
Juvenilná myelomonocytová leukémia asociovaná s neurofibromatózou a komplikovaná hemofagocytovým syndrómom
Juvenile Myelomonocytic Leukemia Associated with Neurofibromatosis and Complicated by Hemophagocytic Syndrome
Objective:
Juvenile Myelomonocytic Leukemia (JMML) is a disease from the spectrum of myelodysplastic/myeloproliferative diseases and it is also included in the category of malignant diseases presently classified in histiocytic diseases. Hemophagocytic syndromes classified in histiocytic diseases are included into the diseases of variable biologic behavior. The association of myelomonocytic leukemia (JMML) with neurofibromatosis type 1 (NF 1) has been known. The objective of this contribution is to demonstrate, in the present case of a patient with JMML and NF 1, complicated by a simultaneous secondary hemophagocytic syndrome – VAHS or MAHS (Virus associated hemophagocytic syndrome, or Malignancy associated hemophagocytic syndrome), the problems in differential diagnosis as well as in the therapy of combinations of these two serious histiocytic diseases.
The case and results:
The disease became manifest in a two and three quarter year old boy with a clinical picture of a febrile state with bronchopneumonia and hepatosplenomegaly, icterus, the blood count with leukocytosis, thrombocytopenia and anemia, the differential blood could being characterized by a marked leukocytosis with marked monocytosis and the finding of younger developmental forms of granulocytes and blasts. The simultaneously present hypoproteinemia, hypofibrinogenemia and global coagulopathy, the presence of active virus infection (Herpes simples and Coxsackie B1) as well as multiplication of macrophages in bone marrow and the signs of hemophagocytosis resulted in the diagnosis of a simultaneous VAHS. In spite of the therapy aimed at influencing malignity as well as hemophagocytic syndrome the course of the disease was rapid and fatal.
Conclusion:
JMML without successful allogenic transplantation is fatal in almost 100% of cases. VAHS, if not therapeutically influenced, may also result in death of the patient. In the situation of a lacking donor and rapid course of the two simultaneous complications, the child could not be saved.
Key words:
juvenile myelomonocytic leukemia, neurofibromatosis type 1, secondary hemophagocytic syndrome – VAHS (Virus associated hemophagocytic syndrome)
Authors:
E. Bubanská 1; L. Plank 2; P. Szépe 2; P. Mesár 1; P. Bician 1
Authors‘ workplace:
Klinika pediatrickej onkológie a hematológie SZU pri DFNsP, Banská Bystrica
prednostka h. doc. MUDr. E. Bubanská, PhD.
1; Ústav patologickej anatómie a Konzultačné centrum bioptickej diagnostiky JLF UK a MFN, Martin
prednosta prof. MUDr. L. Plank, CSc.
2
Published in:
Čes-slov Pediat 2008; 63 (1): 24-32.
Category:
Case Report
Overview
Cieľ:
Juvenilná myelomonocytová leukémia (JMML) je ochorenie zo spektra myelodysplastických/myeloproliferatívnych ochorení a je tiež zahrnutá do kategórie malígnych ochorení v súčasnej klasifikácii histiocytových ochorení. Hemofagocytové syndrómy sa v klasifikácii histiocytových ochorení zaraďujú medzi ochorenia s variabilným biologickým správaním. Známa je asociácia juvenilnej myelomonocytovej leukémie (JMML) s neurofibromatózou typ l (NF1). Cieľom práce je na vlastnom prípade pacienta s JMML a NFl, ktorý bol komplikovaný súčasným sekundárnym hemofagocytovým syndrómom – VAHS, resp. MAHS (Virus associated haemophagocytic syndrome, resp. Malignancy associated haemophagocytic syndrome), poukázať na problémy v diferenciálnej diagnostike, ako aj liečbe kombinácie týchto dvoch závažných histiocytových ochorení.
Vlastný prípad a výsledky:
Ochorenie sa prejavilo u chlapca vo veku 2 a tri štvrte roka v klinickom obraze ako febrilný stav s bronchopneumóniou a hepatosplenomegáliou, ikterom, v krvnom obraze s leukocytózou, trombocytopéniou a anémiou, v diferenciálnom krvnom obraze s výraznou monocytózou a nálezom mladších vývojových foriem granulocytov a blastov. Súčasne prítomná hypoproteinémia, hypofibrinogenémia a globálna koagulopatia, prítomnosť aktívnej vírusovej infekcie (Herpes simplex a Coxsackie B1), ako aj zmnoženie makrofágov v kostnej dreni aj so známkami hemofagocytózy viedli k diagnóze súčasne prebiehajúceho VAHS. Ochorenie malo napriek podávanej liečbe s cieľom ovplyvniť malignitu aj hemofagocytový syndróm rýchly priebeh s fatálnym koncom.
Záver:
JMML je bez úspešne vykonanej alogénnej transplantácie ochorenie s takmer 100% mortalitou. VAHS, ktorý sa nepodarí liečebne ovplyvniť, môže tiež viesť k smrti pacienta. Pri aktuálnej nedostupnosti darcu a rýchlom priebehu kombinácie dvoch tak závažných ochorení sa dieťa nepodarilo zachrániť.
Kľúčové slová:
juvenilná myelomonocytová leukémia, neurofibromatóza typ 1, sekundárny hemofagocytový syndróm – VAHS (Virus associated haemophagocytic syndrome)
Úvod
Juvenilná myelomonocytová leukémia je vzácne ochorenie detského veku, v minulosti sa označovala ako juvenilná chronická myeloidná leukémia (JCML) alebo chronická myelomonocytová leukémia (CMML). Podľa FAB klasifikácie myelodysplastického syndrómu (MDS) bola vedená ako pediatrický variant CMML. JMML tvorí asi 3–5 % detských hematologických malignít, je akýmsi prechodom medzi MDS a myeloproliferatívnymi ochoreniami a zahrnutá je do novej WHO kategórie myelodysplastických/myeloproliferatívnych ochorení (tab. 1) [1].
![WHO diagnostické kategórie myelodysplastických a myeloproliferatívnych ochorení u detí [1].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/9928a34ba26f255ebdc3169d4a4768bf.png)
Ochorenie sa charakteristicky vyskytuje v rannom detskom veku s mediánom 1,8 roka pri diagnóze, prevažne u chlapcov (M : F = 2,1 : 1), v klinickom obraze je charakterizované hepatosplenomegáliou, lymfadenopatiou, bledosťou, teplotami a často postihnutím kože. V krvnom obraze je leukocytóza s výraznou monocytózou (nad 1 x 109/l), nezrelé prekurzory myelopoézy až blasty. Často je zvýšená hodnota fetálneho hemoglobínu. Na 65 % detí s JMML má normálny karyotyp, 25 % detí má nález monozómie 7. 15 % pacientov má klinický obraz neurofibromatózy typu 1. Myeloidné prekurzory majú in vitro excesívny rast a hypersenzitivitu na GM-CSF (tab. 2) [1, 2, 3].
