
Syndromy periodických horeček
Periodic Fever Syndromes
Periodic fever syndromes are clinical entities classified as autoinflammatory diseases. A relatively new group of periodic fever syndromes are characterised by failure of innate immunity mechanisms. Recurrent episodes of fever are accompanied by local inflammation.
They include two autosomal dominant disorders:
CAPS (Cryopyrin Associated Periodic Syndromes) and TRAPS (TNFR-Associated Periodic Syndrome) and autosomal recessive ones: FMF (Familiar Mediterranean Fever) and MAPS (Mevalonate kinase Associated Periodic fever Syndrome). PFAPA (Periodic Fever, Aphthous stomatitis, Pharyngitis a Adenitis) syndrome is an idiopathic disease with unknown aetiology.
Key words:
periodic fever, innate immunity, inflammation
:
P. Król 1; R. Katra 2; P. Doležalová 1
:
Klinika dětského a dorostového lékařství UK 1. LF a VFN, Praha
přednosta prof. MUDr. J. Zeman, DrSc.
1; Klinika ušní, nosní a krční UK 2. LF a FN Motol, Praha
přednosta doc. MUDr. Z. Kabelka
2
:
Čes-slov Pediat 2009; 64 (10): 480-487.
:
Review
Syndromy periodických horeček jsou klinické jednotky patřící do velké skupiny autoinflamatorních chorob. Jde o relativně novou skupinu onemocnění, která jsou charakterizovaná poruchou mechanismů vrozené imunity. Opakované epizody teplot jsou doprovázeny lokálními zánětlivými projevy. Většina syndromů periodických horeček vzniká na podkladě genetické predispozice.
Podle typu dědičnosti je rozdělujeme na autozomálně dominantní:
kryopyrin-asociované periodické syndromy (CAPS; Cryopyrin Associated Periodic Syndromes) a TNF-receptor asociovaný periodický syndrom (TRAPS; Tumor Necrosis Factor Receptor-Associated Periodic Syndrome) a autozomálně recesivní: familiární středomořská horečka (FMF; Familiar Mediterranean Fever) a mevalonátkináza-asociovaný periodický syndrom (MAPS; Mevalonate kinase Associated Periodic fever Syndrome). Mimo tuto klasifikaci stojí periodická horečka s aftózní stomatitidou, faryngitidou a krční adenitidou (PFAPA; Periodic Fever, Aphthous stomatitis, Pharyngitis, Adenitis), u které doposud není definována vyvolávající příčina.
Klíčová slova:
periodická horečka, vrozená imunita, zánět
Úvod
Syndromy periodických horeček se řadí mezi autoinflamatorní onemocnění, která jsou v pediatrii poměrně nově definovanou jednotkou. Pojem periodická nemoc však vznikl mnohem dříve, první popsaný případ byl zaznamenán již v roce 1806 Heberdenem, který pozoroval u pacienta recidivující bolesti břicha, občas doprovázené bolestmi na hrudi a končetinách [1]. V roce 1895 popsali Janeway a Mosenthal případ 16leté dívky trpící od narození opakovanými bolestmi břicha s krátkými epizodami horeček [2]. Na základě těchto a dalších rekurentních doprovodných příznaků byl v roce 1948 Reimannem definován termín „periodická nemoc“ [3]. V roce 1958 byla v odborné literatuře poprvé popsána familiární středomořská horečka (FMF; Familiar Mediterranean Fever) charakterizovaná horečkou, akutní bolestí břicha, pleuritidou a artralgiemi, typicky přítomná v rodinách středomořského původu. V průběhu následujících desetiletí se podařilo popsat další syndromy periodických horeček, u kterých byly díky rozvoji molekulárně-genetických metod definovány genetické příčiny.
Soubor onemocnění byl označen termínem „autoinflamatorní“, který lépe vystihuje předpokládaný mechanismus nemoci. Dysregulace vrozené imunitní odpovědi spolu s absencí autoprotilátek a autoreaktivních T-lymfocytů odlišuje tyto jednotky od autoimunitních onemocnění. Rekurentní febrilie, definované více nebo méně pravidelnými časovými intervaly, jsou doprovázeny lokálními zánětlivými projevy. U většiny syndromů periodických horeček se podařilo definovat vrozenou příčinu. Podle typu dědičnosti lze odlišit syndromy dědičné autozomálně dominantně, ke kterým se řadí kryopyrin-asociované periodické syndromy (CAPS; Cryopyrin Associated Periodic Syndromes) a TNF-receptor asociovaný periodický syndrom (TRAPS; TNFR-Associated Periodic Syndrome) a autozomálně recesivně, familiární středomořská horečka a mevalonátkináza-asociovaný periodický syndrom (MAPS; Mevalonate kinase Associated Periodic fever Syndrome). Mimo tuto klasifikaci stojí periodická horečka s aftózní stomatitidou, faryngitidou a krční adenitidou (PFAPA; Periodic Fever, Aphthous stomatitis, Pharyngitis a Adenitis) u které se doposud nepodařilo definovat vyvolávající příčinu (tab. 1).
