5.
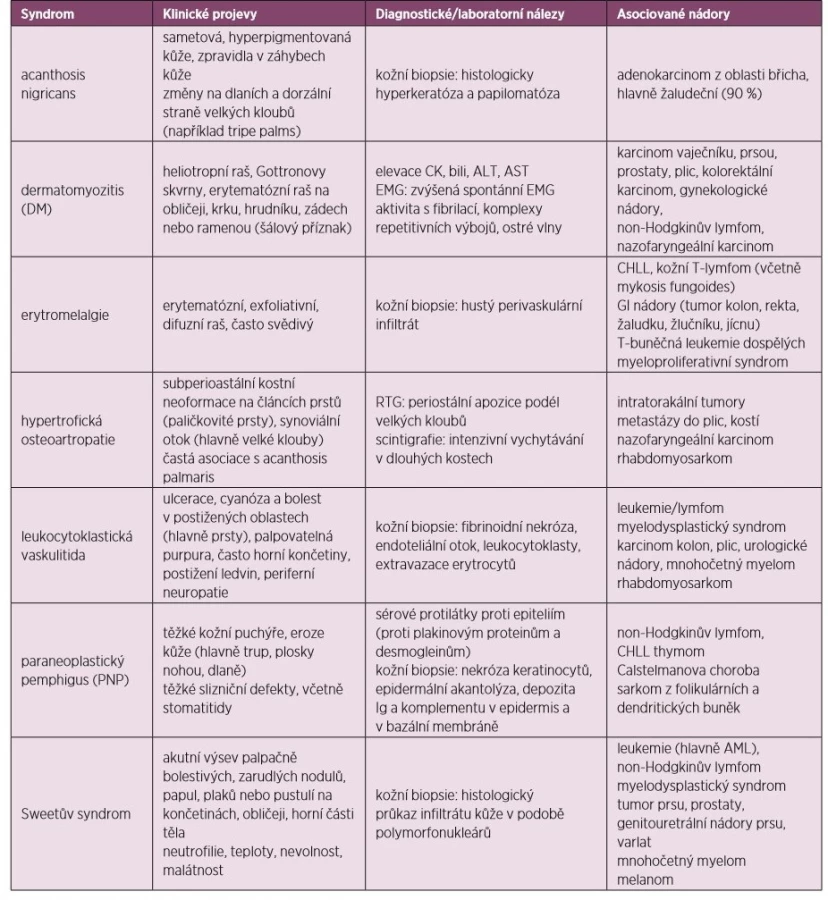
Acanthosis nigricans
Projevuje se ztluštěním hyperpigmentované kůže, která se predominantně vyskytuje v axile a na krku (obr. 10). Většina případů acanthosis nigricans není asociovaná s malignitou, vyskytuje se často u pacientů s inzulinovou rezistencí nebo u nemaligních endokrinních onemocnění. V případě paraneoplastické etiologie je nejčastěji asociovaná s nádorem žaludku. Naopak 90 % případů acanthosis nigricans na dlaních je paraneoplastické etiologie, kdy se kůže v záhybech dlaní podobá sliznici žaludku skotu (dršťky = tripe). Typický obraz „tripe palm“ ukazuje již zmiňovaný obrázek 3. Více než 50 % pacientů má současně slizniční postižení (62). Předpokládá se, že produkce transformujícího růstového faktoru a epidermálního růstového faktoru buňkami nádoru hrají úlohu při mechanismu vzniku tohoto onemocnění (63).

Paraneoplastický pemphigus
Paraneoplastický pemphigus (PNP) je většinou dramaticky probíhající onemocnění, kdy puchýřovité afekce napadají jak kůži, tak i slizniční membrány. V případě, že není pemphigus náležitě léčen, může mít za následek těžké komplikace (především sekundární infekce), dokonce i s následkem smrti. Paraneoplastický pemphigus je charakterizován bolestivými slizničními lézemi a polymorfním rašem, který se vyskytuje na těle, dlaních a chodidlech. Důvodem vzniku PNP je tvorba protilátek proti tumorózním antigenům, které mají zkříženou reaktivitu proti různým epidermálním proteinům. PNP je typicky asociován s lymfoproliferativním onemocněním vycházejícím z B-buněk. Léčba spočívá v podávání imunosupresivní terapie (glukokortikoidy, rituximab), která bývá kombinovaná s léčbou primárního tumoru (64).