![Klinické prejavy a kritériá pre diagnózu JMML [1].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/d6ba4a9049d9f475c30df6261debca95.png)
Neurofibromatóza typu 1, známa tiež ako Recklinghausenova choroba alebo periférna neurofibromatóza, patrí do skupiny fakomatóz. Je to vrodené ochorenie neuroektodermu so vznikom hamartómov na koži, oku, centrálnom nervovom systéme a orgánoch. Vekom prejavov pribúda a zväčšujú sa. Ochorenie je autozómovo dominantné s takmer 100% penetranciou, s vysoko variabilnou expresivitou aj medzi postihnutými súrodencami. 30–50 % prípadov vznikne ako nová mutácia. Výskyt NF1 v populácii je 1 : 3000–5000 bez ohľadu na pohlavie a rasu. Najčastejším prejavom NF1 sú škvrny café-au lait na koži, neurofibrómy na koži, gliómy optického nervu a pigmentové uzlíky na dúhovke (Lischove noduly). Veľmi závažná je asociácia NF1 s niektorými typmi malignít, ako sú: neurofibrosarkóm, gliómy, Wilmsov tumor, rabdomyosarkóm, feochromocytóm a rôzne typy leukémie [4].
S vírusmi asociovaný hemofagocytový syndróm – VAHS – patrí do skupiny sekundárnych hemofagocytových syndrómov združených s infekciou (tab. 3) [8]. Klinické príznaky a laboratórne nálezy sú podobné ako pri vrodenej forme histocytového syndrómu – familiárnej hemofagocytovej lymfohistiocytóze (HLH). S infekciou asociovaný hemofagocytový syndróm (IAHS) je závažné ochorenie, v detskom veku až s 50% mortalitou [5]. Sekundárny hemofagocytový syndróm vzniká najčastejšie pri infekcii uvedenými vírusmi: EBV, CMV, humánny herpes vírus 6, varicella-zoster, parvovírus B-19, adenovírus, môže však vzniknúť aj pri bakteriálnej, protozoálnej alebo mykotickej infekcii [6, 7].
![Súčasná klasifikácia histiocytových ochorení [8].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/961f80aaaaeb91dba9d89f4522037d27.png)
Diagnostické kritériá definuje Histiocyte Society takto: horúčka inak neurčeného pôvodu, splenomegalia, cytopénia, hypofibrinogenémia a/alebo hypertriglyceridémia a známky hemofagocytózy v kostnej dreni, slezine, lymfatických uzlinách alebo iných tkanivách [9]. Uvádzame aktuálnu tabuľku diagnostických kritérií hemofagocytovej lymfohistiocytózy (tab. 4) [6]. Významným diagnostickým kritériom je prítomnosť pečeňovej poruchy. Nález izolovanej hemofagocytózy nie je diagnostický pre hemofagocytový syndróm, nevyhnutná je prítomnosť kompletných klinických a laboratórnych znakov. Dôležitý je však aj moment, že v čase diagnózy hemofagocytového syndrómu nemusia byť prítomné všetky diagnostické kritériá a tieto sa objavujú až v ďalšom priebehu ochorenia [6, 8, 9].
![Diagnostické kritériá pre hemofagocytovú lymfohistiocytózu [6].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/98c8f9f69c9629f140c84799eb2e351c.png)
Vlastné pozorovanie
Nami referovaný chlapec bol z 1. nekomplikovanej gravidity, s pôrodnou hmotnosťou 4300 g a dĺžkou 53 cm. Už v útlom veku mal stanovenú diagnózu M. Recklinghausen – neurofibromatóza typu 1 na základe prítomnosti café-au lait na koži a pozitívnej rodinnej anamnézy tohto ochorenia u otca. Mutácia NF1 génu nebola vyšetrená.
Vo veku 33 mesiacov bol preložený na Onkologické oddelenie DFNsP Banská Bystrica s podozrením na JMML z Kliniky anestézie a intenzívnej medicíny (KAIM) inej fakultnej nemocnice. Ochorenie sa manifestovalo v úvode febrilným stavom s bronchopneumóniou, bez zlepšenia po liečbe antibiotikami. V krvnom obraze (KO) bola v úvode výrazná leukocytóza (až 59,1 x 109/l), v diferenciálnom krvnom obraze monocytóza s výskytom mladších vývojových foriem granulocytového radu aj blastov, trombocytopénia, zistená bola hypoproteinémia, koagulopatia a v klinickom obraze hepatosplenomegália. V punktáte kostnej drene bola v úvode ľahko hypercelulárna dreň so zachovaným pomerom myelopoézy a erytropoézy 2,6 : 1, bez zmnoženia blastov. Počas 5-dňovej hospitalizácie na KAIM malo dieťa podávané antibiotiká, hemostyptiká, nízkomolekulový heparín, substitučnú liečbu čerstvou mrazenou plazmou (ČMP), albumín, antitrombín III, erytrocytovú masu aj trombocytový koncentrát.
Pri prijatí v detskom onkocentre bolo dieťa ešte eutrofické, bledé, dyspnoické, so slabým ikterom a početnými škvrnami café-au lait, bez krvácavých prejavov. Prítomný bol auskultačný nález vlhkých fenoménov aj vrzotov, hepatosplenomegália s mierne prominujúcim bruchom s malým množstvom ascitu, dieťa malo hnačky, vo v. femoralis l. dx. bol zavedený centrálny venózny katéter (CVK).
Vstupné parametre KO: Hb 83 g/l, Ery 2,99 x 1012/l, Ht 0,26, Rt 0,004, Le 17,5 x 109/l, Tr 11 x 109/l, SOEr 86 fl, dif. KO: neutrofilné segmenty 10, neutrofilné paličky 1, lymfocyty 23, monocyty 58 – atypické, neutrofilný metamyelocyt 1, neutrofilný myelocyt 1, blast 1. Z ďalších patologických laboratórnych parametrov uvádzame hyperbilirubinémiu – Bi celkový 61,5 a konjugovaný 36,7 ∝mol/l, prítomná bola hypoproteinémia 43 g/l a globálna koagulačná porucha – Quickov čas 37,17 %, APTT 64,50 sec., fibrinogén 2,58 g/l, trombínový čas 18,90 sec., D-diméry 223,78 ∝g/ml, antitrombín III 38,06 % – výsledky koagulačných parametrov už boli ovplyvnené predchádzajúcou substitučnou liečbou. Vysoká bola hodnota TPS 250,3 U/l, beta-2 mikroglobulínu 5592 ∝g/ml, CRP 78,60 mg/l, v norme bola hodnota LD 3,7 ∝kat/l a feritínu 60,6 ng/ml. V mozgomiešnom moku boli ľahko zvýšené elementy 21/3, v náteri boli monocyty a lymfocyty.
Podľa laboratórnych parametrov aj klinického obrazu bola hematologická malignita – JMML vysoko pravdepodobná, podporovaná aj súčasnou asociáciou s NF1. Hladina fetálneho hemoglobínu bola len mierne zvýšená na 1,8 % (možné ovplyvnenie predchádzajúcou transfúziou). V punktáte kostnej drene bola v celkove hypercelulárnej dreni prítomná hyperplázia myelopoézy s posunom k mladším formám, prevahu tvorili myelocyty a metamyelocyty, podiel blastov bol pod 20 % a mierne bol zvýšený počet eozinofilov. Erytropoéza bola hypoplastická, so známkami dyserytropoézy, dostatočný bol počet megakaryocytov s jadrami buď hypersegmentovanými alebo s hypolobulizáciou. Tento nález podporoval diagnózu JMML, v diferenciálnej diagnostike bolo doporučené pátrať aj po možnej vírusovej infekcii. Imunofenotypizácia buniek kostnej drene vykazovala fenotyp myelo-monocytovej línie.
Vzhľadom na riziko progresie pri danom klinickom a laboratórnom náleze bola už v tomto štádiu diagnostiky započatá liečba merkaptopurínom a nízkymi dávkami cytozín-arabinozidu podľa protokolu EWOG MDS, podávané boli antibiotiká, acyklovir, substitučná liečba a ďalšia komplexná terapia daného klinického stavu.
