
Juvenilní dermatomyozitida – klinický případ
Juvenile Dermatomyositis. Case Report
Juvenile dermatomyositis is the most common idiopathic inflammatory myopathy in children. It primarily affects the skin and the muscles. This article describes a case of 14-year-old boy with predominantly skin symptoms. The authors present up-to-date diagnostic criteria and treatment possibilities.
Keywords:
therapy – juvenile dermatomyositis
Authors:
K. Honzíková; H. Bučková
Authors‘ workplace:
Dětské kožní oddělení PEK FN Brno a LF MU v Brně, primář MUDr. Hana Bučková, Ph. D.
Published in:
Čes-slov Derm, 94, 2019, No. 3, p. 108-112
Category:
Case Reports
Overview
Juvenilní dermatomyozitida je nejčastěji se vyskytující idiopatická zánětlivá myopatie u dětí. Projevuje se kožními příznaky a postižením svalů. Popsán je případ 14letého chlapce, u kterého dominovaly kožní projevy. Autoři uvádí současná diagnostická kritéria a možnosti léčby.
Klíčová slova:
juvenilní dermatomyozitida – terapie
ÚVOD
Juvenilní dermatomyozitida (JDM) je nejčastější zánětlivá myopatie u dětí. Projevuje se symetrickou svalovou slabostí a typickým kožním postižením. Kožní projevy JDM jsou pestré. Onemocnění se nejčastěji vyskytuje u dětí mezi 2. a 5. rokem života a mezi 12. a 13. rokem života. U dospělých se dermatomyozitida vyskytuje mezi 45. až 65. rokem života [6, 7], až 20 % případů v dospělosti je spojeno s výskytem malignity [6]. Spouštěcím faktorem JDM jsou nejčastěji infekce. V diagnostice JDM jsou klíčové kožní projevy, mezi další kritéria patří svalová slabost, elevace svalových enzymů, elektromyografické změny a histologický nález. Dětský dermatolog může přispět k časné diagnóze JDM, neboť kožní projevy tohoto onemocnění jsou typické.
POPIS PŘÍPADU
Čtrnáctiletý chlapec se dostavil na kožní ambulanci s čtyři měsíce trvajícími intermitentními otoky a erytémem v obličeji. Dominoval výrazný erytém celého obličeje, otok horních víček a horního rtu (obr. 1), shluky erytémových makulopapulí nad loketními (obr. 2) a kolenními klouby. Prsty a hřbety rukou byly oteklé, s erytémem a zduřením nehtových valů. Pacient se s ničím neléčil, udával bolesti břicha, které ustoupily po Espumisanu cps. (simeticon). Alergie neudával, léky trvale neužíval. Rodinná anamnéza byla rovněž bez pozoruhodností. Projevy u pacienta byly léčeny praktickým lékařem pro děti a dorost, zprvu krátkodobě celkovými kortikosteroidy (Prednison tbl.), pak antihistaminiky (Zodac tbl.) bez efektu.