PARANEOPLASTICKÉ HEMATOLOGICKÉSYNDROMY
Paraneoplastické hematologické syndromy bývají zpravidla asymptomatické. Syndromy se většinou objevují již při diagnostikovaném nádoru, zpravidla v asociaci s již pokročilým stavem onemocnění. Jen málokdy vyžadují specifickou léčbu, mohou se ovšem zlepšit s léčbou základního onemocnění (65, 66). Přehled paraneoplastických hematologických syndromů uvádí tabulka 6.

Eozinofilie
Paraneoplastická eozinofilie představuje subset sekundárních eozinofilií, které vznikají díky nádorové produkci eozinofilního růstového faktoru, interleukinu (IL)-3, IL-5 a GM-CSF (granulocyte-macrophage colony-stimulating factor). Oproti tomu primární eozinofilie se objevuje v hematologicko-onkologické praxi jako důsledek klonální proliferace při hematologickém onkologickém procesu. V diferenciální diagnóze sekundární eozinofilie zvažujeme alergické nebo parazitární onemocnění, eventuálně některé typy vaskulitid. Jako paraneoplastický syndrom provází především lymfomy a leukemie, pojí se ovšem i s gastrointestinálními, gynekologickými nebo plicními nádory. Paraneoplastická eozinofilie je zpravidla asymptomatická, v některých případech se může projevovat dušností a sípáním. Léky, které typicky užíváme pro léčbu klonální eozinofilie, při léčbě paraneoplastické eozinofilie nemají efekt. Podává se symptomatická léčba, jako inhalační nebo perorální glukokortikoidy, jinak nejdůležitější je samozřejmě terapie základního nádorového onemocnění.
Granulocytóza
Paraneoplastická granulocytóza se objevuje přibližně u 15 % pacientů se solidními nádory. Počet leukocytů se zpravidla pohybuje mezi 12–30 × 109/l, někdy přesahuje ovšem i počet 50 × 109/l. V etiologii granulocytózy hraje roli produkce hematopoetických růstových faktorů, může ovšem přispívat i infekce, léčba kortikoidy, zasažení kostní dřeně nádorem atd.
Paraneoplastická granulocytóza provází nejvíce nádory plic (hlavně velkobuněčný karcinom), gastrointestinálního traktu, mozku, prsou, ledvin a dále gynekologické tumory (67). Zpravidla nevyžaduje samostatnou léčbu. Na rozdíl od chorob při myeloproliferaci, kdy leukocytóza způsobuje hyperviskózní syndrom při hyperprodukci blastů, zralé a deformovatelné neutrofily u paraneoplastické granulocytózy nezpůsobují leukostázu, a stav tedy nevyžaduje obvykle provedení leukoferézy.
Čistá aplazie červené krevní řady
Čistá aplazie červené krevní řady je nejčastěji asociovaná s thymomem. Neefektivní eradikace autoreaktivnních T-lymfocytů v neoplastické tkáni thymu má za následek autoimunitní napadení prekurzorů červené krevní řady. Čistou aplazii červené krevní řady můžeme pozorovat i u jiných typů nádorů, především u lymfomů a leukemií, kdy zvýšení počtu velkých granulárních T-lymfocytů způsobuje dysfunkci erytropoézy. V diferenciální diagnóze stejný syndrom ovšem může vznikat při infekcích různými viry (HIV, herpes virus, parvovirus B19 a viry hepatitidy) (68).
Terapie spočívá v léčbě základního nádorového onemocnění a podávání imunosupresivní terapie. Užívají se glukokortikoidy, antithymocytový globulin, azathioprin, cyklosporin A, cyklofosfamid a dále monoklonální protilátky, jako je alemtuzumab nebo rituximab.