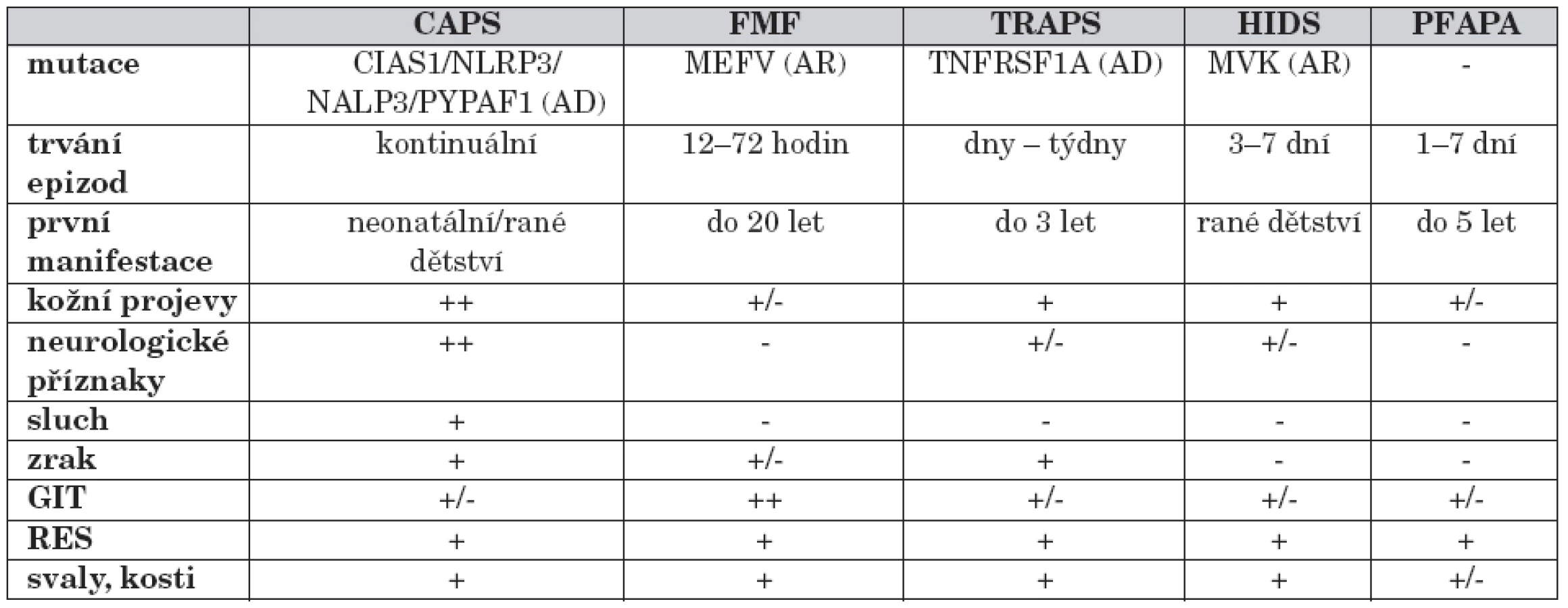
Pro stanovení diagnózy periodické horečky je nutná široká diferenciálně diagnostická rozvaha. V první řadě je třeba vyloučit infekce (bakteriální, virové, mykotické a parazitární), případně na pozadí imunodeficitního stavu.
Rekurentní horečky mohou být součástí příznaků u řady systémových onemocnění (např. systémová forma juvenilní idiopatické artritidy, systémový lupus erythematosus, morbus Crohn, systémové vaskulitidy atd.). Opakované horečky mohou být asociované s vrozenou nebo získanou poruchou imunity, protilátkové i buněčné, např. syndrom cyklické neutropenie. Protrahovaná či opakující se horečka může být také příznakem generalizovaných malignit (akutní lymfoblastická leukémie, lymfomy, neuroblastom atd.).
Jednotlivé syndromy periodických horeček pomůže rozlišit pečlivá rodinná a osobní anamnéza a přesný popis klinického obrazu. Další diagnostické schéma je pro podobnost klinických projevů často obtížné. Navíc specifické odchylky není možné rozlišit ani vyšetřením základních laboratorních ukazatelů, jako jsou CRP, sedimentace, krevní obraz atd., které bývají obvykle pouze nespecificky zánětlivě změněny.
Dominantní je zánětlivá aktivace imunitního systému (zvýšené parametry zánětu – C-reaktivní protein (CRP), sedimentace (FW), leukocytóza, sérový amyloid A (SAA)) bez mikrobiologického průkazu vyvolávajícího agens. Analýza přítomnosti známých mutací DNA je významnou pomocnou diagnostickou metodou, negativní nález však nevylučuje přítomnost periodického syndromu na podkladě zatím nepopsané mutace.
Periodická horečka s aftózní stomatitidou, faryngitidou a krční adenitidou
Periodická horečka s aftózní stomatitidou, faryngitidou a krční adenitidou (PFAPA syndrom; Periodic Fever, Aphthous stomatitis, Pharyngitis a Adenitis, Marshallův syndrom) je idiopatické onemocnění charakterizované periodickou horečkou s trváním obvykle 3–5 dnů, faryngitidou/tonzilitidou, krční adenitidou a často aftózní stomatitidou, která je přítomna přibližně ve 40 % případů [4].
