
Klinické projevy a výsledky laboratorních vyšetření u čtyř pacientů s alfa-manosidózou
Clinical Manifestations and Results of Laboratory Examinations in Four Patients with Alpha-Mannosidosis
Alpha-mannosidosis is a slow progressive lysosomal storage disease resulting from the deficiency of lysosomal alpha-mannosidase. The aims of the study were to describe the clinical course of the disease and the results of biochemical and molecular analyses in all four Czech patients with alpha-mannosidosis.
First symptoms of the disease manifested within the first year of life and included psychomotor retardation, repeated respiratory and skin infections, hearing loss and speech problems, progressive development of skeletal changes with coarse facial features, sternum protrusion and scoliosis. The further neurological impairment with the pyramidal and extrapyramidal symptomatology developed since the second year of life.
The activity of alpha-mannosidase was profoundly decreased in isolated leukocytes and all patients are in the gene for alpha-mannosidase homozygous for the mutation p.R750W. The mild hypogamaglobulinaemia was present in all patients.
Conclusion:
Better understanding of the natural course of alpha mannosidosis in childhood will help to identify affected children earlier in the course of the disease. It is important not only for the genetic counseling and prenatal diagnostics in affected families but also for the successful use of new therapeutic approaches including enzyme replacement therapy.
Key words:
alpha-mannosidosis, psychomotor delay, lysosomal storage disease
Authors:
M. Magner 1*; M. Buganová 1*; B. Asfaw 2; H. Poupětová 2; J. Ledvinová 2; O. Brantová 1; H. M. Stesland 3; J. Zeman 1,2
Authors‘ workplace:
Klinika dětského a dorostového lékařství VFN a UK 1. LF, Praha
1; Ústav dědičných poruch metabolismu VFN a UK 1. LF, Praha
2; Department of Medical Genetics, University Hospital of North Norway, Tromso, Norway
3
Published in:
Čes-slov Pediat 2008; 63 (12): 677-682.
Category:
Original Papers
Overview
Alfa-manosidóza je pomalu progredující onemocnění s lyzozomálním střádáním způsobené poruchou lyzozomální alfa-manosidázy. Cílem sdělení je snaha popsat klinický průběh onemocnění a výsledky laboratorních vyšetření u 4 pacientů, u kterých autoři diagnózu alfa-manosidózy potvrdili na enzymatické i molekulární úrovni.
První příznaky onemocnění se objevily již v kojeneckém věku a zahrnovaly zpomalení psychomotorického vývoje, časté respirační a kožní infekty, poruchu sluchu, poruchu vývoje řeči a kostní deformity se zhrubělými rysy obličeje s protruzí sterna a skoliózou. V batolecím věku progredovaly neurologické příznaky ve smyslu extrapyramidové a pyramidové symptomatologie.
U všech pacientů byla přítomná mírná hypogamaglobulinemie. Enzymatickým vyšetřením v izolovaných leukocytech byla zjištěna nízká aktivita alfa-manosidázy a molekulární analýza v genu pro alfa-manosidázu prokázala u všech pacientů přítomnost homozygotní mutace p.R750W, která je ve slovanské populaci zřejmě prevalentní.
Závěr:
Znalost klinického průběhu onemocnění pomáhá identifikovat postižené děti v časnějších fázích onemocnění. Včasná diagnostika je důležitá nejen pro genetické poradenství a eventuální prenatální diagnostiku v postižených rodinách, ale i pro léčbu. Transplantace hematopoetických kmenových buněk pupečníkové krve a především nové metody enzymové substituční terapie rozšiřují spektrum léčitelných lyzozomálních onemocnění.
Klíčová slova:
alfa-manosidóza, psychomotorická retardace, lyzozomální střádavé onemocnění
Úvod
Alfa-manosidóza je pomalu progredující lyzozomální střádavé onemocnění, které vzniká v důsledku poruchy aktivity alfa-D-manosidázy (LAMAN, EC 3.2.1.24). LAMAN je kyselá exoglykosidáza, která štěpí α-1,2, α-1,3 a α-1,6 glykosidovou vazbu manózy v N-vázaných oligosacharidech. Při enzymové poruše dochází ve většině buněk k postupnému nahromadění oligosacharidů obsahujících manózu.