Trombocytóza
Odhaduje se, že přibližně 35 % pacientů s trombocytózou, která je obvykle definována jako počet trombocytů > 400 × 109/l, má současně maligní onemocnění (69). Diferenciálně diagnosticky trombocytózu způsobuje infekce, stav po splenektomii, akutní krevní ztráta nebo deficit železa. Paraneoplastická trombocytóza je obvykle způsobena zvýšenou sekrecí IL-6 tumorózními buňkami. Vysoká sérová hladina IL-6 odliší paraneoplastickou trombocytózu nebo jiné reaktivní trombocytózy od vysoké hladiny trombocytů, která je způsobena klonální proliferací, jako je esenciální trombocytemie, polycythaemia vera, myelodysplazie, akutní nebo chronická leukemie atd. Mutace JAK2 V61F je přítomna u 50 % případů esenciálních trombocytemií, nikoliv však u reaktivní trombocytózy. Vazomotorické symptomy a trombohemoragické komplikace, které obvykle provázejí až polovinu pacientů s esenciální trombocytózou, se u pacientů s paraneoplastickou trombocytózou nevyskytují.
ZÁVĚR
Tak, jako s růstem průměrného věku obyvatelstva celkově roste počet pacientů s nádorovým onemocněním a prodlužuje se i doba jejich přežití, zvyšuje se výskyt paraneoplastických syndromů. V některých případech může být průběh paraneoplastických syndromů asymptomatický, především pak u hematologickým paraneoplastických syndromů, u mnoha pacientů však může komplikující paraneoplastický syndrom (například neurologické syndromy, dermatomyozitida, vaskulitida atd.) značně zhoršovat kvalitu života i s možným rizikem zvýšení mortality. Schopnost rozeznat včas paraneoplastické syndromy s následným zahájením jejich léčby tak může v mnoha případech značně zlepšit celkovou prognózu pacienta i kvalitu jeho života. Paraneoplastický syndrom také často předchází diagnóze vlastního tumoru o řadu měsíců i let. Jeho včasné rozpoznání pak může přispět k dřívější diagnóze vlastního tumoru, což ve svém důsledku vede k včasnějšímu zahájení léčby malignity, a tak celkovému snížení morbidity i mortality postižených.
Některé obrázky mají horší kvalitu, ale pro ilustraci jednotlivých onemocnění jsme je i přesto ponechali.
Konflikt zájmů: žádný.
MUDr. Šárka Forejtová
Revmatologický ústav
Na Slupi 4, 128 50 Praha 2
e-mail: forejtova@revma.cz
Sources
1. Oppenheim H. Über Hirn Symptome bei Carcinamatóse ohne nachweisbare Veränderungen in Gehirn. Charité-Annalen (Berlin) 1888; (13): 335–344.
2. Kankeleit H. Über primare nichteirige Polymyositis. Dtsch Arch Klin Med 1916; 120 : 335–349.
3. Guichard A, Vignon G. La Polyradiculonéurite cancéreuse métastatique. J Med Lyon 1949; 30 : 197–207.
4. Baijens LW, Manni JJ. Paraneoplatic syndromes in patients with primary malignancies of the head and neck: four cases and review of the literature. Eur Arch Otorhinolaryngol 2006; 263 : 32–36.
5. Pines A, Kaplinsky N, Olchovsky D, et al. Rheumatoid arthritis-like syndrom: s presenting symptom of malignancy. Report of 3 cases and review of the literature. Eur J Inflamm 1984; 7 : 51–55.
6. Kisacik B, Onat AM, Kasifoglu T, et al. Diagnostic dilemma of paraneoplastic arthritis: case series. Int J Rheum Dis 2014; 17 : 640–645.
7. Morel J, Deschamps V, Toussirot E, et al. Charakteristics and survival of 26 patients with paraneoplatic arthritis. Ann Rheum Dis 2008; 67 : 244–247.