Klinický obraz tonzilitidy s krční adenitidou připomíná streptokokovou angínu, proto v diferenciální diagnostice hraje významnou roli výtěr z krku k vyloučení bakteriálního zánětu. Méně častými příznaky jsou zvracení, průjem, bolesti břicha, artralgie, artritida, konjunktivitida a kožní vyrážka. Mezi atakami jsou děti zcela bez příznaků a dobře prospívají. První manifestace je zpravidla před dosažením 5. roku věku. Diagnóza je stanovena per exclusionem. Laboratorní nálezy jsou nespecifické (zvýšení CRP, FW, leukocytóza), během afebrilního mezidobí se normalizují. Kultivační vyšetření výtěrů z krku je i při klinickém obraze tonzilitidy negativní. Dosud se nepodařilo objasnit etiologii PFAPA syndromu. Někdy lze pozorovat rodinný výskyt, který by mohl poukazovat na hereditární původ [5].
Syndrom byl poprvé popsán Marshallem et al. a byl považován za vzácně se vyskytující onemocnění. Ukazuje se, že incidence tohoto onemocnění je mnohem vyšší a pravděpodobně se jedná o nejčastější syndrom periodických horeček v kavkazské populaci [5, 6]. Důvod pro malý počet pacientů diagnostikovaných s PFAPA syndromem je malé všeobecné povědomí o této jednotce, a proto stále zůstává mnoho pacientů, u kterých není diagnóza dlouho, nebo vůbec stanovena. K nepoznání PFAPA syndromu přispívá i to, že opakované tonzilitidy jsou indikací k tonzilektomii, která může u řady pacientů vést k remisi [7, 8].
V současné době jsou k dispozici diagnostická kritéria navržená Marshallem. Tato kritéria jsou i nadále málo senzitivní, proto evropská pracovní skupina pro studium PFAPA syndromu navrhla na základě informací z registru pacientů nový soubor kritérií, který bude třeba prospektivně validizovat (tab. 2) [9].
![Diagnostická kritéria pro PFAPA syndrom [9].](https://www.prelekara.sk/media/cache/resolve/media_object_image_large/media/image/42a3ec579e637fb514b84ef4a9dbd67d.png)
V současné době neexistuje kauzální terapie pro PFAPA syndrom. Léčba se opírá o jednorázové podání prednisonu v dávce 1 mg/kg v začátku typických klinických příznaků, nejčastěji horečky, s efektem účinku většinou do 2–4 hodin. V poslední době byl prokázán pozitivní efekt tonzilektomie s/bez adenotomie u dětí, které byly pro opakované tonzilitidy indikované k odstranění mandlí. V zatím největší evropské studii byl pozitivní efekt tonzilektomie u 28/35 tonzilektomovaných dětí [5].
TNF-receptor asociovaný periodický syndrom
TNF-receptor asociovaný periodický syndrom (TRAPS; TNFR-Associated Periodic Syndrome) byl poprvé popsán v roce 1982 v rodině irsko-skotského původu. Byl označen jako familiární hibernační horečka (Familial Hibernian Fever), později, pro opakované pozorování rodinného výskytu, familiární periodická horečka (Familial Periodic Fever).
Jedná se o autozomálně dominantní syndrom způsobený mutací genu pro receptor tumor-nekrotizujícího faktoru (TNFRSF1A) ležícího na krátkém raménku 12. chromozomu [10]. Bylo již popsáno přes 40 mutací asociovaných s klinickým obrazem TRAPS. TNFRSF1A je gen kódující protein p55, který představuje extracelulární podjednotku membránového TNF receptoru, tzv. TNFR1. TNFR1 spolu s proteinem p75 vytváří membránový TNF receptor (TNFR). Mutace genu TNFRSF1A vedou ke strukturálním změnám zasahujícím do procesu uvolňování (tzv. shedding) TNFR1 [10]. Za fyziologických podmínek je TNFR aktivován TNFα, přičemž dojde k odštěpení TNFR1 do cirkulace. Zde působí TNFR1 jako solubilní receptor vychytávající volný TNFα z cirkulace. Tímto homeostatickým dějem je regulována koncentrace TNFα v oběhu.
TNFα je důležitý prozánětlivý cytokin, který prostřednictvím TNFR, exprimovaného na povrchu mnoha typů buněk, spouští intracelulární signální kaskádu, vedoucí k aktivaci NF-kappa B a aktivačního proteinu 1 (AP-1). Tyto proteiny regulují transkripci řady genů pro další prozánětlivé cytokiny (např. IL-6, IL-8). Výsledkem je přítomnost protrahovaného zánětu.
Patogenezi TRAPS však nelze vysvětlit pouze defektním sheddingem, protože již byly popsány i mutace, u kterých není porucha v odštěpování TNFR1, jako např. nedostatečná proapoptotická aktivace [11].
TRAPS se zpravidla manifestuje v dětství, výjimečně i v pozdějším věku. Klinický obraz je charakterizován opakovanými horečkami s variabilním trváním, zpravidla řadu dnů až týdny trvajícími. Ataky horečky mohou být provázeny migrujícím rašem, konjunktivitidou, periorbitálním edémem, bolestí břicha, artralgiemi a artritidou. Laboratorně se prokazují zvýšené reaktanty akutní fáze zánětu (CRP, FW, leukocytóza, SAA). Nejzávažnější dlouhodobou komplikací onemocnění je systémová amyloidóza, přítomná asi u 10 % pacientů.