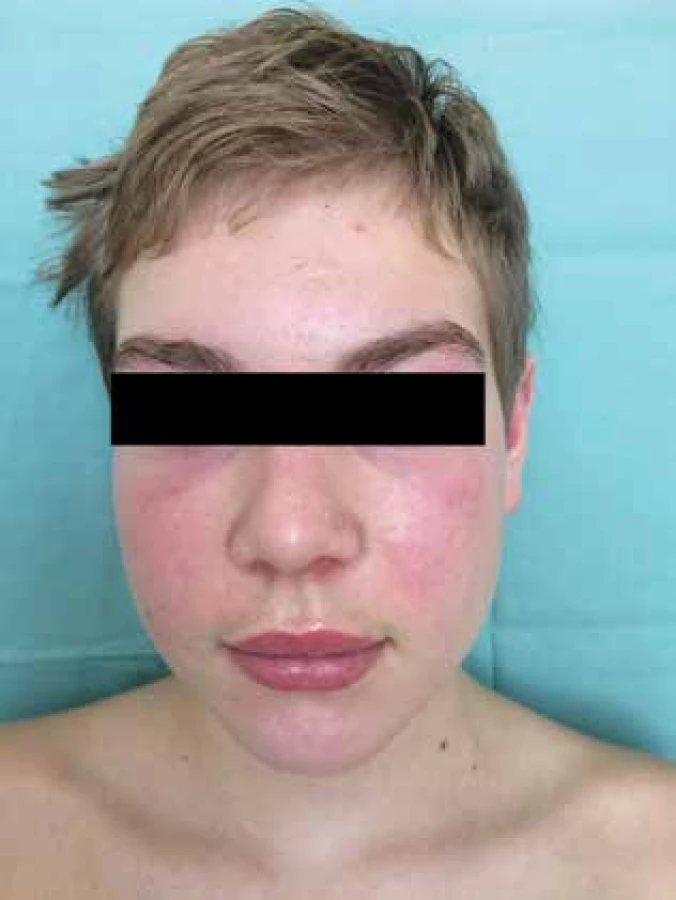
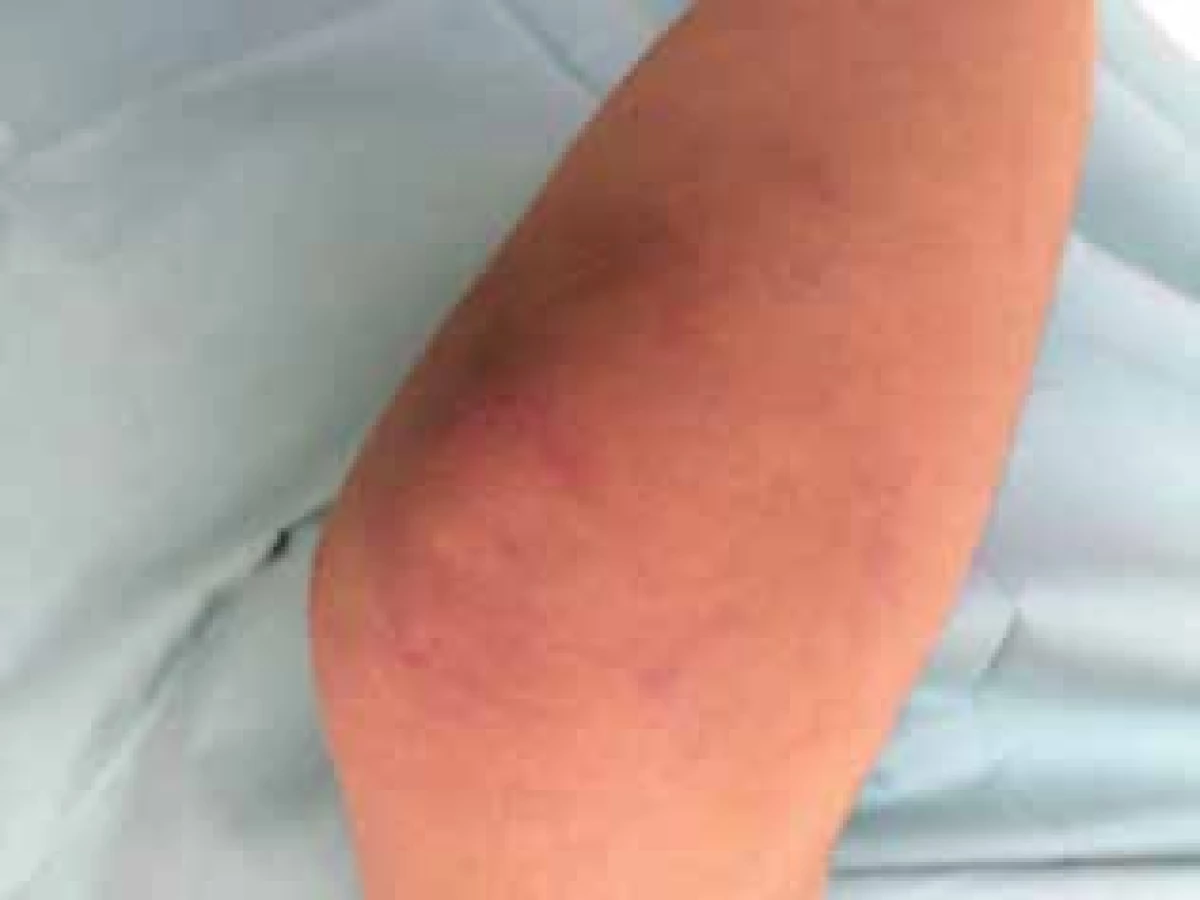
Vysloveno podezření na JDM, pacient byl přijat na lůžkovou část Dětského kožního oddělení FN Brno (DKO) k celkovému došetření a nastavení terapie. Z provedených vyšetření: krevní obraz v normě, v biochemickém vyšetření zachycena elevace svalových enzymů – kreatinkináza 2,51 µkat/l (norma do 2,27), laktátdehydrogenáza 5,95 µkat/l (norma do 5,0), myoglobin 84,4 µg/l (norma do 72), troponin T 82 ng/l (norma do 14), jinak základní biochemické vyšetření bez patologie, v moči jednorázově zachycena bílkovina, ve screeningu autoprotilátek pozitivní protilátky IgG anti Ku, které patří mezi protilátky asociované s myozitidou.