Cytogenetické vyšetrenie kostnej drene bolo v norme (karyotyp 46,XY), vylúčila sa prítomnosť translokácie t(9;21), FISH metodikou boli vylúčené aj najčastejšie chromozómové aberácie asociované s MDS – cytogenetický syndróm 5q-, monozómia 5, resp. 7 a trizómia 8.
V pátraní po možnej vírusovej infekcii sa potvrdila prítomnosť aktívnej infekcie vírusom Herpes simplex s prítomnou dynamikou titra protilátok triedy IgM 1 : 11 000...1 : 22 000...l:8900, ako aj prítomnosť aktívnej infekcie vírusom Coxsackie B1, potvrdenej sérokonverziou a prítomnosťou vírus-špecifickej IgM protilátky.
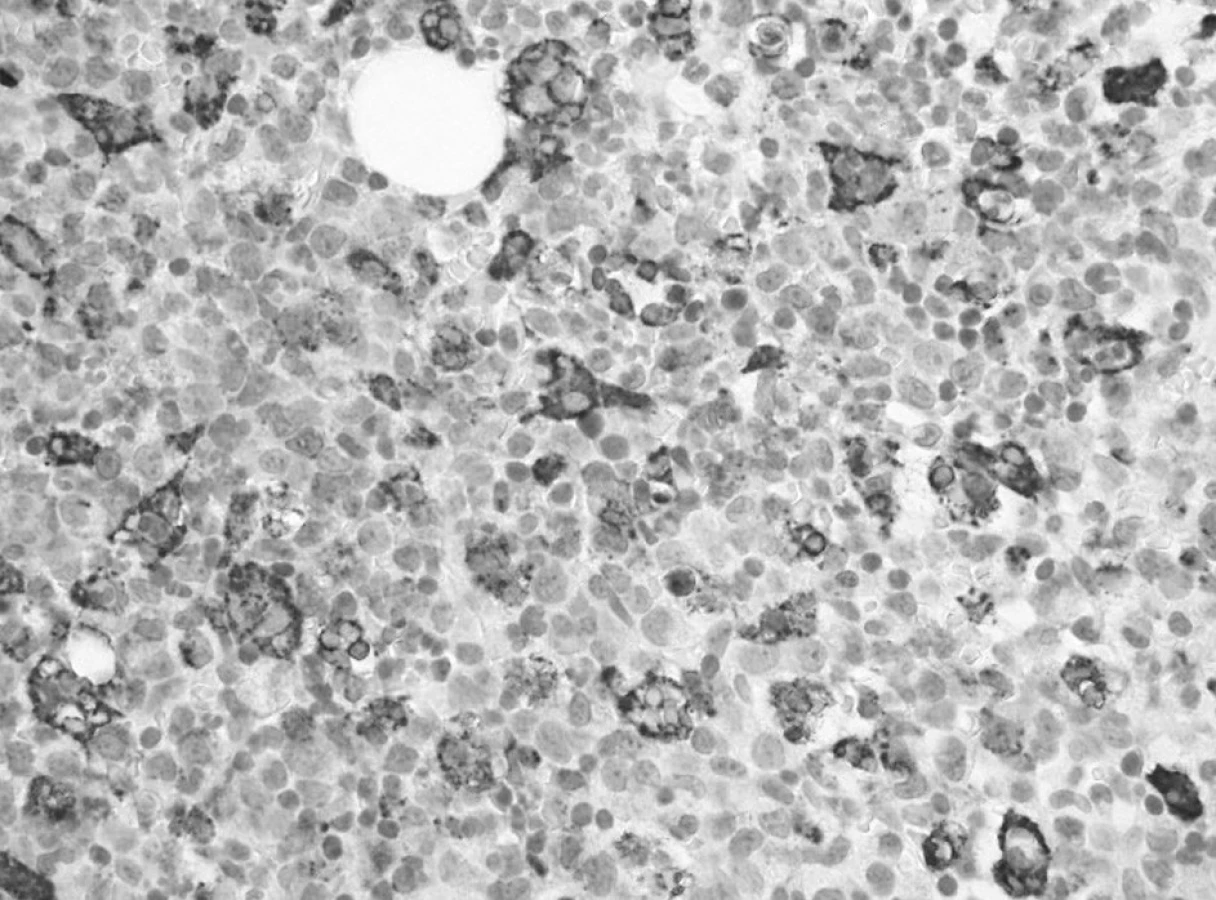
Trepanobioptické vyšetrenie kostnej drene ukázalo obraz hypercelulárnej krvotvorby s prevahou proliferácie bieleho radu so zadržanou maturáciou, pričom celkový počet myeloblastov dosahoval minimálne 50 % (obr. 1). Prítomné boli črty dyserytropoézy a bioptický nález upozornil aj na črty hemofagocytózy (obr. 2), ktoré nútili zvážiť najmä možnosť IAHS (Infection associated haemophagocytic syndrome). Záver vyšetrenia bol formulovaný rezervovane v zmysle potreby zvážiť diferenciálnu diagnostiku reaktívnej myeloproliferácie v rámci klinicky známeho septického stavu versus AML s tým, že definitívna diagnóza závisí od aktuálneho klinického stavu.
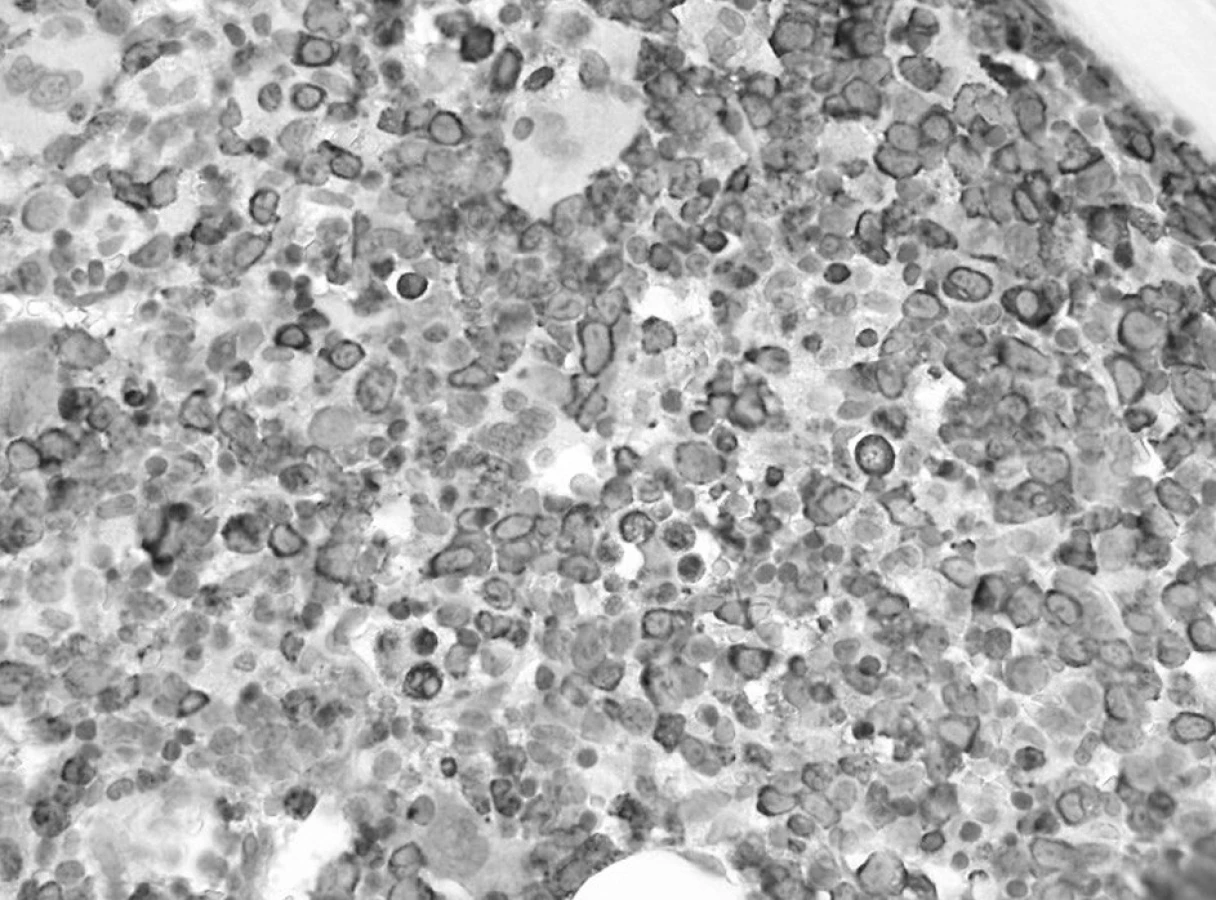
V kontrolnom punktáte kostnej drene s odstupom 2 týždňov od začiatku liečby pokleslo zastúpenie blastických buniek, prítomná bola hypoplázia erytropoézy s kvalitatívnymi zmenami (megaloblastoidia), už zreteľný výskyt makrofágov aj so známkami hemofagocytózy a početné zastúpenie monocytov. Tento obraz nútil naďalej diferenciálne diagnosticky uvažovať o reaktívnych zmenách pri infekcii (obr. 3).