K diagnóze TRAPS může pomoci stanovení nízké sérové hladiny TNFR1, diagnóza je pak potvrzena genetickou analýzou.
Celosvětově bylo dosud diagnostikováno asi 100 nosičů TNFRSF1A mutací, přičemž nejvíce pacientů pochází ze Skandinávie. Předpokládá se, že toto onemocnění je nejčastější příčinou hereditárních syndromů periodických horeček ve vyspělých zemích a, po familiární středomořské horečce, druhou nejčastější příčinou celosvětově.
V terapii TRAPS se u většiny pacientů uplatňuje etanercept – lidský rekombinantní fúzní protein, složený ze solubilního TNFR2 spojeného s Fc komponentou lidského IgG1, který redukuje klinické a laboratorní známky zánětu svou vazbou na buněčný receptor, čímž blokuje biologickou aktivitu TNFα [12]. Podávání vysokých dávek kortikoidů je také účinné, ale jejich nežádoucí účinky jsou limitujícím faktorem.
Mevalonátkináza-asociovaný periodický syndrom
Mevalonátkináza-asociovaný periodický syndrom (MAPS; Mevalonate kinase Associated Periodic fever Syndrome) byl poprvé popsán v roce 1984 Jos van der Meerem et al. jako syndrom periodické horečky doprovázený lokalizovanými formami zánětlivé reakce a byl označen jako Hyper IgD syndrom (HIDS; Hyperimmunoglobulinemia D syndrome) [13]. HIDS byl jistou dobu považován za variantu Stillovy nemoci [14, 15], nebo také nazýván etiocholanolonová horečka [16].
Jedná se o autozomálně recesivní onemocnění způsobené mutací genu pro mevalonátkinázu (MVK) na dlouhém raménku 12. chromozomu [17]. Nejčastěji se jedná o missense mutace (bodová mutace, při které dochází k záměně aminokyseliny vedoucí k tvorbě abnormálního proteinu), u 80 % pacientů byla prokázaná mutace genu V377I [18, 19]. Vzniklý produkt, enzym mevalonátkináza, za fyziologických podmínek katalyzuje fosforylaci kyseliny mevalonové na kyselinu 5-fosfomevalonovou, důležitou pro vznik cholesterolu [20]. Cholesterol je důležitý prekurzor žlučových kyselin, vitaminu D, steroidních hormonů a nonsterolových isoprenoidů [21]. Mutace genu MVK u HIDS pacientů vede k produkci mevalonátkinázy s reziduální enzymatickou aktivitou (1–7 %). Významnou roli v patogenezi hraje zřejmě zvýšená akumulace kyseliny mevalonové a snížení tvorby isoprenoidových produktů, včetně geranylu a farnesylu [18, 20]. Následkem nedostatečné koncentrace těchto produktů dochází k zvýšené sekreci IL-1beta v mevalonátkináza-deficientních buňkách a tudíž k prozánětlivé stimulaci [22].
Onemocnění bývá často vyvoláno očkováním, infektem nebo jiným stresem. In vitro studie poukázaly na zvýšenou aktivitu HIDS-defektní mevalonátkinázy při nižší teplotě (30 °C). Se zvyšováním teploty na 37–39 °C docházelo k progresivnímu snížení funkce enzymu. Tento in vitro experiment by mohl potvrzovat hypotézu asociace doby nástupu HIDS s očkováním nebo infektem a vysvětlit tak epizodicky zvýšený odpad kyseliny mevalonové v moči [23]. Existuje varianta mutace MVK genu, tzv. mevalonová acidurie, u které je defekt enzymu výrazný (<1 %) s obvykle nedetekovatelnou aktivitou mevalonátkinázy. Toto onemocnění má na rozdíl od HIDS závažný klinický dopad.
Příznaky HIDS se objevují obvykle v prvním roce života s typickou horečkou, trvající <7 dní, s faryngitidou, krční lymfadenopatií, bolestí břicha, artralgiemi, artritidou, makulopapulárním rašem, bolestí hlavy [24, 25]. V dutině ústní mohou být přítomny i afty. Ataky horeček se střídají s asymptomatickými obdobími. Interval mezi jednotlivými atakami je většinou 4–6 týdnů. Zvýšené laboratorní parametry v krvi (CRP, leukocytóza, FW, SAA) ukazují zánětlivou aktivaci v průběhu horečky. V moči je zvýšená exkrece neopterinu jako markeru aktivované buněčné imunitní odpovědi, který je jedním z ukazatelů aktivity onemocnění [26]. Kontinuálně zvýšené hladiny IgD a u víc než 80 % pacientů také zvýšené hladiny IgA slabě korelují se závažností onemocnění, dokonce jsou popsáni pacienti s prokázanou MVK mutací a normálními hladinami IgD. Zvýšené IgD bylo pozorováno i u části pacientů s FMF a TRAPS. Specificita tohoto nálezu je tedy nízká. Navíc je známo, že zejména u malých dětí (kojenců a batolat) bývají koncentrace IgD po dlouhou dobu v normě a zvýší se až v pozdějším věku. V moči je zejména v době ataky horečky přítomná vysoká hladina mevalonátu a jeho metabolitů.