Vzhledem k elevaci troponinu T bylo doplněno kardiologické vyšetření, které bylo negativní. Na RTG plic byla zachycena zmnožená plicní kresba oboustranně. Ultrazvuk břicha byl s normálním nálezem, na ultrazvuku střev byla zachycena folikulární hyperplazie lymfatické tkáně. Kalprotektin ze stolice byl negativní. Oční vyšetření prokázalo exkavaci zrakového nervu obou očí.
Utrazvukové vyšetření svalů bylo v normě, při magnetické rezonanci svalů stehen a hýždí byly zachyceny některé svalové skupiny s patologickým signálem odpovídající myozitidě. Funkční vyšetření plic potvrdilo lehkou restrikční poruchu, HRCT plic v mezích normy, popsána minimální zvýšená denzita podkožní tukové tkáně hrudníku v rámci základní diagnózy. Na revmatologii testy svalové síly Kendall a CMAS prokázaly svalovou slabost. Kapilaroskopie nehtových valů ukázala lehké větvení kapilárních kliček s avaskulárními zónami a drobné hemoragie (obr. 3). Cévním vyšetřením byl potvrzen Raynaudův fenomén.
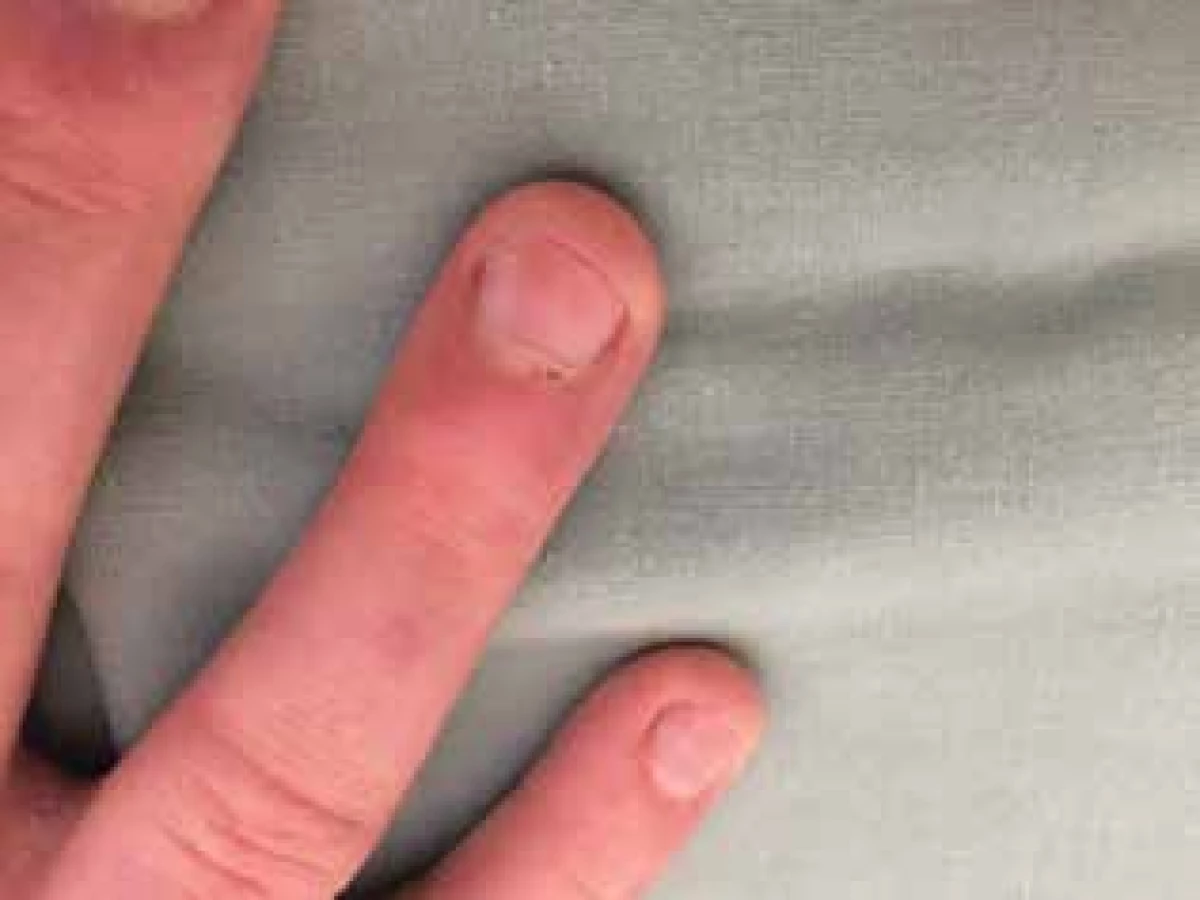
Po domluvě s revmatology bylo před zahájením terapie vysokými dávkami kortikosteroidů provedeno denzitometrické vyšetření s normálním nálezem. Terapie byla zahájena pulzy kortikosteroidů (Solu-Medrol v dávce 1000 mg i. v., celkem 3krát), následně byl pacient převeden na perorální formu (Medrol tbl.) v dávce 48 mg/den a zároveň byla zahájena celková terapie metotrexátem v iniciální dávce 15 mg, dále 20 mg/týden. V podpůrné terapii bylo podáváno Acidum folicum, Helicid, Caltrate, Vigantol, KCl, probiotika. Z lokální terapie