8. von Bamberger E. Veränderungen der Röhrenknochen bei Bronchiaktasie. Wien Klin Wochenschr 1889; 2 : 226–240.
9. Craig JW. Hypertrophic pulmonary osteoartropathy as the first symptom of pulmonary neoplasm. Br Med J 1937; 1 : 750–752.
10. Azar L, Khasnis A. Paraneoplastic rheumatology syndromes. Curr Opin Rheumatol 2013; 25 : 44–49.
11. Jayakar BA, Abison AG, Yao O. Treatment of hypertrophic osteoartropathy with zolendronic acid: case report and review of literature. Semin Arthritis Rheum 2011; 41 : 291–296.
12. Emmerson BT. The management of gout. N Eng J Med 1996; 334 : 445–451.
13. Dylewski J, Luterman L. Septic arthritis and Clostridium septicum: a clue to colon cancer. CMAJ 2010; 182 : 1446–1447.
14. Garcia-Porrua C, Gonzales-Gay MA, Monterroso JT, et al. Septic arthritis due to Streptococcus bovis as presenting sign of „silent“ colon carcinoma. Rheumatology 2000; 39 : 338–339.
15. Hashefi M. Rheumatologic manifestation of malignancy. Clin Geriatr Med 2017; 33 : 73–86.
16. Vencovský J. Revmatické projevy nádorových onemocnění. In: Pavelka K, Vencovský J, Horák P, Šenolt L, Mann H, Štěpán J. Revmatologie, druhé vydání. Praha: Maxdorf 2018; 744–747.
17. Buchbinder R, Forbes A, Hall S, et al. Incidence of malignant disease in biopsy-proven inflammatory myopathy. A population-based cohort study. Ann Intern Med 2001; 134 : 1087–1092.
18. Sparsa A, Liozon E, Herrmann F, et al. Routine vs extensive malignancy search for adult dermatomyositis and polymyositis: a study of 40 patients. Arch Dermatol 2002; 138 : 885–890.
19. Schoser B, Eymard B, Datt J, Mantegazza R. Lambert-Eaton myasthenic syndrome (LEMS): a rare autoimmune presynaptic disorder often associated with cancer. J Neurol 2017; 264 : 1854–1863.
20. Carpenter TO. Oncogenic osteomalacia: a complex dance of factores. N Eng J Med 2003; 348 : 1705–1708.
21. Fain O, Hamidou M, Cacoub P, et al. Vaskulitides associated with malignancies: analysis of sixty patients. Arthritis Rheum 2007; 57 : 1473–1480.
22. Solans-Laque R, Bosch-Gil JA, Perez-Bccanerga C, et al. Paraneoplastic vaskulitis in patients with solid tumors: report of 15 cases. J Rheumatol 2008; 35 : 294–304.
23. Park HJ, Ranganathan P. Neoplastic a paraneoplastic vaskulitis, vaskulopathy, and hyperkoagulability. Rheum Dis Clin North Am 2011; 37 : 593–606.
24. Hasler P, Kistler P, Gerber H. Vaskulitides in hairy cell leukemia. Semin Arthritis Rheum 1995; 25 : 134–142.
25. Han JH, Lee JB, Kim SJ, et al. Paraneoplastic erytromelalgia associated with breast carcinoma. Int J Dermatol 2012; 51 : 878–880.
26. Skeik N, Rooke TW, Davis MD, et al. Severe case and literature review of primary erythromelalgia: novel SCN9A gene mutation. Vasc Med 2012; 17 : 44–49.
27. Bermer C. Shloulder hand syndrome. A case of unusual etiology. Ann Phys Med 1967; 9 : 168–171.
28. Medsger TA, Dixon JA, Garwood VF. Palmar fascitis a polyarthritis associated with ovarian carcinoma. Ann Intern Med 1982; 96 : 424–431.
29. Lakhanpal S, Ginsburg WW, Michet CJ, et al. Eosinophlic fascitis: clinical spectrum and therapeutic response in 52 cases. Semin Arthritis Rheum 1988; 17 : 221–231.