V diagnostice je důležitá podrobná anamnéza horeček, přidružených symptomů a doba nástupu onemocnění. Laboratorní průkaz vysokého IgD event. spolu s IgA a prokázanou mevalonaturií jsou indikacemi ke genetické analýze. Z dlouhodobého hlediska mají pacienti dobrou prognózu, v porovnání s FMF a TRAPS je výskyt amyloidózy velmi raritní. Horečky doprovázejí člověka s HIDS po celý život, ale v dospělosti bývá klinický obraz mírnější.
V terapii neexistuje kauzální lék. Někteří pacienti mohou dobře reagovat na Simvastatin [27]. Anti-TNF terapie (Etanercept) má zatím rozdílné výsledky [28–30], zkouší se také efekt blokády IL-1 (Anakinra). K terapii těmito léky se přistupuje pouze u pacientů s klinicky závažně probíhajícím onemocněním, které výrazně ovlivňuje jejich kvalitu života nebo má orgánové projevy, jako např. výraznou hepatosplenomegalii.
Familiární středomořská horečka
Familiární středomořská horečka (FMF; Familiar Mediterranean Fever) je autozomálně recesivní onemocnění způsobené mutací v MEFV genu na krátkém raménku 16. chromozomu, který kóduje protein pyrin/marenostrin. Gen byl poprvé popsán v roce 1997, predominantně je exprimován v neutrofilech, eozinofilech a monocytech. Nejčastější mutace genu jsou lokalizované v B30.2 (SPRY) doméně, která funguje jako ligand nebo signál-transdukční doména. Mutace domény mohou způsobovat opoždění apoptózy a prodloužení zánětlivé odpovědi buňky [31]. Exprese pyrinu je stimulovaná prozánětlivými cytokiny, např. IL-4, TNFα a IFNγ. Pro vysvětlení patogeneze onemocnění existují 2 odlišné hypotézy.
První teorie, nazývaná sekvestrační, podporuje inhibiční efekt poškozené proteinové domény na kaspaza-1-mediovanou aktivaci IL-1β jako prozánětlivého cytokinu [32, 33]. Druhá teorie, tzv. pyrin inflamazomová hypotéza, ukazuje schopnost pyrinu vytvořit vlastní „prozánětlivou jednotku“, tzv. pyrinový inflamazom, výsledkem čehož je aktivace IL-1β [34].
U FMF je začátek onemocnění do 20 let s trváním ataky horečky obvykle 12–72 hodin. Po odeznění příznaků může přetrvávat inaparentní zánět v organismu, který podporuje další ukládání AA amyloidu, hlavně u neléčených pacientů. Doprovodné příznaky jsou pro toto onemocnění typické, zejména serozitidy (v první řadě peritonitida). Raš připomínající erysipeloid je lokalizovaný na nohách, mohou být také akutní artritidy (obvykle oligoartritida) a artralgie. Kromě zhodnocení klinického obrazu sledujeme vzestup zánětlivých parametrů.
Jeden z nejsenzitivnějších a nejobjektivnějších laboratorních markerů v monitoraci rozvoje komplikací zánětu u hereditárních periodických horeček je sérový amyloid A (SAA), patřící do skupiny plazmatických lipoproteinů. V porovnání s CRP bývá často zvýšen i u méně závažných infekcí, včetně virových, a jeho stanovení tak umožňuje lepší rozlišení minimální odpovědi akutní zánětlivé fáze od normálního stavu. Déletrvající zvýšená hladina SAA proteinů vyvolává vznik málo rozpustné AA amyloidové fibrily, která se ukládá v mezibuněčném prostoru predisponovaných tkání, např. ledvin, a podmiňuje klinický obraz sekundární amyloidózy. Možnosti ovlivnění sekundární amyloidózy jsou omezené, nicméně potlačením zánětlivé odpovědi organismu dochází k snížení produkce cytokinů vedoucích k aktivaci tvorby SAA v játrech. Sekundární amyloidóza je nejvíc zastoupená u FMF, častá je u neléčených pacientů. U CAPS se u jednotlivých syndromů liší procentuální zastoupení (2–25 %), u TRAPS je >10 %. Jen raritně se objevuje u HIDS a PFAPA.
V průběhu ataky horečky je u FMF kromě SAA zvýšeno i CRP, fibrinogen a další parametry zánětu. Pokud je v moči zaznamenaná proteinurie, jde pravděpodobně o signál alarmující přítomnost amyloidózy. Mutace v genu pro pyrin/marenostrin u FMF umožňuje rozlišit genetická analýza.
V léčbě se uplatňuje kolchicin jako prevence febrilních atak u 60 % pacientů a signifikantní redukce počtu atak u dalších 20–30 % dětí [35–37]. Kolchicin je třeba užívat celoživotně včetně období gravidity a laktace. Většinou pacientů je velmi dobře tolerován. Na velkých souborech pacientů v endemických oblastech se ukázalo, že špatný terapeutický efekt kolchicinu často souvisí se špatnou compliance pacientů. U skutečných non-responderů je zkoumán terapeutický efekt blokády IL-1 (Anakinra) [38, 39].