Začátek klinických projevů onemocnění i jeho průběh závisí na progresi lyzozomálního střádání. Mezi nejčastější příznaky patří porucha růstu, hepatomegalie, zpomalení, zástava a regres psychomotorického vývoje, poruchy sluchu, progredující změny fenotypu doprovázené hrubými rysy obličeje, kostní deformity s radiologickými projevy dysostosis multiplex a opakované infekty pro sekundární poruchy imunity [1, 2, 3]. Dědičnost onemocnění je autozomálně recesivní. Gen pro alfa - -manosidázu je na krátkém raménku chromozomu 19 [4], má 24 exonů a funkční protein alfa-manosidázy je tvořen 1011 aminokyselinami [5, 6]. Výskyt alfa-manosidózy v populaci není přesně znám, ale jedná se stejně jako u většiny ostatních lyzozomálních poruch o onemocnění, které patří do skupiny tzv. „rare diseases“. Například v Norsku se 4 miliony obyvatel bylo popsáno šest pacientů [2], v Austrálii uvádějí incidenci pro alfa-manosidózu 1 : 500 000 [7].
V poslední době dochází v oblasti lyzozomálních onemocnění k rychlému rozvoji nových léčebných metod, a to nejen pomocí transplantace hematopoetických kmenových buněk z pupečníkové krve, ale i pomocí enzymové substituční terapie (ERT), při které se postiženým osobám celoživotně podává chybějící enzym infuzí, a to obvykle v týdenních nebo dvoutýdenních intervalech. Dlouhodobé zkušenosti již existují s léčbou pacientů s Gaucherovou a Fabryho nemocí, jako příklad nedávno zahájené ERT lze uvést mukopolysacharidózu typu I [8], mukopolysacharidózu typu II [9], mukopolysacharidózu typu VI [10] nebo morbus Pompe [11]. Předpokládá se, že v krátké době bude k dispozici i ERT pro další nemoci včetně alfa-manosidózy. Úspěch ERT léčby však závisí i na včasné diagnostice.
Cílem našeho sdělení je snaha popsat klinický průběh onemocnění a výsledky laboratorních vyšetření u 4 pacientů, u kterých jsme diagnostikovali alfa-manosidózu na enzymatické i molekulární úrovni.
Metody
Oligosacharidy v moči byly analyzovány metodou tenkovrstevné chromatografie (TLC) s orcinolovou detekcí [12]. Leukocyty z periferní krve byly izolovány metodou sedimentace v dextranu [13]. Koncentrace proteinu v buněčném homogenátu byla následně stanovena modifikací Lowryho metody [14].
Aktivita alfa-manosidázy (LAMAN) byla stanovena v homogenátu leukocytů a fibroblastů metodou s využitím fluorogenního substrátu 4-methylumbelliferyl-alpha-D-manopyranosidu (Glycosynth Limited, Anglie) [15]. Pro zjištění mutace v genu (MANB2) kódujícího alfa-manosidázu byla z krve nebo fibroblastů izolována RNA a následně byla připravena cDNA pomocí PCR. Takto připravená cDNA byla použita přímo k sekvenaci [16].
Klinický průběh onemocnění
V uplynulých 10 letech jsme v ČR diagnostikovali alfa-manosidózu u čtyř pacientů ze tří rodin. Jedná se o tři chlapce a jednu dívku. Diagnóza byla stanovena ve věku 3–13 let.
Pacient č. 1
Chlapec se narodil z 1. fyziologické gravidity nepříbuzných rodičů. Rodinná anamnéza je nevýznamná, perinatální období bez komplikací. Psychomotorický vývoj byl od počátku mírně opožděn, první úsměv až ve 3. měsíci, chlapec seděl v 10. měsíci, chodil od 14. měsíce a první slova používal kolem 15. měsíce. Ve dvou letech byl operován pro oboustrannou inguinální hernii. V pěti letech byl doporučen k metabolickému vyšetření pro hypotonický syndrom, makrocefalii, zhrubělé črty obličeje, hepatomegalii, gibbus v torakolumbální oblasti a středně těžkou oboustrannou poruchu sluchu s převodní složkou (tab. 1).