pacient používal glycerinový krém a fotoprotektivní krémy. Dávka kortikoidů byla po měsíci terapie snížena na 40 mg/den (Medrol tbl.). Došlo k přechodné elevaci ALT (3,63) i AST (1,13), proto byla přechodně snížena dávka metotrexátu na 10 mg/ týden. Jaterní testy se v průběhu dvou měsíců normalizovaly a dávka metotrexátu byla opět navýšena na 20 mg/týden. Po šesti týdnech byla do medikace přidána celková terapie Plaquenilem v dávce 200 mg denně, dávka Medrolu byla postupně snižována na 16 mg/den. Po dvou měsících terapie se pacientovi výrazně zlepšila svalová síla, ustoupily otoky v obličeji, zůstal drobný erytém v oblasti loktů a kolen. Pacient je nadále pravidelně sledován v dermatologické a revmatologické ambulanci.
DISKUSE
JDM je nejčastější idiopatická zánětlivá myopatie u dětí [3]. Mendez et al. udávají incidenci 3,2/1mil. pro děti ve věku 2–17let v USA [4]. Nejčastější výskyt u dětí je mezi 2. a 5. rokem života a mezi 12. a 13. rokem života [6, 7]. Etiopatogeneticky se nejpravděpodobněji jedná o reakci spuštěnou zevními vlivy (nejčastějším spouštěčem jsou infekce) u geneticky predisponovaného jedince. Ve více než 50 % případů předcházela projevům JDM infekce respiračního traktu, ve 30 % to byla infekce gastrointestinálního traktu [6]. Jako agens se nejčastěji udávají Coxsackie viry, parvoviry, EBV, borrelie [5,7]. V dospělosti je až 20 % případů spojeno s výskytem malignity [6]. Genetická predispozice je vázána na oblast HLA molekul. Přítomnost molekul HLA-B*08, DRB1*0301, DQA1*0501 je asociována s JDM [7].