Kryopyrin-asociované periodické syndromy
Kryopyrin-asociované periodické syndromy (CAPS; Cryopyrin Associated Periodic Syndromes) je skupina autoinflamatorních onemocnění způsobená mutací v CIAS1 (Cold Induced Autoinflammatory Syndrome 1) genu, jinak známého jako NLRP3/ NALP3, nebo-li PYPAF1 gen [40–42]. Onemocnění je autozomálně dominantní. Mutace může vznikat spontánně v průběhu koncepce nebo je přítomná u jednoho z rodičů. Gen kódující kryopyrin patří do rodiny NLRs (NOD-like receptory). U zdravého jedince je kryopyrin součástí inflamazomu jako makromolekulární jednotky podílející se na zánětlivé kaskádě, složené z několika interagujících molekul. Inflamazom vede přes kaspázu-1 k aktivaci IL-1β a NF-κB. Kromě IL-1β jako prozánětlivého cytokinu je významná i role NF-κB, který se podílí na produkci dalších prozánětlivých cytokinů (např. IL-6) [43]. Mutace v CIAS1 genu vede ke strukturálnímu porušení inflamazomu a tudíž ke kontinuální nadprodukci IL-1β a ostatních zánětlivých cytokinů. Nadprodukce IL-1β způsobuje mnohé symptomy typické pro toto onemocnění.
CAPS je soubor klinických jednotek s různou intenzitou závažnosti a podle toho se dělí na rodinnou chladovou kopřivku (FCAS; Family Cold Autoinflammatory Syndrome), Muckle-Wellsův syndrom (MWS; Muckle-Wells Syndrome) a chronický infantilní neurologický, kožní a kloubní syndrom (CINCA/NOMID; Chronic Infantile Neurologic Cutaneous Articular Syndrome nebo-li Neonatal Onset Multisystem Inflammatory Disease) [44]. U CAPS se může vyskytovat překrývání projevů jednotlivých syndromů. FCAS je považován za nejméně závažné onemocnění, u kterého zánětlivá aktivace nezpůsobuje permanentní poškození jednotlivých systémů.
Projevuje se epizodami horeček s rašem, artralgiemi a konjunktivitidou 1–2 hodiny po expozici chladem. Symptomy trvají minimálně 12–24 hodin. Dalším zástupcem je MWS, charakterizovaný horečkami, rašem, artralgiemi, nauzeou, bolestí břicha, konjunktivitidou. Příznaky trvají 1–3 dny. U pacientů se často vyvine progresivní senzorineurální hluchota [45]. Nejzávažnější formou v této skupině je CINCA/NOMID s kontinuálním zánětem se začátkem horeček a rašem často již v novorozeneckém období [44]. Nejzávažnějším příznakem je neurologické postižení s chronickou aseptickou meningitidou, bolestmi hlavy, zvýšeným nitrolebním tlakem, edémem papily a progresivní senzorineurální ztrátou sluchu spojenou s mentálním deficitem. Další příznaky zahrnují krátkou postavu, hepatosplenomegalii, kloubní deformity.
Dlouhodobou komplikací onemocnění může být amyloidóza vedoucí až k renálnímu selhání. Diagnostika je založená na podrobné anamnéze, klinickém obraze a genetické analýze. U většiny pacientů se jako první příznak objeví makulopapulární urtika – like raš již při narození nebo krátce po něm. V krvi jsou nespecificky zvýšené parametry akutní fáze zánětu, objevuje se i vysoký SAA.
V léčbě se uplatnil efekt blokády IL-1 [46, 47].
Závěr
V české populaci dětí jsou periodické horečky zatím rozpoznávány jen vzácně, přesnější incidence a prevalence geneticky podmíněných syndromů není známá. Lze však předpokládat analogickou frekvenci jako u kavkazské populace jiných evropských zemí. PFAPA syndrom je v ČR jako syndrom periodické horečky bez jasné etiologie mnohem častější. Tato klinická jednotka patří mezi relativně častou příčinu rekurentních horeček v časném dětství. Její diagnostika není lehká i vzhledem k nízké specificitě určených kritérií, ale její znalost může výrazně obohatit širokou diferenciální diagnostiku recidivujících teplot nejasné etiologie. V ambulanci pro periodické horečky na pracovišti autorů je v současné době sledováno 89 dětí s rekurentními febriliemi, z nichž u 75 se pravděpodobně jedná o PFAPA syndrom a u 14 o geneticky podmíněné horečky. V rámci evropské iniciativy EuroFever, na které se autoři podílejí, je k dispozici bezplatné genetické vyšetření u indikovaných pacientů, kteří prošli specializovaným klinickým vyšetřením.
U dětí s autoinflamatorními syndromy, zejména PFAPA, časná diagnostika a vhodný terapeutický management mohou zamezit zbytečným vyšetřením a chránit pacienty před nadměrným užíváním antibiotik, stejně jako zlepšit kvalitu života postižených rodin.