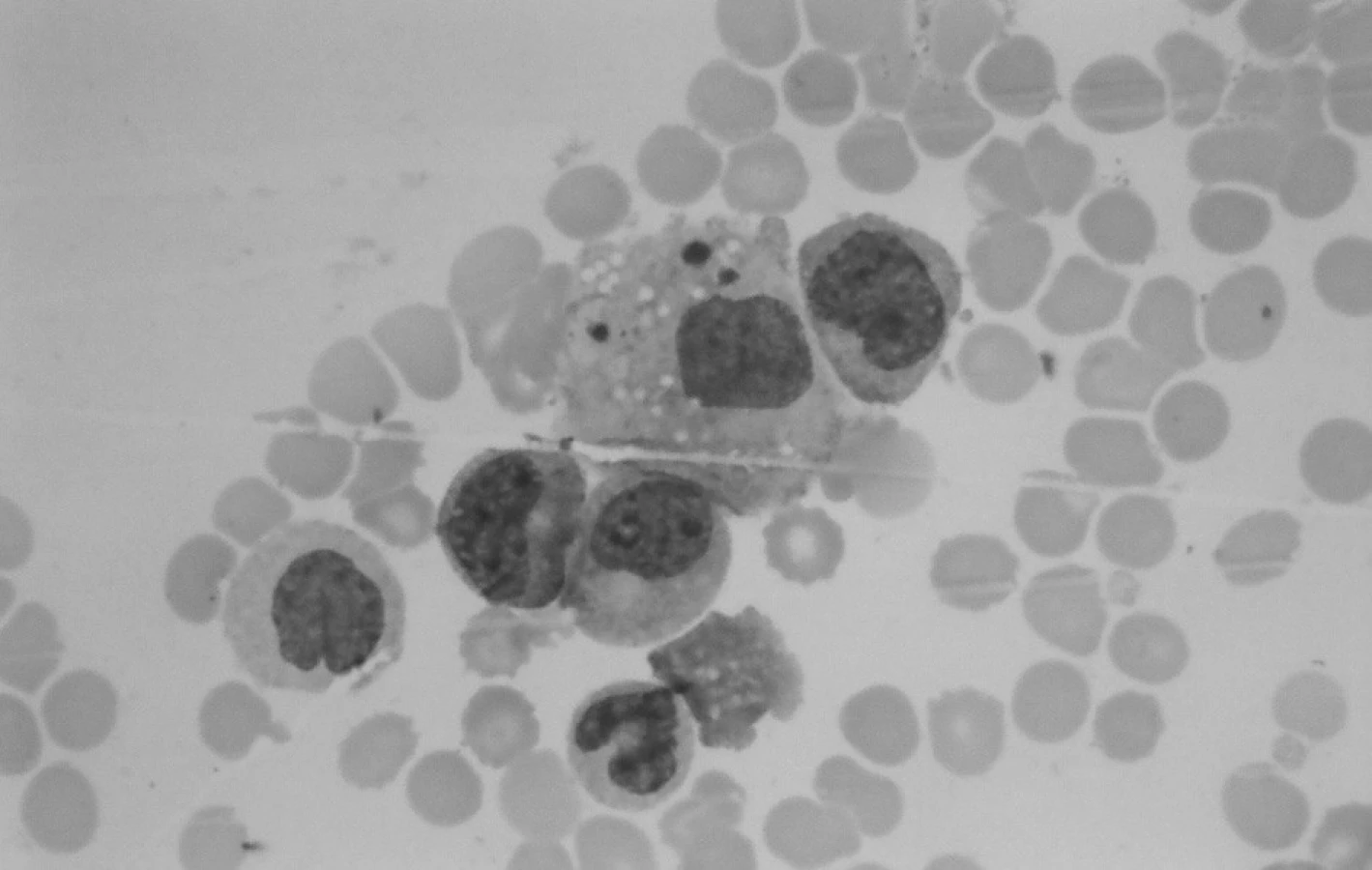
Aj záver druhého trepanobioptického vyšetrenia v tom čase bol formulovaný rezervovane a patológ pri neznalosti aktuálnych hodnôt dif. KO (vrátane Mo, Tr) a výsledku cytogenetického vyšetrenia odporučil nález korelovať so známymi parametrami klinického obrazu (organomegália, extramedulárne prejavy?). V dreni bola prítomná zmes myeloproliferatívnych a myelodysplastických zmien a v kontexte prvej biopsie nebol pozorovaný nárast počtu blastov (obr. 4).
Pacient pokračoval v liečbe antimetabolitmi, prechodne došlo k zmenšeniu organomegálie a vzostupu trombocytov až na 46 x 109/l. Poklesla zápalová aktivita, teploty. Stav bol komplikovaný trombózou v oblasti v. femoralis l. dx. v mieste zavedeného CVK, zvládnutý podávaním terapeutickej dávky nízkomolekulového heparínu. Dieťa vyžadovalo opakovanú substitúciu ČMP pre hypoproteinémiu s edémami a liečbu diuretikami, intermitentne podávanie transfúzie erytrocytovej masy a trombocytov. Po krátkodobej relatívnej stabilizácii stavu sa objavila znova ťažká trombocytopénia, pristúpila aj leukopénia a postupujúca hepatosplenomegália, ikterus, dieťa aj napriek dlhodobej parenterálnej výžive kachektizovalo.