Kritéria ke stanovení diagnózy juvenilní dermatomyozitidy uvedli autoři Bohan a Peter v roce 1975. Zahrnují typický exantém, proximální svalovou slabost, zvýšené svalové enzymy, elektromyografické změny a histopatologické změny ve smyslu zánětlivé myopatie (tab. 1). V současnosti nahrazuje nebo doplňuje elektromyogram ultrazvuk a magnetická rezonance. Zvažuje se začlenit do kritérií i další parametry (nález při kapilaroskopii, přítomnost kalcinóz) [1]. Přítomnost exantému a 3 dalších příznaků je podmínkou ke stanovení diagnózy JDM [7, 10].

Pro stanovení diagnózy je nutná přítomnost exantému a 3 dalších kritérií.
Počátek onemocnění se většinou manifestuje slabostí, nevolností, horečkou, únavou, ztrátou hmotnosti. Typické kožní změny zahrnují heliotropní erytém – červenofialový otok víček (podle barvy rostliny Heliotropium), symetrické změny nad klouby (typicky nad lokty, koleny, klouby rukou) – erytém, ploché, lividní papuly označované jako Gottronovy papuly. U 35–90 % pacientů nacházíme změny periunguálně, a to keříčkovitě se větvící kapiláry, avaskulární zóny, drobné hemoragie. Obdobné cévní změny můžeme zaznamenat i na gingivách, kde může dojít i k ulceracím. Erytém v celém obličeji, na krku a v dekoltu se označuje jako šálový příznak. Asi v 50 % případů bývá udávána fotosenzitivita. Kalcifikace se objevují u 12–47 % dětí s JDM. Riziko rozvoje kalcifikací se zvyšuje s věkem pacienta a dobou trvání JDM do zahájení terapie. Častěji se kalcifikace vyskytují u mužů [7]. Kalcifikace se nejčastěji objevují na loktech, kolenou, prstech, hýždích. V chronickém stadiu nemoci vidíme na kůži atrofii, poikiloderma, dyspigmentace, lipodystrofii. Ta je provázena hypertriglyceridémií, inzulinovou rezistencí, hypertenzí a steatózou jater, která zvyšuje riziko rozvoje metabolického syndromu [6, 7]. Viscerální vaskulopatie je spojována s horší prognózou. Projevuje se bolestmi břicha, melénou, hematemézou, pankreatitidou, střevním infarktem [7]. Klinické projevy jako obtížná chůze do schodů či potíže při česání jsou způsobeny postižením proximálních svalových plexů. U dětí je často pozitivní Gowersovo znamení – neschopnost postavit se z kleku bez pomoci horních končetin, což jsme u našeho pacienta nezaznamenali. Při postižení faryngeálních a hypofaryngeálních svalů dochází k polykacím obtížím, u dětí hrozí riziko aspirace. Svalová slabost se hodnotí pomocí testů svalové síly Childhood myositis assessment scale (CMAS) a Kendallův manuální svalový test. V laboratorním vyšetření se opíráme o hodnoty svalových enzymů – AST, CK, LD, aldoláza. Hodnoty kreatinkinázy mohou nabývat až patnáctinásobek normálních hodnot [7]. U našeho pacienta byly hodnoty svalových enzymů zvýšeny jen mírně, nedosahovaly násobků normy. Zvláštní formou JDM je amyopatická dermatomyozitida, u které svalové postižení chybí, označuje se proto jako dermatomyositis sine myositis [10]. Elektromyografické vyšetření (EMG) pomůže odlišit případnou neuropatii od myopatie [3]. U pacientů, kde je diagnóza nejistá, je indikováno provedení biopsie svalu, který by měl vykazovat patologii, ale zároveň by neměl být atrofický. Určit reprezentativní vzorek proto může pomoci magnetická rezonance. Histologicky nacházíme perivaskulárně a v perimyziu zánětlivý infiltrát hlavně z lymfocytů a makrofágů a perifascikulární atrofii a nekrózu [7]. Asymptomatické plicní postižení se objevuje až u 50 % dětí a je výsledkem snížené kapacity a difuze, proto má být plicní vyšetření provedeno u všech pacientů s diagnózou JDM. Postižení kardiovaskulárního systému je v dětském věku vzácné. Nejčastěji se manifestuje jako šelest či kardiomegalie [3, 6, 7].