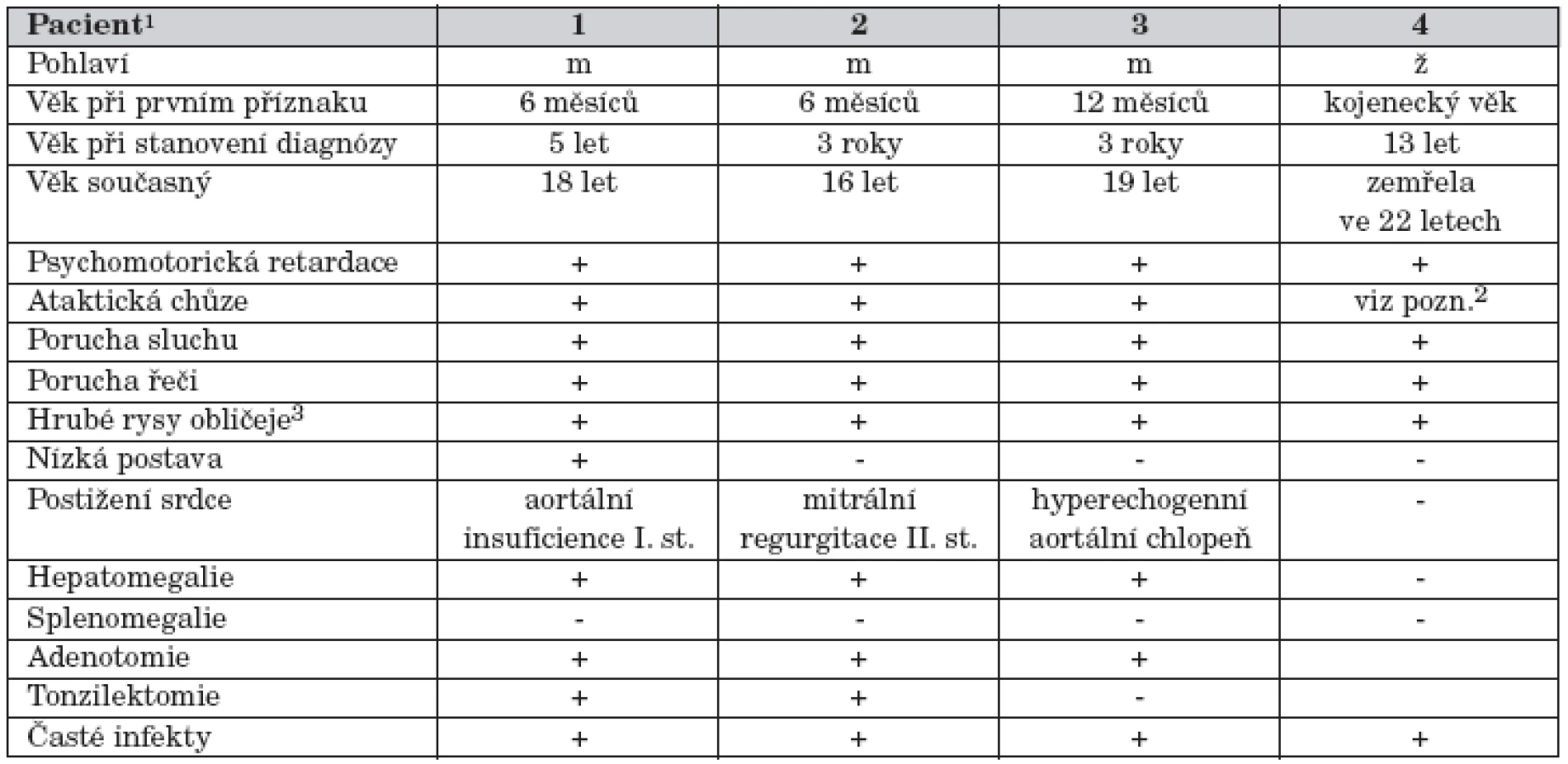
Pro opakované katary horních cest dýchacích byla ve věku sedm let provedena adeno - a tonzilektomie. Při hyperplastických gingivách byla vzhledem k opakovaným cheilitidám a gingivitidám nutná plastika dásní. Imunologické vyšetření prokázalo snížené hladiny IgG. Objevily se kožní projevy charakteru atopické dermatitidy s opakovanými bakteriálními a mykotickými superinfekcemi, tvorbou furunklů a abscesů. Zmírnění kožních projevů nastalo po opakovaných aplikacich Endobulinu. Chlapec byl operován pro genua valga. V současné době ve věku 18 let se u chlapce pro problémy s ataktickou chůzí zvažuje indikace artrodézy talokrurálních kloubů.