Diagnózu PFAPA syndromu je možné udělat v 1. linii kontaktu s pacientem. Typický klinický obraz, vyloučení bakteriální infekce (opakované kultivace z lege artis odebraného materiálu – výtěry, moč) a případného imunodeficitu (anamnéza, laboratorní screening), opakovaná rychlá normalizace stavu i laboratorních nálezů a plné zdraví v afebrilním mezidobí prakticky vylučují přítomnost systémových onemocnění zánětlivých i nádorových. V případě pochybností je vhodné odeslat pacienta k vyšetření na specializované pracoviště. K takovému vyšetření je vhodné dodat podrobnou anamnézu nemocnosti dítěte, vakcinací, přehled prodělaných epizod horečky a jejich léčby, výsledky provedených vyšetření a růstovou křivku dítěte. Pacienty s periodickými syndromy, které klinicky neodpovídají syndromu PFAPA, je vhodné odeslat ke specializovanému vyšetření přímo.
Grant GA UK (Grantová agentura Univerzity Karlovy) č. 52608/2008.
Grant GA ČR (Grantová agentura ČR) 305/08/H037.
Došlo: 6. 8. 2009
Přijato. 2. 9. 2009
Prof. MUDr. Jiří Zeman, DrSc.
Klinika dětského a dorostového lékařství
UK 1. LF a VFN
Ke Karlovu 2
121 00 Praha 2
e-mail: petrakrol@seznam.cz
Sources
1. Heberden W. Commentaries on history and care of disease. London, 1806 : 151.
2. Janeway TC, Mosenthal HO. An unusual paroxysmal syndrome, probably allied to recurrent vomiting, with a study of the nitrogen metabolism. Trans. Ass. Am. Phys. 1908;23 : 504–518.
3. Reimann HA. Periodic disease. Probable syndrome including periodic fever, benign paroxysmal peritonitis, cyclic neutropenia and intermittent arthralgia. JAMA 1948;141 : 239–244.
4. Marshall GS, Edwards KM, Lawton AR. PFAPA syndrome. Pediatr. Infect. Dis. J. 1989;8(9): 658–659.
5. Hofer M, et al. PFAPA (Periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis) syndrome registry: analysis of a cohort of 214 patients, in FMFSAID. Clin. Exp. Rheumatol. 2008;23(2): 273–274.
6. Hofer MF, et al. International PFAPA syndrome registry: cohort of 214 patients. Pediatric Rheumatology 2008;6(Suppl 1): P182.
7. Renko M, et al. A randomized, controlled trial of tonsillectomy in periodic fever, aphthous stomatitis, pharyngitis, and adenitis syndrome. J. Pediatr. 2007;151(3): 289–292.
8. Licameli G, et al. Effect of adenotonsillectomy in PFAPA syndrome. Arch. Otolaryngol. Head Neck Surg. 2008;134(2): 136–140.
9. Hofer MGM, Cochard M, Anton J, et al. PFAPA (periodic fever, oral aphtae, pharyngitis and cervical adenitis) syndrome: a new consensus on diagnostic criteria. Ann. Rheum. Dis. 2009;68(Suppl 3): 705.
10. McDermott MF, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999;97(1): 133–144.
11. Siebert S, et al. Reduced tumor necrosis factor signaling in primary human fibroblasts containing a tumor necrosis factor receptor superfamily 1A mutant. Arthritis Rheum. 2005;52(4): 1287–1292.
12. Drewe E, et al. Prospective study of anti-tumour necrosis factor receptor superfamily 1B fusion protein, and case study of anti-tumour necrosis factor receptor superfamily 1A fusion protein, in tumour necrosis factor receptor associated periodic syndrome (TRAPS): clinical and laboratory findings in a series of seven patients. Rheumatology (Oxford) 2003;42(2): 235–239.
13. van der Meer JWVM, Radl J, van Nieuwkoop JA, Meyer CJ, Lobatto S, van Furth R. Hyperimmunoglobulinaemia D and periodic fever: a new syndrome. Lancet 1984;19(1): 1087–1090.
14. Prieur AM, Griscelli C. Nosologic aspects of systemic forms of very-early-onset juvenile arthritis. Apropos of 17 cases. Sem. Hop. 1984;60(3): 163–167.
15. Geny B, Griscelli C, Mozziconacci P. Immunoglobulin D (IgD) in childhood. II. Serum IgD levels in juvenile rheumatoid arthritis. Biomedicine 1974;20(2): 125–130.
16. Driesen O, Voute PA Jr, Vermeulen A. A description of two brothers with permanently raised non-esterified aetiocholanolone blood level. Acta Endocrinol. (Copenh.) 1968;57(2): 177–186.
17. Drenth JP, et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat. Genet. 1999;22(2): 178–181.
18. Houten SM, Wanders RJ, Waterham HR. Biochemical and genetic aspects of mevalonate kinase and its deficiency. Biochim. Biophys. Acta 2000;1529(1–3): 19–32.
19. Cuisset L, et al. Molecular analysis of MVK mutations and enzymatic activity in hyper-IgD and periodic fever syndrome. Eur. J. Hum. Genet. 2001;9(4): 260–266.
20. Houten SM, Frenkel J, Waterham HR. Isoprenoid biosynthesis in hereditary periodic fever syndromes and inflammation. Cell Mol. Life Sci. 2003;60(6): 1118–1134.