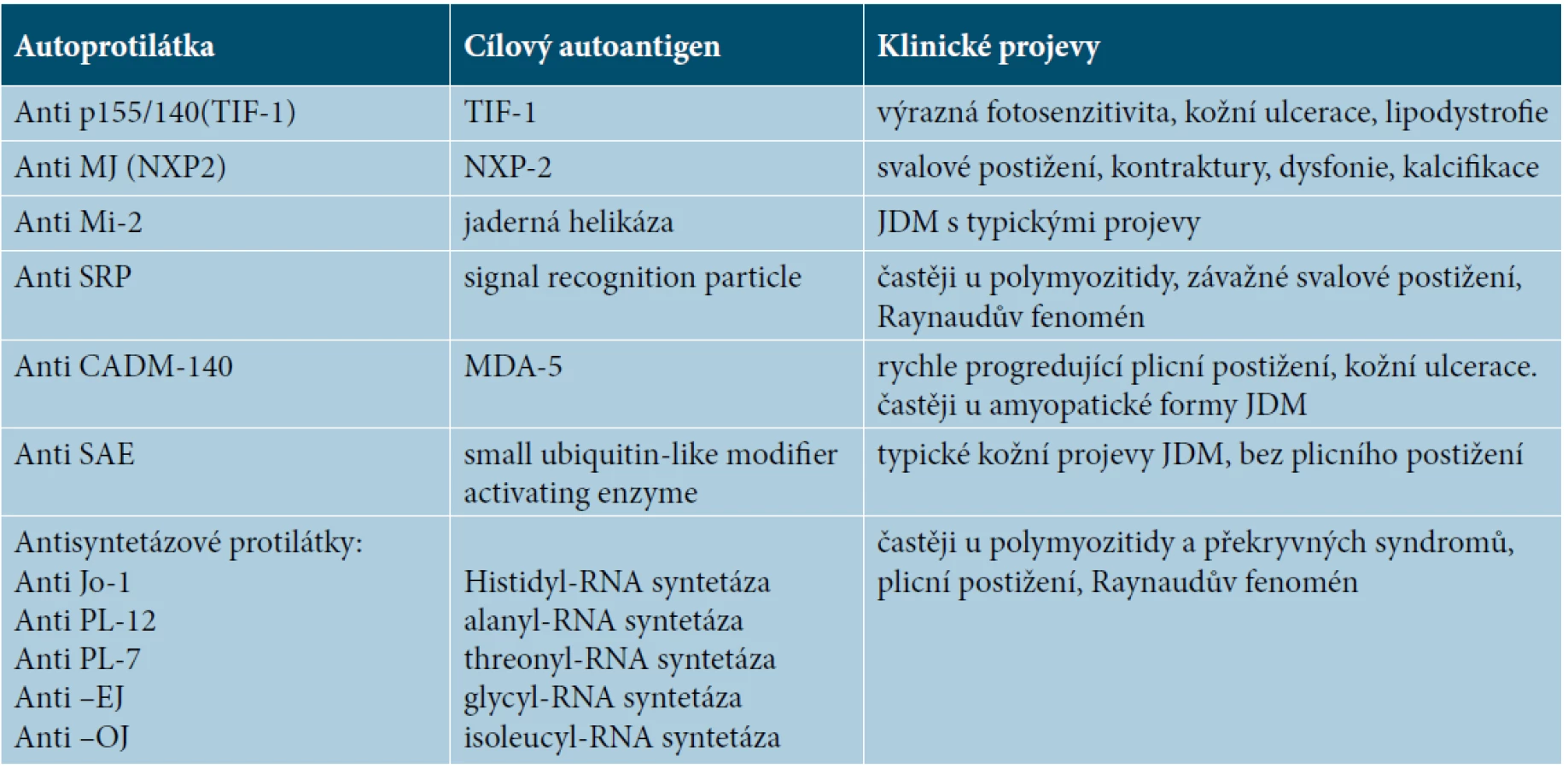
Antinukleární protilátky jsou pozitivní u 41–72 % pacientů s JMD. Mezi nejčastěji prokázané autoprotilátky specifické pro myozitidu patří anti TIF 1-γ(p155/140), anti NXP2 (p140/MJ) a anti MDA5. Přítomnost protilátky anti TIF1-γ bývá spojována s horším kožním postižením a častěji s chronickým průběhem onemocnění. Je nalézána u 23–30 % dětských pacientů [7]. Anti NXP2 protilátka bývá asociována s vyšším výskytem svalových křečí, kalcifikací a krvácením z gastrointestinálního traktu. Vyšší riziko ulcerací je spojeno s přítomností protilátky anti MDA5 [3, 7]. Antisyntetázové protilátky (nejčastěji anti Jo) jsou nalézány zhruba u 5 % dětských pacientů, tedy významně méně často než u dospělých, kde je nalézáme u 20 % pacientů s myozitidou. Mezi další autoprotilátky specifické pro myozitidu patří anti Mi2, anti SAE, anti SRP. Anti CADM 140 bývá nacházena u amyopatické dermatomyozitidy. Autoprotilátka antiKu nalezená u našeho pacienta patří mezi protilátky asociované s myozitidou, kromě JDM bývá pozitivní u překryvných syndromů. Mezi další autoprotilátky asociované s myozitidou patří anti-U1-RNP, anti Ro, anti PM-Scl [7] (tab. 2, 3).

Náš pacient splnil kritéria ke stanovení diagnózy JDM, měl typický kožní nález – heliotropní exantém, Gottronovy papuly, změny na nehtových valech průkazné při kapilaroskopii, svalovou slabost a postižení svalů podle magnetické rezonance, elevaci svalových enzymů, pozitivní protilátku anti Ku. Svalová biopsie nebyla vzhledem k zjištěným diagnostickým nálezům u našeho pacienta provedena.