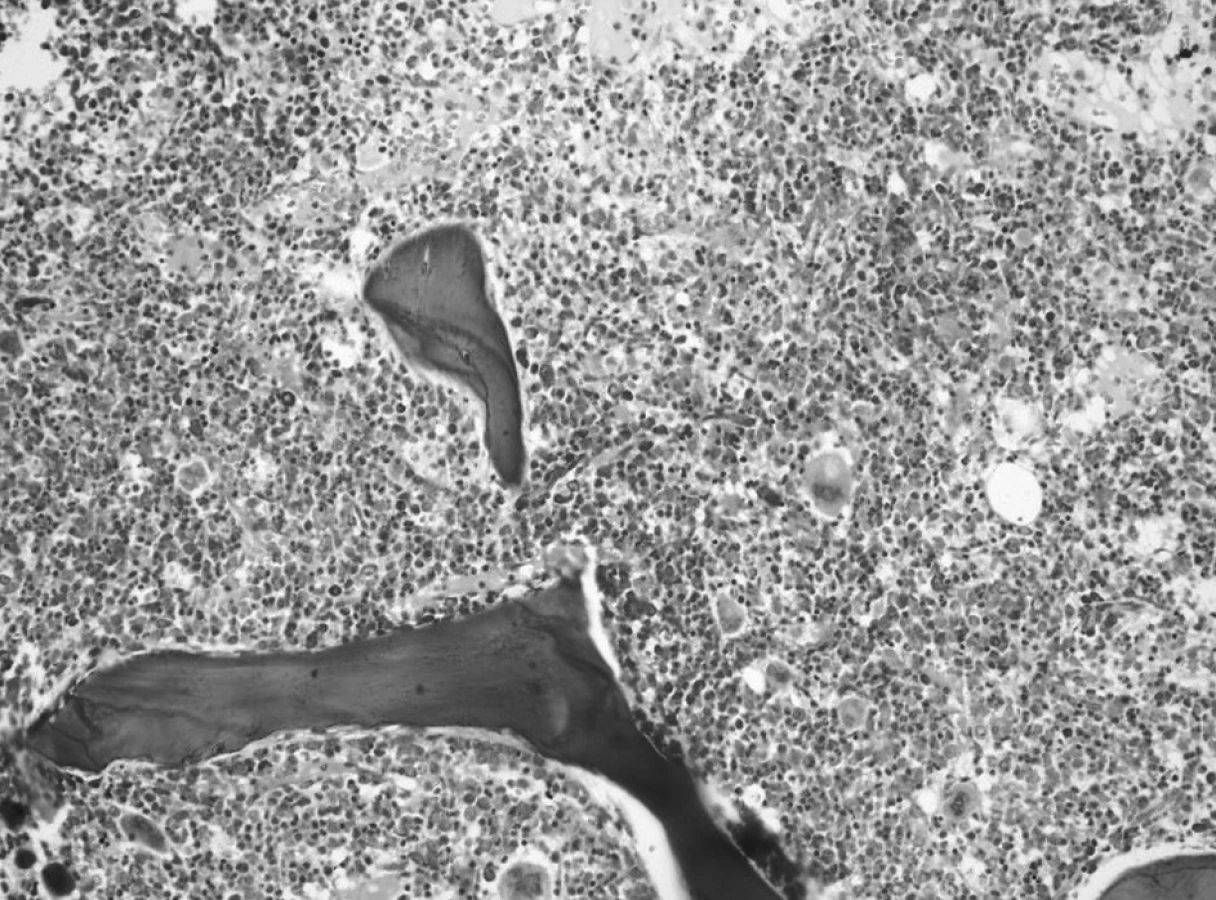
V kontrolnom punktáte kostnej drene mesiac od začiatku liečby bola v spleti makrofágov a monocytov zastúpená myelopoéza prevažne v mladších formách – myeloblasty a nezrelé myelocyty, vývojový rad bol zastavený v štádiu metamyelocytu, bohatá bola erytropoéza s kvalitatívnymi zmenami po CHT. Záver: Maturačná porucha v granulocytovom rade s hraničným počtom blastov, zmnožené makrofágy a monocyty (obr. 5).
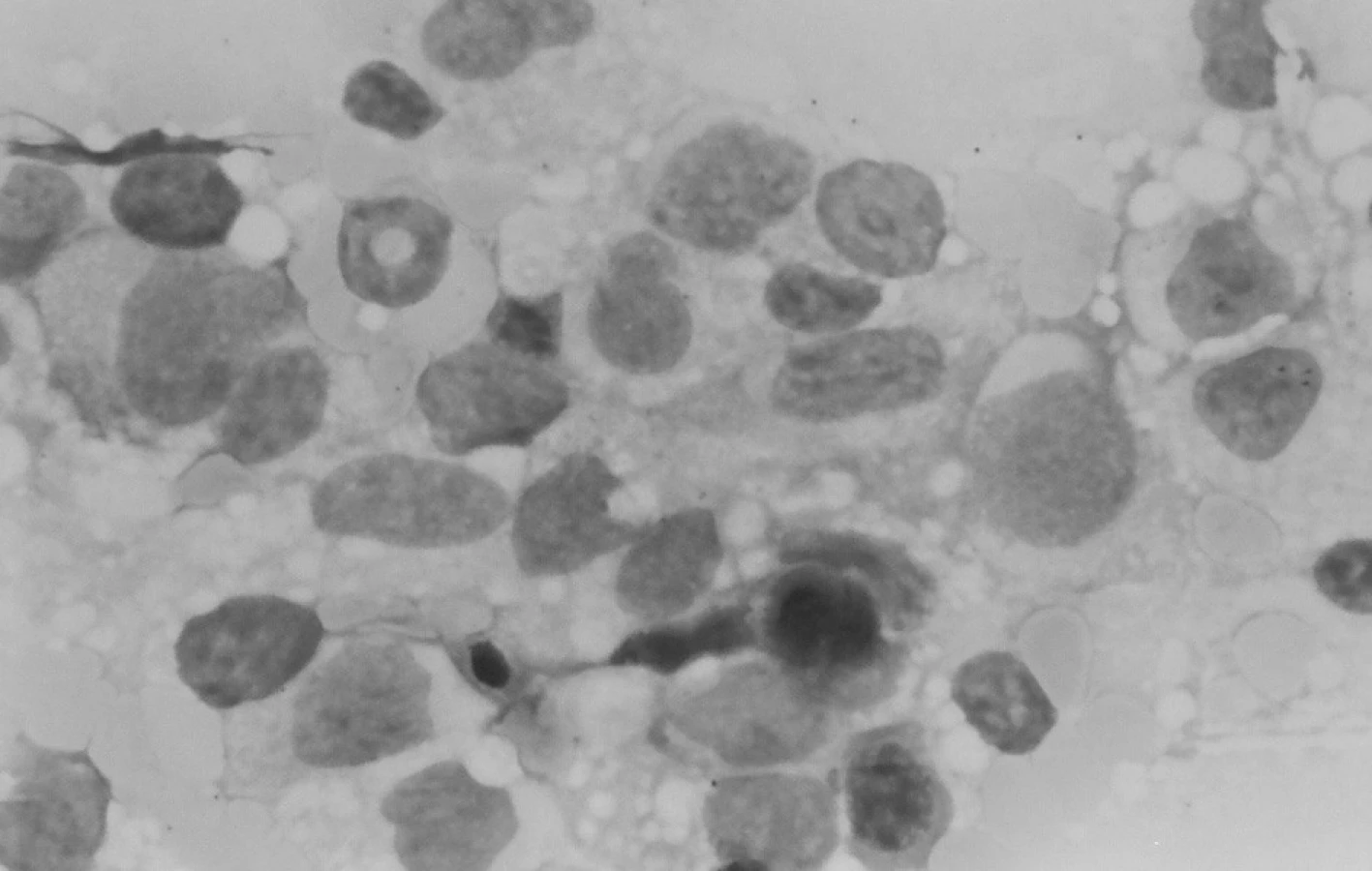
Klinický aj laboratórny nález splnil kritériá na VAHS (Virus associated haemophagocytic syndrome), resp. MAHS? (malignancy associated haemophagocytic syndrome). Do liečby bol znova pridaný acyklovir (Herpesin), súčasne aj izoprinozín. Klinický aj laboratórny nález progredoval. Predpokladanú patologickú cytokínovú reakciu pri terapeuticky nezvládnutom VAHS sme sa snažili ovplyvniť kombinovanou imunosupresívnou liečbou podľa protokolu HLH 94 (Dexamethason, Etoposid, Cyclosporin A). Ani táto liečba neviedla k zlepšeniu, naopak, zvýraznila sa organomegália, pancytopénia, hepatopatia, stúpol bilirubín a dieťa začalo mať krvácavé prejavy. Zhoršený klinický stav s alteráciou vedomia a prítomnosťou početnej orgánovej dysfunkcie vyžadoval hospitalizáciu na oddelení anestézie a intenzívnej medicíny (OAIM). Na 16. deň od začiatku liečby histiocytového syndrómu začal progresívne stúpať počet leukocytov. Indikovaná bola punkcia kostnej drene, ktorá odhalila 83% blastickú infiltráciu kostnej drene AML-M4 (resp. M5) FAB. V dreni bola v spleti makrofágov prítomná blastická infiltrácia a zbytková kvalitatívne zmenená erytropoéza (obr. 6). Podľa cytochémie mali blasty fenotyp M4, podľa imunofenotypizácie skôr M5.