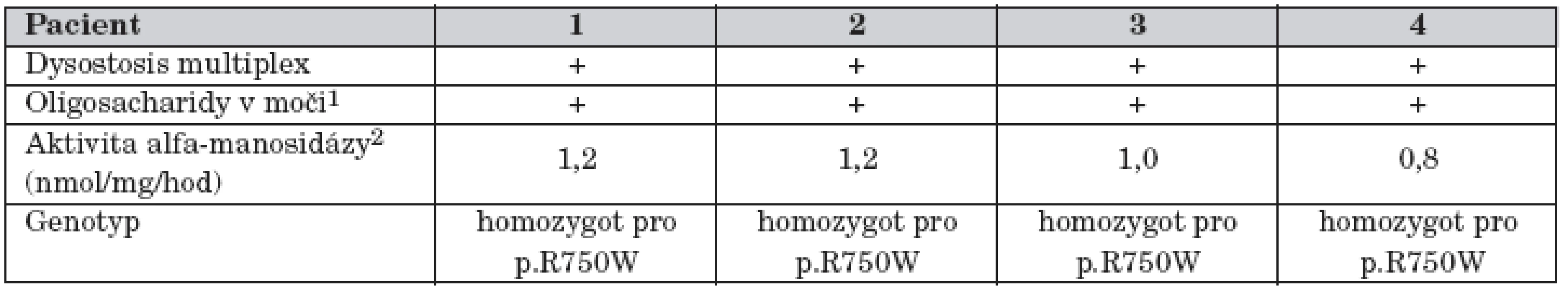
Metabolické vyšetření ukázalo patologický profil oligosacharidů v moči a diagnóza alfa-manosidózy byla potvrzena nálezem deficitu aktivity alfa-manosidázy v izolovaných leukocytech (tab. 2). Molekulární vyšetření ukázalo, že chlapec je v genu pro alfa-manosidázu homozygot pro mutaci p.R750W. Oba rodiče jsou zdraví heterozygoti.
Pacient č. 2
Šestnáctiletý chlapec je mladší bratr pacienta č. 1. Klinický průběh onemocnění je u obou sourozenců obdobný, mladší bratr má pouze výraznější ataxii, naopak frekvence jeho akutních infekcí je nižší. Rodičům bylo poskytnuto genetické poradenství a byla nabídnuta prenatální diagnostika. Při další graviditě byl ve 13. týdnu gestace proveden odběr choriových klků. Enzymatická vyšetření ukázala normální aktivitu alfa-manosidázy. V termínu se narodila zdravá dcera.
Pacient č. 3
U nyní 19letého chlapce byl psychomotorický vývoj od narození zpomalen, a proto byl sledován na dětské neurologii a rehabilitoval. V kojeneckém věku byl operován pro oboustrannou dysplazii kyčelních kloubů, stavět se začal po 14. měsíci, ale další motorický vývoj pokračoval jen pomalu, chodit začal až v 5 letech, trvala i porucha vývoje řeči. Při foniatrickém vyšetření byla zjištěna porucha sluchu (ztráta cca o 70 dB), chlapec dostal naslouchadla. Ve 4 letech se u chlapce objevily opakované respirační infekty a otitidy. Opakovaně byla provedena adenotomie, zaváděny gromety a později došlo i k tonzilektomii. Metabolické vyšetření ukázalo patologické spektrum oligosacharidů v moči a enzymatické vyšetření potvrdilo poruchu aktivity alfa-manosidázy. Poslední dva roky došlo k výraznějšímu zhoršení chůze – chlapec ztrácí stabilitu, zakopává, podvrtávají se mu nohy. Kardiologické vyšetření svědčí pro počínající projevy střádání na chlopních (tab. 1). Spirometrické vyšetření ukazuje restriktivní plicní poruchu. Současná úroveň mentálních schopností je na úrovni středního mentálního deficitu.