Zavedení terapie kortikosteroidy výrazně zlepšilo prognózu pacientů s JDM. V současné době představují kortikosteroidy základ 1. terapeutické linie JDM. Schémata užití kortikosteroidů se různí, preferován je třídenní pulz methylprednisolonu 15–30 mg/kg/dávka, následovaný prednisonem v dávce 1–2 mg/kg/den během prvního měsíce léčby, poté se dávka snižuje v závislosti na klinické a laboratorní odpovědi (pokles hodnot svalových enzymů). Obtížné je určit čas, kdy můžeme léčbu kortikosteroidy bezpečně ukončit, aniž by došlo k exacerbaci onemocnění. Svalové enzymy by měly signifikantně poklesnout v průběhu prvních dvou týdnů terapie, zlepšení svalové síly můžeme pozorovat až po 1–2 měsících. Za exacerbaci onemocnění může být ale také zaměněna steroidní myopatie, která nejčastěji zahrnuje slabost a atrofii v oblasti pánevního pletence. Steroidní myopatie nevykazuje změny na EMG a není spojena s elevací svalových enzymů. Hydroxychloroquine v komedikaci se steroidy je doporučován k léčbě kožních projevů JDM [7]. Evropská studie ukázala, že prednison v kombinaci s metotrexátem nebo cyklosporinem vede k dřívější remisi v porovnání s prednisonem v monoterapii [8]. Kombinace KS a MTX se jeví jako bezpečnější z hlediska možných nežádoucích účinků. U našeho pacienta byla zvolena kombinace KS s MTX v dávce 20 mg/týden. K ústupu projevů onemocnění došlo za dva měsíce.
Mezi další terapeutické možnosti závažných a refrakterních forem onemocnění patří cyklosporin, obvykle v dávce 2,5–7,5 mg/kg/den nebo mykofenolát mofetil, azathioprin, cyklofosfamid, anti TNFα, imunoglobuliny [7]. Dobrý terapeutický efekt vykazuje i rituximab [2]. Důležitou součástí léčebného režimu je důsledná fotoprotekce. Celkovou terapii lze kombinovat s lokální léčbou kožních projevů kortikosteroidy nebo topickými imunomodulátory.