30. Lebeaux D, Francès C, Barete S, et al. Eosinophilic fascitis (Shulman disease): new insights into the therapeutic management from a series of 34 patients. Rheumatology (Oxford) 2012; 51 : 557–561.
31. Narvaez J, Bianchi MM, Santo P, et al. Pancreatitis, panniculitis, and polyarthritis. Semin Arthritis Rheum 2010; 39 : 417–423.
32. Radin DR, Colleti PM, Forrester DM, Tang WW. Pancreatic acinar cell carcinoma with subcutaneous and intraosseous fat necrosis. Radiology 1986; 158 : 67–68.
33. Tannenbaum H, Anderson LG, Schur PH. Association of polyarthritis, subcutaneous nodules, and pancreatic disease. J Reumatol 1975; 2 : 15–20.
34. Racanelli V, Prete M, Minoia C, et al. Rheumatic disorders as paraneoplastic syndromes. Autoimmun Rev 2008; 7 : 352–358.
35. Pontifex EK, Hill CL, Roberts-Thomson P. Risk factors for lung cancer in patients with scleroderma: a nested case-control study. Ann Rheum Dis 2015; 66 : 551–553.
36. Evans KG, Heymann WR. Paraneoplastic subacute cutaneous lupus erythematosus: an under–recognized entity. Cutis 2013; 91 : 25–29.
37. Cohen PR, Kurzrock R. Sweet’s syndrome revisited, a review of disease concepts. Int J Dermatol 2003; 42 : 761–778.
38. Cossins, L, Okell RW, Cameron H, et al. Treatment of complex regional pain syndrome in adults: a systematic review of randomized controlled trials published from June 2000 to February 2012. Eur J Pain 2013; 17 : 158–73.
39. Li H, Altman RD, Yao Q. RS3PE: Clinical and Research Development. Curr Rheumatol Rep 2015; 17(8): 49.
40. Toz B, Büyükbabani N, Inanc M. Multicentric reticulohistiocytosis: Rheumatology perspective. Best Pract Res Clin Rheumatol 2016; 30 : 250–260.
41. El-Haddad B, Hammoud D, Shaver T, Shahouri S. Malignancy associated multicentric histiocytosis. Rhematol Int 2011; 31 : 1235–1238.
42. Snow JL, Muller SA. Malignancy-associated multicentric reticulohistiocytosis: a clinical, histological and imunophenotypic study. Br J Deramtol 1995; 133 : 71–76.
43. Dispenzieri A. Diagnosis and treatment of POEMS syndrome. In: Rajkumar SV, Kyle RA. Treatment of multiple myeloma and related disorders. New York, NY, USA: Cambridge University Press 2009; 182–195.
44. Dispenzieri A. POEMS syndrome. Blood Rev 2007; 21 : 285–299.
45. Dispenzieri A, Kyle RA, Lacy MQ, et al. POEMS syndrome: definitions and long–term outcome. Blood 2003; 101 : 2496–2506.
46. Kuwabara S, Misawa S, Kanai K, et al. Autologous peripheral blood stem cell transplantation for POEMS syndrome. Neurology 2006; 66 : 105–107.
47. Pelosof LC, Gerber DE. Paraneoplastic syndromes: an approach to diagnosis and treatment. Mayo Clin Proc 2010; 85 : 838–854.
48. Spinazze S, Schrijvers D. Metabolic emergencies. Crit Rev Oncol Hematol 2006; 58 : 79–89.
49. Raftopulous H. Diagnosis and management of hyponatremia in cancer patients. Support Care Cancer 2007; 15 : 1341–1347.
50. Ellison DH, Berl T. The syndrome of inappropriate antidiuresis. N Engl J Med 2005; 356 : 2064–2072.
51. Wysolmerski JJ. Parathyroid hormone-related protein: an update. J Clin Endocrinol Metab 2012; 97 : 2947–2956.
52. Lumachi F, Brunello A, Roma A, Basso U. Medical treatment of malignancy-associated hyperkalcemia. Curr Med Chem 2008; 15 : 415–421.