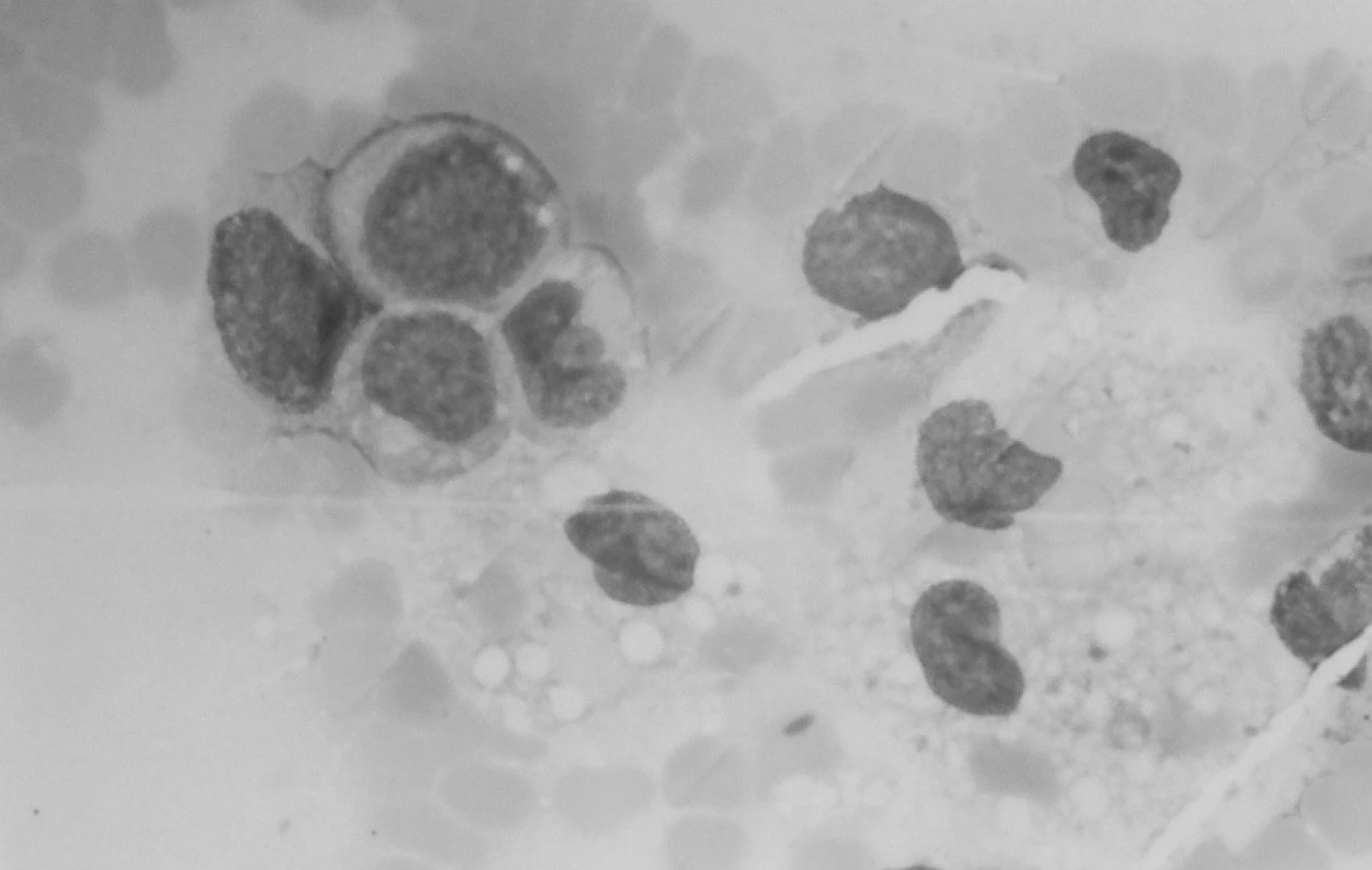
Liečba podľa protokolu AML BFM 98 interim 2003 (Indukcia AIE: Cytosin-Arabinosid, Idarubicin, Etoposid) už priebeh ochorenia neovplyvnila. Pacient exitoval po 6 dňoch indukčnej liečby so známkami generalizovaného slizničného krvácania a multiorgánového zlyhania, po 2,5 mesiaci od prijatia na oddelenie. Pitvu matka odmietla.
Diskusia
Asociácia JMML s NF1 je známa, popisuje sa u 14–15 % detí s JMML. Malé deti s NF1 majú 200–500-krát vyššie riziko vzniku malígnych myeloidných ochorení, hlavne JMML, zatiaľ čo dospelí s NF1 nemajú zvýšený sklon k leukémii [10]. NF1 gén kóduje proteín neurofibromín, 327-kD proteín s doménou, ktorá funguje ako GTP-ázový aktivačný proteín (GAP) pre p21ras(RAS) rodinu. Predpokladá sa, že NF1 gén pôsobí ako tumor supresorový gén na nezrelé myeloidné prekurzory. Mutácia v NF1 géne u pacientov s NF1 má za následok, že neurofibromín je chybný alebo nie je prítomný [10, 11, 12]. Klinické známky neurofibromatózy typ l, ako sú makuly café-au lait, kožné neurofibrómy a Lischove noduly, sa nemusia objaviť v prvých rokoch života, kedy sa diagnostikuje väčšina prípadov JMML. Škvrny café-au lait sú niekedy jediným klinickým prejavom NF1, pretože deti s JMML zomrú skôr, ako by boli klinicky zrejmé ostatné prejavy neurofibromatózy. JMML však môže byť iniciálnym klinickým prejavom NF1 u detí v dojčenskom a batolivom veku a teda prítomnosť mutácie v NF1 géne je u detí s JMML pravdepodobne vyššia. Ďalší pozoruhodný údaj je, že deti s NF1 a súčasnými prejavmi juvenilnej xantogranulomatózy (JXG) majú 20–30-krát vyššie riziko vzniku JMML ako pacienti s NF1, ktorí nemajú prejavy JXG [12].