21. Goldstein JL, Brown MS. Regulation of mevalonate pathway. Nature 1990;(343): 425–430.
22. Frenkel J, et al. Lack of isoprenoid products raises ex vivo interleukin-1beta secretion in hyperimmunoglobulinemia D and periodic fever syndrome. Arthritis Rheum. 2002;46(10): 2794–2803.
23. Houten SM, et al. Temperature dependence of mutant mevalonate kinase activity as a pathogenic factor in hyper-IgD and periodic fever syndrome. Hum. Mol. Genet. 2002;11(25): 3115–3124.
24. Drenth JP, et al. Cutaneous manifestations and histologic findings in the hyperimmunoglobulinemia D syndrome. International Hyper IgD Study Group. Arch. Dermatol. 1994;130(1): 59–65.
25. Drenth JP, Haagsma CJ, van der Meer JW. Hyperimmunoglobulinemia D and periodic fever syndrome. The clinical spectrum in a series of 50 patients. International Hyper-IgD Study Group. Medicine (Baltimore) 1994;73(3): 133–144.
26. Drenth JP, et al. Interferon-gamma and urine neopterin in attacks of the hyperimmunoglobulinaemia D and periodic fever syndrome. Eur. J. Clin. Invest. 1995;25(9): 683–686.
27. Simon A, et al. Simvastatin treatment for inflammatory attacks of the hyperimmunoglobulinemia D and periodic fever syndrome. Clin. Pharmacol. Ther. 2004;75(5): 476–483.
28. Takada K, et al. Favorable preliminary experience with etanercept in two patients with the hyperimmunoglobulinemia D and periodic fever syndrome. Arthritis Rheum. 2003;48(9): 2645–2651.
29. Arkwright PD, et al. Hyper IgD syndrome (HIDS) associated with in vitro evidence of defective monocyte TNFRSF1A shedding and partial response to TNF receptor blockade with etanercept. Clin. Exp. Immunol. 2002;130(3): 484–488.
30. Marchetti F, et al. Inefficacy of etanercept in a child with hyper-IgD syndrome and periodic fever. Clin. Exp. Rheumatol. 2004;22(6): 791–792.
31. Gumucio DL, et al. Fire and ICE: the role of pyrin domain-containing proteins in inflammation and apoptosis. Clin. Exp. Rheumatol. 2002;20(4 Suppl 26): S45–53.
32. Chae JJ, et al. Targeted disruption of pyrin, the FMF protein, causes heightened sensitivity to endotoxin and a defect in macrophage apoptosis. Mol. Cell 2003;11(3): 591–604.
33. Chae JJ, et al. The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc. Natl. Acad. Sci. (USA) 2006;103(26): 9982–9987.
34. Yu JW, et al. Cryopyrin and pyrin activate caspase-1, but not NF-kappaB, via ASC oligomerization. Cell Death Differ. 2006;13(2): 236–249.
35. Ozkaya N, Yalcinkaya F. Colchicine treatment in children with familial Mediterranean fever. Clin. Rheumatol. 2003;22(4-5): 314–317.
36. Livneh A, et al. Colchicine prevents kidney transplant amyloidosis in familial Mediterranean fever. Nephron 1992;60(4): 418–422.
37. Zemer D, et al. Colchicine in the prevention and treatment of the amyloidosis of familial Mediterranean fever. N. Engl. J. Med. 1986;314(16): 1001–1005.
38. Roldan R, et al. Anakinra: new therapeutic approach in children with Familial Mediterranean Fever resistant to colchicine. Joint Bone Spine 2008;75(4): 504–505.
39. Moser C, et al. Successful treatment of familial Mediterranean fever with Anakinra and outcome after renal transplantation. Nephrol. Dial. Transplant. 2009;24(2): 676–678.
40. Aksentijevich I, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 2002;46(12): 3340–3348.
41. Boschan C, et al. Neonatal-onset multisystem inflammatory disease (NOMID) due to a novel S331R mutation of the CIAS1 gene and response to interleukin-1 receptor antagonist treatment. Am. J. Med. Genet. A 2006;140(8): 883–886.
42. Feldmann J, et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am. J. Hum. Genet. 2002;71(1): 198–203.
43. O’Connor W Jr, et al. Cutting edge: CIAS1/cryopyrin/ PYPAF1/NALP3/CATERPILLER 1.1 is an inducible inflammatory mediator with NF-kappa B suppressive properties. J. Immunol. 2003;171(12): 6329–6333.
44. Prieur AM, et al. A chronic, infantile, neurological, cutaneous and articular (CINCA) syndrome. A specific entity analysed in 30 patients. Scand. J. Rheumatol. 1987;66(Suppl): 57–68.
45. Hawkins PN, et al. Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra. Arthritis Rheum. 2004;50(2): 607–612.
46. Callejas JL, et al. Anakinra in mutation-negative CINCA syndrome. Clin. Rheumatol. 2007;26(4): 576–577.
47. Hawkins PN, et al. Response to anakinra in a de novo case of neonatal-onset multisystem inflammatory disease. Arthritis Rheum. 2004;50(8): 2708–2709.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2009 Issue 10
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Periodic Fever Syndromes
- Trichophyton verrucosum as an Unusual Cause of Wound Infection in the Hairy Part of Head
- Alcohol Use in Czech Adolescents
- Experience of Young School Aged Children with Legal Drugs