Pacientka č. 4
Dívka s nevýznamnou perinatální anamnézou byla od kojeneckého věku sledována pro psychomotorickou retardaci. Prodělávala časté respirační infekty, pro které byla opakovaně hospitalizována. V předškolním a školním věku se postupně vyvíjely a progredovaly kostní deformity – skolióza, gibbus a genua valga. Ortopedické konzilium u dívky ve věku 6 let doporučilo korzet, ale pro sekundárně infikovaný ekzém jej dívka nemohla nosit. Organomegalie přítomná nebyla. Diagnóza alfa-manosidózy byla stanovena ve věku 13 let, kdy již dívka byla připoutaná na invalidní vozík. V té době měla strabismus, hrubé rysy obličeje a radiologické projevy dysostosis multiplex. Dívka zemřela ve věku 22 let po chirurgickém výkonu na infekční komplikace.
U všech pacientů se první příznaky onemocnění objevily již v kojeneckém období. Vedle opožďování psychomotorického vývoje to byly zejména kostní projevy onemocnění s deformitami páteře a hrudníku. Nápadný je vzhled obličeje se zhrubělými rysy. Hepatomegalie je jen mírná a není doprovázená splenomegalií. V pozdějším věku se objevily i další neurologické příznaky ve smyslu extrapyramidové a pyramidové symptomatologie. U všech pacientů byla přítomná mírná hypogamaglobulinemie, pro opakované infekty horních cest dýchacích byly prováděny adenotomie a tonzilektomie. Psychomotorický vývoj se u postižených chlapců nachází v pásmu lehké až středně těžké mentální retardace. Pro poruchu sluchu mají všichni naslouchadla. Vybrané klinické údaje jsou uvedeny v tabulce 1.
Diskuse
Lyzozomální střádavá onemocnění představují skupinu více než 40 dědičných poruch metabolismu, které vznikají pro poruchu katalytické funkce některého z lyzozomálních hydrolytických enzymů [17] nebo v důsledku poruchy transportu přes lyzozomální membránu. Ačkoliv výskyt jednotlivých lyzozomálních onemocnění je poměrně nízký (cca 1 : 25–500 tisíc), celkový výskyt všech lyzozomálních poruch v populaci se odhaduje na více než 1 : 3500, čímž se lyzozomální poruchy řadí mezi nejčastější metabolická onemocnění. První zmínka o manosidóze pochází z roku 1967 od Ockermana [18], od té doby již bylo popsáno více než sto pacientů [19]. Alfa-manosidóza byla nalezena i u krav [20], koček [21] a morčat [22], k vědeckým účelům byly připraveny i geneticky upravené myši s alfa-manosidózou [23].
Podle rychlosti progrese klinických příznaků se rozlišují dvě formy onemocnění. Pacienti s infantilní formou alfa-manosidózy (typ I) jsou charakterizováni rychlým zhoršováním celkového klinického stavu, progresí mentální retardace, organomegalií, těžkou dysostosis multiplex a úmrtím ve věku mezi 3 a 12 roky [2]. Juvenilně-adultní forma (typ II), která postihuje i naše pacienty, probíhá pomaleji. Dominuje psychomotorická retardace, poruchy sluchu a opakované infekce, někteří pacienti se dožívají i čtvrté dekády. Mezi klinické projevy onemocnění patří i svalová hypotonie, opacity čočky, makroglosie, gingivální hyperplazie a prognatie. Z radiografických změn v rámci dysostosis multiplex je u pacientů s alfa-manosidózou nápadné ztluštění lebečních švů, časté jsou ovoidní tvary obratlových těl s předozadní klínovitou deformací [1]. Na pánevních kostech byla popsána mělká acetabula, coxa valga a hypoplazie iliackých kostí. V současné době prochází klinické dělení na dva podtypy onemocnění řadou přehodnocení, a to nejen s ohledem na širší škálu spektra klinických projevů (spontánní potraty v rodinné anamnéze nebo nástup prvních projevů onemocnění až v dospělosti), ale i s ohledem na typy mutací v genu pro alfa-manosidázu [6].