Náš pacient nemal v čase diagnózy prítomnú žiadnu klonálnu abnormalitu, ktorá by v diferenciálnej diagnostike ochorenia bola potvrdzujúcim nálezom. JMML sa často spája s prítomosťou monozómie 7. Ochorenie s nálezom monozómie 7 sa v literatúre často označuje ako syndróm juvenilnej monozómie 7. Klinicky a laboratórne sa tento syndróm prakticky nelíši od JMML a predpokladá sa, že JMML a syndróm monozómie 7 predstavujú spektrum toho istého ochorenia [1, 2]. Hladina fetálneho hemoglobínu (HbF) je u pacientov s monozómiou 7 väčšinou v norme, čo má pomocný diagnostický význam, u pacientov bez monozómie 7 je často nad 10 % [2]. Klinický obraz je u pacientov bez aj s monozómiou 7 prakticky identický [1].
JMML je ochorenie s veľmi zlou prognózou. Pravdepodobnosť 10-ročného prežitia u netransplantovaných pacientov je len 6 %. Rysuje sa skupina detí s lepšou prognózou. 70 % detí s počtom trombocytov nad 33 x 109/l, s vekom pod 2 roky pri diagnóze a s hodnotou HbF pod 15 % žije 3 roky od diagnózy. Účinnou liečbou je len alogénna transplantácia kostnej drene [2].
Klinický a morfologický obraz podobný JMML môžu mať niektoré vírusové infekcie, ktoré je nutné mať na zreteli v diferenciálnej diagnostike tohto závažného hematologického ochorenia. Ide hlavne o herpetické vírusy – EBV, CMV, HHV-6 [2, 13].
Prítomnosť vírusovej infekcie sme potvrdili aj u nášho pacienta, a to súčasne infekciu vírusom Herpes simplex a Coxsackie Bl. Komplikujúcou situáciou v diferenciálnej diagnostike bol aj fakt, že pri vyšetrení kostnej drene sa našli popri známkach myeloproliferácie a dysplastických zmenách aj známky hemofagocytózy. Na tento nález upozornil najskôr patológ pri vyšetrení 1. trepanobioptickej vzorky kostnej drene odobratej hneď po prijatí pacienta (obr. 2). Morfologický obraz zmnoženia makrofágov inak benígneho vzhľadu sa stal neskôr zreteľný v nasledujúcich punktátoch kostnej drene v priebehu liečby JMML (obr. 3, 5) a napriek súčasne podávanej virostatickej liečbe progredoval.
Do laboratórneho obrazu JMML u nášho pacienta od začiatku nezapadala prítomnosť závažnej hepatálnej lézie s pretrvávajúcou hypoproteinémiou, hypofibrinogenémiou, resp. globálnou poruchou koagulácie v zmysle DIC a hyperbilirubinémiou. Splnené boli diagnostické kritériá pre sekundárny hemofagocytový syndróm.
Infekcie, hlavne vírusovej etiológie a malígne ochorenia (najčastejšie T-bunkové lymfómy), môžu byť asociované so sekundárnym hemofagocytovým syndrómom, hlavne u imunokompromitovaných jedincov [8, 14]. Sekundárny hemofagocytový syndróm sa môže vyskytnúť aj pri iných ochoreniach – napr. pri Chediakovom-Higashiho, Griscelliho syndróme, vzácne pri juvenilnej idiopatickej artritíde, popísaný je výskyt opakovaného HS v spojitosti s vírusovou infekciou pri histiocytóze z Langerhansových buniek [8, 15]. Odlíšenie HLH od JMML môže robiť značné diagnostické problémy (cit. v [16]). Sekundárny HS v asociácii s JMML bol prvýkrát opísaný v literatúre v roku 2004, vtedy známky hemogafocytového syndrómu predchádzali diagnóze JMML [12]. V prípade nášho pacienta sa klinické aj laboratórne prejavy VAHS a JMML manifestovali súčasne.
Za klinické aj laboratórne prejavy hemofagocytového syndrómu, či už primárneho alebo sekundárneho, je zodpovedná vysoká hladina prozápalových cytokínov (hypercytokinémia), ako sú tumor necrosis factor alfa (TNFα), interleukíny IL-1, IL-2, IL-6, interferon gama a ďalšie, ako aj početné hemopoetické rastové faktory, uvoľnené zo stimulovaných T-lymfocytov a makrofágov [6, 17, 18]. Zistilo sa, že pri primárnom hemogafocytovom syndróme (familiárna HLH) mutácia v géne pre perforín vedie k zníženej expresii perforínu na lymfocytoch, makrofágoch a iných dreňových prekurzoroch. Jeho hlavnou úlohou v cytolytickom procese je vytvárať póry v bunkovej membráne, čo vedie k osmotickej lýze cieľových buniek. Predpokladá sa tiež, že perforín môže kontrolovať lymfocytovú proliferáciu. Porucha perforínu môže teda viesť k perzistujúcej aktivácii lymfocytov, tým k zvýšenej produkcii interferonu gama a GM-CSF, čo vedie k aktivácii makrofágov. Makrofágy infiltrujú tkanivá a produkujú vyššie uvedené cytokíny [17]. Pri získanom, sekundárnom hemofagocytovom syndróme môžu k perzistujúcej stimulácii lymfocytov viesť exogénne faktory – infekcie, toxíny (VAHS, IAHS), endogénne produkty (poškodenie tkanív, oxidačný stres, metabolické produkty), či reumatické ochorenie alebo malignita [6]. Ďalším defektom pri hemofagocytových syndrómoch je znížená alebo chýbajúca aktivita NK buniek [6]. Je známe, že mutácia perforínu a SAP (signaling lymphocytic activation molecule {SLAM} – associated protein) génu môže byť príčinou fatálneho VAHS. Nové štúdie tiež predpokladajú, že Tool-like receptory (TLRs) môžu hrať dôležitú rolu pri indukcii zápalových cytokínov ako odpoveď na vírusovú infekciu (cit. v [19]).
Primárna hemofagocytárna lymfohistiocytóza je fatálne ochorenie a liečiteľné je len alogénnou transplantáciou. Bez liečby je medián prežitia 2 mesiace [9]. Pred ňou sa na stlmenie klinicky závažnej cytokínovej reakcie podáva imunosupresívna, imunomodulačná aj cytostatická liečba. HLH Study Group otvorila v januári 1995 prvú medzinárodnú liečebnú štúdiu pre liečbu familiárnej HLH – HLH-94 [9]. Otázkou je, ako liečiť prejavy pri sekundárnom hemofagocytovom syndróme, určité doporučenia sú zrejmé z tabuľky 5 [6]. Nie sú žiadne jasné doporučenia na liečbu HS v priebehu malígneho ochorenia, pri ťažkom priebehu sa tiež doporučuje protokol ako pri primárnej HLH s etoposidom a steroidmi [5]. Tak ako pri pochybnosti o diagnóze familiárneho HLH pri chýbaní niektorého špecifického markera ochorenia a rozhodnutí klinika pri závažnosti klinického priebehu pre započatie prokolárnej liečby [8], tak aj rozhodnutie o začatí imunosupresívnej, imunomodulačnej alebo cytostatickej liečby pri sekundárnom hemofagocytovom syndróme by malo závisieť od klinického stavu pacienta a prítomných laboratórnych zmien. Pacient, ktorý je kriticky chorý, má pretrvávajúce teploty, progresívnu bi - alebo pancytopéniu, poruchu koagulácie alebo známky hepatálnej lézie a tiež pacient pod 1 rok veku, by sa mal liečiť podľa HLH protokolu [6, 20]. Rozlíšenie medzi primárnou (familiárnou) a sekundárnou formou HLH má zásadný význam pre voľbu liečebnej stratégie, ale hlavne na začiatku ochorenia nie je často možné. Pacienti s primárnou HLH sú indikovaní na transplantáciu kostnej drene zavčasu v priebehu ochorenia, čo znamená bezodkladné začatie vyhľadávania vhodného darcu. Pacienti so sekundárnou HLH nie sú indikovaní na trasplantáciu. Infekcia môže byť spúšťacím momentom aj pri familiárnej forme HLH. Pri rozpakoch, o akú formu ide, je vždy vhodné začať liečbu kortikoidmi, etoposidom a cyklosporínom A. Po dosiahnutí remisie je vhodné liečbu prerušiť. Ak vznikne relaps, diagnóza familiárnej HLH je veľmi pravdepodobná a pacient by mal byť transplantovaný [16]. Ak HLH komplikuje priebeh malígneho ochorenia, je na individuálnom rozhodnutí prerušiť chemoterapiu nádoru a cielene liečiť HLH. Liekmi prvej voľby HLH, ktorá komplikuje priebeh systémového autoimunitného ochorenia, sú kortikoidy a cyklosporín A [16].