ZÁVĚR
Juvenilní dermatomyozitida je nejčastější zánětlivá myopatie u dětí. Průběh JDM může být monofázický, s dosažením remise do 2 let od stanovení diagnózy, polyfázický s rekurencí, či chronický s aktivním onemocněním trvajícím více než 2 roky, jak je tomu až u 60 % pacientů [7]. Perzistující kožní projevy, zvláště Gottronovy papuly a abnormality kapilár nehtového lůžka, stejně jako přítomnost gastrointestinální vaskulopatie, kalcifikací nebo ulcerací jsou spojeny s horší prognózou [9]. Z prognostického hlediska je důležitá včasná diagnóza a časné zahájení léčby. Kožní projevy u dermatomyozitidy jsou typické, proto dermatolog může přispět k včasné diagnóze onemocnění. Časná a adekvátní terapie zabrání možným komplikacím onemocnění. Péče o pacienty s juvenilní dermatomyozitidou vyžaduje mezioborovou spolupráci.
Prohlášení o konfliktu zájmů
Autorka v souvislosti s tématem práce nespolupracovala v posledních 12 měsících s žádnou farmaceutickou firmou.
Do redakce došlo dne 21. 12. 2018.
Adresa pro korespondenci:
MUDr. Klára Honzíková, Ph.D.
Dětské kožní oddělení
Pediatrická klinika FN Brno
Černopolní 9
613 00 Brno
e-mail: Honzikova.Klara@fnbrno.cz
Sources
1. ROWN, V. E., PILKINGTON, C. A., FELDMAN, B. M., DAVIDSON, J. E. An international consensus survey of the diagnostic criteria for juvenile dermatomyositis. Rheumatology, 2006, 45(8), p. 990–993.
2. COOPER, M. A. WILLINGHAM, D. L., BROWN, D. E. et al. Rituximab for the treatment of juvenile dermatomyositis: A report of four pediatric patient. Arthritis & Rheumatism, 2007, 56(9), p. 3107–3111.
3. ENDERS, F. B, BADER-MEUNIER, B., BAILDAM, E. et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis published online first: August 11, 2016 doi: 10.1136/annrheumdis-2016-209247.
4. MENDEZ, E. P., LIPTON, R., RAMSAY-GOLDMAN, R., ROETTCHER, P. et al. US incidence of juvenile dermatomyositis, 1995–1998: Results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Registry. Arthritis care & research, 2003, 49(3), p. 300–305.
5. PACHMAN, L. M., LIPTON, R., RAMSEY-GOLDMAN, R., SHAMIYEH, E. et. al. History of infection before the onset of juvenile dermatomyositis: Results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Research Registry. Arthritis care & research, 2005, 53(2), p. 166–172.
6. PALLER, A. S., MANCINI, A. J. Hurwitz Clinical Pediatric Dermatology. 4th edition, 2011, p. 509–514.
7. PETTY, R. E., LAXER, R. M., LINDSLEY, C. B., WEDDERBURN L. Textbook of Pediatric Rheumatology, 7th edition, 2016, p. 350–382.
8. RUPERTO, N., PISTORIO, A., OLIVEIRA, S. et al. Prednisone versus prednisone plus ciclosporin versus prednisone plus methotrexate in new-onset juvenile dermatomyositis: a randomised trial for the Paediatric Rheumatology International Trials Organisation (PRINTO), 2016, 387(10019), p. 671–678.
9. STRINGER, E., SINGH-GREWAL, D., FELDMAN, B. M. Predicting the course of juvenile dermatomyositis: Significance of early clinical and laboratory features. Arthritis & rheumatology, 2008, 58(11), p. 3585–359.
10. ŠTORK, J. et al. Dermatovenerologie. 2. vydání, Praha, Galén, 2013, p. 225-227, ISBN 978-80-7262-898-8.
Labels
Dermatology & STDs Paediatric dermatology & STDsArticle was published in
Czech-Slovak Dermatology

2019 Issue 3
Most read in this issue
- CHRONICKÁ KOPŘIVKA
- Juvenilní dermatomyozitida – klinický případ
- Liekové hypersenzitívne reakcie: klasifikácia a patogenéza (1. časť)
- Pigmentovaná varianta morbus Bowen v agminátním uspořádání – popis případu