Pacienti s alfa-manosidózou mívají často dlouhodobé nebo chronické infekční komplikace, především na kůži a sliznicích. Sérové koncentrace cirkulujících leukocytů včetně B-lymfocytů jsou obvykle v normě, ale hladiny specifických humorálních protilátek vytvořených po očkování bývají snížené. Jen část nemocných popsaných v literatuře mělo hypogamaglobulinemii, ale u všech našich pacientů jsme pozorovali mírnou hypogamaglobulinemii. V buněčné složce imunity bývá respirační vzplanutí bez výraznějších odchylek, ale neoxidativní složka dráhy nitrobuněčné degradace bakterií je narušena. Popsán byl i kombinovaný imunodeficit se snížením jak humorální, tak i buněčné složky [24].
Laboratorní diagnostika u pacientů s klinickým podezřením na alfa-manosidózu je založena na přítomnosti vakuol v lymfocytech v nátěru z periferní krve a na nálezu patologického profilu oligosacharidů v moči. Potvrzení diagnózy se provádí stanovením aktivity alfa-manosidázy v izolovaných leukocytech nebo v kultivovaných fibroblastech [2]. Prenatální diagnostika tohoto onemocnění je možná stanovením aktivity alfa - -manosidázy v nativních nebo kultivovaných choriových klcích a v kultivovaných amniocytech [25]. Elektronovou mikroskopií lze zobrazit nespecifický retikulogranulární nebo pěnový charakter cytoplazmy hepatocytů, Kupferových buněk a významné zvýšení objemu nervových buněk [26]. Obraz je morfologií i distribucí podobný jiným střádavým onemocněním [27].
V poslední době lze diagnózu alfa-manosidózy potvrdit i na molekulární úrovni. U pacientů byla nalezena řada mutací, většina z nich je „privátních“, pouze „missense“ mutace p.R750W, která byla nalezena i u všech našich pacientů, se ve slovanské populaci vyskytuje častěji – cca u pětiny nemocných s alfa-manosidózou [16].
Variabilita klinických projevů onemocnění u pacientů s alfa manosidózou nekoreluje s reziduální aktivitou alfa-manosidázy ani s typem mutace [16]. Podobné profily oligosacharidů v moči byly nalezeny u pacientů s mírnějším i těžkým průběhem onemocnění [27]. Různé klinické projevy onemocnění byly nalezeny u pacientů s obdobně minimální reziduální aktivitou enzymu (<1 %) [16] a odlišný fenotyp byl nalezen i u sourozenců se stejnou mutací [28]. Tato diskrepance dává prostor k úvahám o významu zevního prostředí na klinický průběh geneticky determinovaných onemocnění či o vlivu dalších zatím neupřesněných genetických faktorů účastnících se na degradaci oligosacharidů.
Kauzální terapie pro pacienty s alfa-manosidózou zatím neexistuje. Cílem symptomatické terapie je snaha zlepšit kvalitu života nemocných pomocí ortopedické péče nebo pomocí naslouchadel, nutná je včasná antibiotická terapie při bakteriálních infekcích, eventuálně opakovaná aplikace imunoglobulinů. Důležitá je rehabilitace a pedagogické vedení. Nově se připravuje enzymatická substituční terapie (ERT) pomocí rekombinantně připravené alfa-manosidázy, která již byla vyzkoušena u myší [22, 29]. Po ERT došlo u myší s alfa-manosidózou k ústupu vakuolizace lyzozomů v játrech, slezině, pankreatu i v gangliích trigeminu. Snížilo se i vylučovaní oligosacharidů bohatých na manózu, ale zatím se nepodařilo uspokojivě vyřešit nedostatečný přestup enzymu přes hematoencefalickou bariéru do CNS [29].
Metodou volby u pacientů s alfa-manosidózou tak zůstává transplantace hematopoetických kmenových buněk z pupečníkové krve. U pacientů po úspěšné transplantaci bylo pozorováno zmenšení organomegalie, normalizace aktivity alfa-manosidázy v leukocytech, poklesla frekvence infektů a zlepšil se sluch, popsáno bylo i částečné zlepšení kostních projevů a stabilizace neurokognitivních funkcí [30], mírné zlepšení v oblasti adaptačních dovedností a verbální paměti [31]. Léčba je však zatížena nezanedbatelnou morbiditou a mortalitou, hlavní podmínkou pro úspěch je včasná diagnostika. Včasná diagnostika navíc umožní i genetické poradenství v postižené rodině.