![Doporučená liečba pri hemofagocytových syndrómoch [6].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/d887a5445ca497269f4af8844e176922.png)
Progresia ochorenia do AML nám v úvode nie celkom jednoznačnú diagnózu JMML potvrdila. Priebeh už aj tak závažného ochorenia bol komplikovaný sekundárnym hemofagocytovým syndrómom a zvýrazňujúce klinické aj laboratórne prejavy sa nepodarilo ovplyvniť podávanou antivírusovou liečbou ani infúziou intravenóznych imunoglobulínov. O efekte následne podanej liečby podľa protokolu HLH-94 sa nedá vyjadriť, pretože základné ochorenie – JMML – prešlo veľmi rýchlo do AML a skončilo exitom.
Záver
U nášho pacienta ide o raritný prípad JMML komplikovanej sekundárnym hemofagocytovým syndrómom, či už bol asociovaný s prítomnou vírusovou infekciou alebo malignitou. Obe diagnózy – VAHS aj JMML – sa radia podľa súčasnej klasifikácie k histiocytovým ochoreniam. VAHS (resp. MAHS) je sekundárny hemofagocytový syndróm združený s makrofágmi. Ochorenie je pôvodom benígne, svojim priebehom však často fatálne. JMML je už ochorenie jednoznačne malígne, bez úspešne vykonanej alogénnej transplantácie takmer 100% fatálne. Náš pacient nemal súrodenca. Indikovaná aj odsúhlasená bola nepríbuzenská transplantácia. Komplikovaný a veľmi rýchly priebeh kombinácie dvoch závažných ochorení však dieťaťu nedávali prakticky žiadnu šancu.
Došlo: 12. 5. 2007
Přijato: 31. 8. 2007
h. doc. MUDr. Eva Bubanská, PhD.
Klinika pediatrickej onkológie
a hematológie SZU pri DFNsP
Nám. L. Svobodu 4
974 09 Banská Bystrica
Slovenská republika
e-mail: ebubanska@dfnbb.sk
Sources
1. Hasle H, Niemeyer CM, Chessells JM, et al. A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia 2003;17(2): 277–282.
2. Mayer J, Starý J. Leukemie. 1. vyd. Praha: Grada Publ., 2002 : 236–237.
3. Niemeyer CM, Arico M, Basso G, et al. Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG – MDS). Blood 1997;89(10): 3534–3543.
4. Chang AE, Ganz PA, Haydes DF, et al. (eds.). Oncology: an evidence-based approach. 1st ed. New York: Springer, 2006 : 517–519.
5. Janka G, Imashuku S, Elinder G, et al. Infection - and malignancy-associated hemophagocytic syndromes. Secondary hemophagocytic lymphohistiocytosis. Hematol. Oncol. Clin. North Am. 1998;12(2): 435–444.
6. Janka GE, Schneider EM. Modern management of children with haemophagocytic lymphohistiocytosis. Br. J. Haematol. 2004;124(1): 4–14.
7. Sugita K, Kurumada H, Eguchi M, et al. Human herpesvirus 6 infection associated with hemophagocytic syndrome. Acta Haematol. 1995;93(2–4): 108–109.
8. Favara BE, Feller AC, Pauli M, et al. Contemporary classification of histiocytic disorders. The WHO Committee On Histiocytic/Reticulum Cell Proliferations. Reclassification Working Group of The Histiocyte Society. Med. Pediatr. Oncol. 1997; 29(3): 157–166.
9. Henter JI, Arico M, Egeler RM, et al. HLH-94: a treatment protocol for hemophagocytic lymphohistiocytosis. HLH Study Group of the Histiocyte Society. Med. Pediatr. Oncol. 1997;28(5): 342–347.
10. Side LE, Emanuel PD, Taylor B, et al. Mutations of the NF1 gene in children with juvenile myelomonocytic leukemia without clinical evidence of neurofibromatosis, type 1. Blood 1998;92(1): 267–272.
11. Shannon KM, O’Connell P, Martin GA, et al. Loss of the normal NF1 allele from the bone marrow of children with typ 1 neurofibromatosis and malignant myeloid disorders. N. Engl. J. Med. 1994;330(9): 597–601.
12. Shin HT, Harris MB, Orlow SJ. Juvenile myelomonocytic leukemia presenting with features of hemophagocytic lymphohistiocytosis in association with neurofibromatosis and juvenile xantogranulomas. J. Pediatr. Hematol. Oncol. 2004;26(9): 591–595.
13. Pinkel D. Differentiating juvenile myelomonocytic leukemia from infectious disease. Blood 1998;91(1): 365–367.
14. Craig FE, Clare CN, Sklar JL, et al. T-cell lymphoma and the virus-associated hemophagocytic syndrome. Am. J. Clin. Pathol. 1992;97(2): 189–194.
15. Klein A, Corazza F, Demulder A, et al. Recurrent viral associated hemophagocytic syndrome in a child with Langerhans cell histiocytosis. J. Pediatr. Hematol. Oncol. 1999;21(6): 554–556.
16. Starý J, Housková J, Špíšek R, et al. Hemofagocytující lymfohistiocytóza – diagnostické a léčebné dilema. Čes.-slov. Pediat. 2004;59(2): 70–82.
17. Filipovich AH. Hemophagocytic lymphohistiocytosis: a lethal disorder of immune regulation. J. Pediatr. 1997;130(3): 337–338.
18. Ravelli A. Macrophage activation syndrome. Curr. Opin. Rheumatol., 14, 2002;14(5): 548–552.
19. Kashiwagi Y, Kawashima H, Sato S, et al. Virological and immunological characteristics of fatal virus-associated haemophagocytic syndrome (VAHS). Microbiol. Immunol. 2007;51(1): 53–62.
20. Starý J, Hrodek O, Housková J. Histiocytární syndromy v dětském věku. Čs. Pediat. 1990,45(10): 577–581.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2008 Issue 1
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Aspirovaná cizí tělesa u dětí
- Časná diagnostika Aspergerova syndromu a její specifické aspekty
- Rázštepy nervovej trubice – súčasné pohľady na etiopatogenézu a možnosti prevencie kyselinou listovou
- Transkutánna bilirubinometria u nedonosených novorodencov