53. Stewart AF. Hypercalcemia associated with cancer. N Eng J Med 2005; 352 : 373–379.
54. Nayar MK, Lombard MG, Furlong NJ, et al. Diagnosis a management of nonislet cell tumor hypoglycemia: case series and review of literature. Endocrinologist 2006; 16 : 227–230.
55. Hoff AO, Vassilopoulou-Sellin R. The role of glucagon administration in the diagnosis and treatment of patients with tumor hypoglycemia. Cancer 1998; 82 : 1585–1592.
56. Nimalasena S, Freeman A, Harland S. Paraneoplastic Cushing's syndrom in prostate cancer: a difficult management problem. BJU Int 2008; 101 : 424–427.
57. Nieman LK. Medical therapy of Cushing’s disease. Pituitary 2002; 5 : 77–82.
58. Didelot A, Honnorat J. Update on paraneoplastic neurological syndromes. Curr Opin Oncol 2009; 21 : 566–592.
59. Shams’ili S, Grefkens J, de Leeuw B, et al. Paraneoplastic cerebellar degeneration associated with antineuronal antibodies: analysis of 52 patients. Brain 2003; 126 : 1409–1418.
60. Ali N, Abbasi AN, Karsan F, et al. A case of finger clubbing associated with nasopharyngel carcinoma in a young girl, and review of pathophysiology. J Pak Med Assoc 2009; 59 : 253–254.
61. Garganese MC, De Sio L, Serra A, et al. Rhabdomyosarcoma associated hypertrophic osteoartropathy in a child: detection by bone scintigraphy. Clin Nucl Med 2009; 34 : 155–157.
62. Mekhail TM, Markman M. Acanthosis nigricans with endometrial carcinoma: case report and review of the literature. Gynecol Oncol 2002; 84 : 332–334.
63. Anderson SH, Hudson-Peacock M, Muller AF. Malignant acanthosis nigricans: potential role of chemotherapy. Br J Dermatol 1999; 141 : 714–716.
64. Thiers BH, Sahn RE, Callen JP. Cutaneous manifestation of internal malignancy. CA Cancer J Clin 2009; 59 : 73–98.
65. Jameson JL, Johnson BE. Paraneoplastic syndromes: endokrinologic/hematologic. In: Fauci AS, Braunwald E, Kasper DL, Huaser SL, Longo DL, Jameson JL, Loscalzo J, etc. Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw Hill Medical 2008; 617–622.
66. Kessler CM. The link between cancer and venous tromboembolism: a review. Am J Clin Oncol 2009; 32: S3–S7.
67. Ganger JM, Kontoyiannis DP. Etiology and outcome of extreme leukocytosis in 758 nonhematologic cancer patients: a retospective, single – institution study. Cancer 2009; 115 : 3919–3923.
68. Sawada K, Hirokawa M,Fujishima N. Diagnosis and managment of acquired pure red cell aplasia. Hematol Oncol Clin North Am 2009; 23 : 249–259.
69. Buss DH, Cashell AW, O’Connor ML, Richards F, Case LD. Occurence, etiology, and clinical signifikance of extreme thrombocytosis: a study of 280 cases. Am J Med 1994; 96 : 247–253.
70. Imboden JB. Rheumatic manifestation of malignany. In: Imboden JB, Hellmann DB, Stone JH. Current Diagnosis & Treatment: Rheumatology, 3rd edition. The McGraww-Hill Companies 2013; 419–422.
Labels
Dermatology & STDs Paediatric rheumatology RheumatologyArticle was published in
Czech Rheumatology

2019 Issue 4
Most read in this issue
- Paraneoplastic syndrom
- Recommendations of the Czech Society for Rheumatology for the treatment of gout
- Impact of inflammatory granulomas in rheumatic diseases
- The importance of achieving remission in patients with rheumatoid arthritis