Práce vznikla s podporou 6. rámcového projektu HUE-MAN, VZ 64165, MSM 0021620849.
Došlo: 4. 3. 2008
Přijato: 20. 5. 2008
Prof. MUDr. Jiří Zeman, DrSc.
Klinika dětského a dorostového lékařství
UK 1. LF a VFN
Ke Karlovu 2
128 08 Praha 2
e-mail: jzem@lf1.cuni.cz
Sources
1. Spranger J, Gehler J, Cantz M. The radiographic features of mannosidosis. Radiology 2000;119 : 401–407.
2. Malm D, Tollersrud OK, Tranebjaerg L, Månsson JE. Alpha-mannosidosis. Tidsskr. Nor. Laegeforen. 1995 Feb 20;115(5):594–597.
3. Thomas GH, Beaudet AL. Disorders of glycoprotein degradation and structure-α: mannosidosis-β, mannosidosis, sialidosis, aspartylglucosaminuria, and carbohydrate-deficient glycoprotein syndrome. In Scriver CR, Beaudet AL, Sly WS, Valle D. (eds). Metabolic Basis of Inherited Disease. 7th ed. New York: McGraw Hill, 1995 : 2529–2561.
4. Kaneda Y, Hayes H, Uchida T, Yoshida MC, Okada Y. Regional assignment of five genes on human chromosome 19. Chromosoma 1987;95 : 8–12.
5. Riise HM, Berg T, Nilssen O, Romeo G, Tollersrud OK, Ceccherini I. Genomic structure of the human lysosomal alpha-mannosidase gene (MANB). Genomics 1997;42 : 200–207.
6. Nilssen O, Berg T, Riise HM, Ramachandran U, Evjen G, Hansen GM, Malm D, Tranebjaerg L, Tollersrud OK. Alpha-mannosidosis: functional cloning of the lysosomal alpha-mannosidase cDNA and identification of a mutation in two affected siblings. Hum. Mol. Genet. 1997 May;6(5):717–726.
7. Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA 1999 Jan 20;281(3): 249–254.
8. Hlavatá A, Kovács L. Skúsenosti s enzýmovou substitučnou liečbou u mukopolysacharidózy. Čes.-slov. Pediat. 2006;10 : 593–598.
9. Wraith JE, Scarpa M, Beck M, Bodamer OA, De Meirleir L, Guffon N, Meldgaard Lund A, Malm G, Van der Ploeg AT, Zeman J. Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur. J. Pediatr. 2008 Mar; 167(3): 267–277.
10. Harmatz P, Ketteridge D, Giugliani R, Guffon N, Teles EL, Miranda MC, Yu ZF, Swiedler SJ, Hopwood JJ; MPS VI Study Group. Direct comparison of measures of endurance, mobility, and joint function during enzyme-replacement therapy of mucopolysaccharidosis VI (Maroteaux-Lamy syndrome): results after 48 weeks in a phase 2 open-label clinical study of recombinant human N-acetylgalactosamine 4-sulfatase. Pediatrics 2005 Jun;115(6): e681–689.
11. Klinge L, Straub V, Neudorf U, Voit T. Enzyme replacement therapy in classical infantile pompe disease: results of a ten-month follow-up study. Neuropediatrics 2005 Feb;36(1): 6–11.
12. Sewell AC. Urinary oligosaccharides. In Hommes FA. (ed). Techniques in Diagnostic Human Biochemical Genetics. New York: Willey-Liss, 1991 : 219–231.
13. Skoog WA, Beck WS. Studies on fibrinogen, dextran and phytohemagglutinin methods of isolating leukocytes. Blood 1956;11 : 436–454.
14. Hartree EF. Determination of protein: A modification of the Lowry method that gives a linear photometric response. Analytical Biochemistry 1972;48 : 422–427.
15. Berg T, Frostad Riise MH, Hansen GM, Malm D, Tranebjærg L, Tollersrud OK, Nilssen O. Spectrum of mutations in α-mannosidosis. Am. J. Hum. Genet. 1999;64 : 77–88.
16. Wenger DA, Williams C. Screening for lysosomal disorders. In Hommes FA. (ed). Techniques in Diagnostic Human Biochemical Genetics. New York: Willey-Liss, 1991 : 587–617.
17. Neufeld EF. Lysosomal storage diseases. Annu. Rev. Biochem. 1991;60 : 257–280.
18. Ockerman PA. A generalized storage disorder resembling Hurler’s syndrome. Lancet 1967;2 : 239–241.
19. Project HUE-MAN: Towards the development of an effective enzyme replacement therapy for human alpha-mannosidosis. LSHM-CT-2006-018692 : 12.
20. Hocking JD, Jolly RD, Batt RD. Deficiency of alpha-mannosidase in Angus cattle. An inherited lysosomal storage disease. Biochem. J. 1972 Jun;128(1): 69–78.
21. Burditt LJ, Chotai K, Hirani S, Nugent PG, Winchester BG, Blakemore WF. Biochemical studies on a case of feline mannosidosis. Biochem. J. 1980;189 : 467–473.
21. Crawley AC, Jones MZ, Bonning LE, Finnie JW, Hopwood JJ. Alpha-mannosidosis in the guinea pig: a new animal model for lysosomal storage disorders. Pediatr. Res. 1999;46 : 501–509.
22. Stinchi S, Lüllmann-Rauch R, Hartmann D, Coenen R, Beccari T, Orlacchio A, von Figura K, Saftig P. Targeted disruption of the lysosomal alpha-mannosidase gene results in mice resembling a mild form of human alpha-mannosidosis. Hum. Mol. Genet. 1999 Aug;8(8): 1365–1372.
23. Malm D, Halvorsen DS, Tranebjaerg L, Sjursen H. Immunodeficiency in alpha-mannosidosis: a matched casecontrol study on immunoglobulins, complement factors, receptor density, phagocytosis and intracellular killing in leucocytes. Eur. J. Pediatr. 2000;159 : 699–703.
24. Fukuda M, Tanaka A, Isshiki G. Variation of lysosomal enzyme activity with gestational age in chorionic villi. J. Inherit. Metab. Dis. 1990;13(6): 862–866.
25. Kjellman B, Gamstorp I, Brun A, Oeckerman PA, Palmgren B. Mannosidosis: a clinical and histopathologic study. J. Pediatr. 1969;75 : 366–373.
26. Monus Z, Konyar E, Szabo L. Histomorphologic and histochemical investigations in mannosidosis. A light and electron microscopic study. Virchows Arch. B Cell Pathol. 1977 Dec 30;26(2): 159–173.
27. Warner TG, Mock AK, Nyhan WL, O’Brien JS. Alpha-mannosidosis: analysis of urinary oligosaccharides with high performance liquid chromatography and diagnosis of a case with unusually mild presentation. Clin. Genet. 1984 Mar;25(3): 248–255.
28. Michelakakis H, Dimitriou E, Mylona-Karayanni C, Bartsocas CS. Phenotypic variability of mannosidosis type II: report of two Greek siblings. Genet. Couns. 1992;3(4): 195–199.
29. Crawley AC, King B, Berg T, Meikle PJ, Hopwood JJ. Enzyme replacement therapy in [alpha]-mannosidosis guinea-pigs. Molecular Genetics and Metabolism 2006;89(1–2): 48–57.
30. Wall DA, Grange DK, Goulding P, Daines M, Luisiri A, Kotagal S. Bone marrow transplantation for the treatment of alpha-mannosidosis. J. Pediatr. 1998 Aug;133(2): 282–285.
31. Grewal SS, Shapiro EG, Krivit W, Charnas L, Lockman LA, Delaney KA, Davies SM, Wenger DA, Rimell FL, Abel S, Grovas AC, Orchard PJ, Wagner JE, Peters C. Effective treatment of alpha-mannosidosis by allogeneic hematopoietic stem cell transplantation. J. Pediatr. 2004 May;144(5): 569–573.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2008 Issue 12
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Výsledky léčby neonatální hydronefrózy
- Klinické projevy a výsledky laboratorních vyšetření u čtyř pacientů s alfa-manosidózou
- Problematika HIV/AIDS v pediatrii
- Výskyt a rizikové faktory alergických ochorení u detí predškolského veku v priemyselnom a vidieckom regióne Slovenska