
Geneticky podmíněné formy nefrotického syndromu u dětí
Genetically Determined Forms of Nephrotic Syndrome in Children
Nephrotic syndrome (NS) is a common glomerulopathy in childhood. Beside idiopathic forms with unclear etiology, genetically determined forms occur. These monogenic forms essentially differ in treatment and patient’s prognosis from the idiopathic ones. They are also clinically and histologically indistinguishable. The only option of differential diagnosis is molecular genetic testing.
The most prominent genes causing genetically determined nephrotic syndrome are NPHS1, NPHS2, WT1 and LAMB2. Most of the cases of genetically caused NS are resistant to initial steroid therapy. In all patients with steroid-resistant nephrotic syndrome (SRNS) genetic background should be examined and genetically caused NS should be taken to differential diagnosis. Patients with SRNS should be molecularly genetically tested, at best at the time of renal biopsy.
Key words:
steroid-resistant nephrotic syndrome, podocin, WT1, NPHS2
Authors:
M. Malina; J. Janda; T. Seeman
Authors‘ workplace:
Pediatrická klinika UK 2. LF a FN Motol, Praha
přednosta prof. MUDr. J. Lebl, CSc.
Published in:
Čes-slov Pediat 2009; 64 (2): 77-82.
Category:
Review
Overview
Nefrotický syndrom (NS) je častým glomerulárním onemocněním u dětí. Vedle forem idiopatických, jejichž přesná etiologie není ještě plně objasněna, existují formy geneticky podmíněné. Tyto monogenní formy se zcela zásadně liší v léčbě a v prognóze od forem idiopatických. Klinicky i histologicky jsou ovšem tyto formy onemocnění zcela neodlišitelné. Jedinou možností diferenciální diagnostiky tedy zůstává molekulárně genetické vyšetření.
V současnosti je známo několik genů způsobujících nefrotický syndrom. V dětském věku jsou nejdůležitější geny NPHS1, NPHS2, WT1 a LAMB2. Většina geneticky podmíněných nefrotických syndromů nereaguje na iniciální léčbu kortikoidy. Proto by u všech pacientů s kortikorezistentním nefrotickým syndromem mělo být pomýšleno na geneticky podmíněný NS a pacienti by měli být molekulárně geneticky vyšetřeni.
Klíčová slova:
kortikorezistentní nefrotický syndrom, podocin, WT1, NPHS2
Úvod
Nefrotický syndrom (NS) je souborem příznaků, jejichž etiologickým podkladem je porušení filtrační bariéry ledvin. Tato bariéra je tvořena komplexem fenestrovaného endotelu glomerulární kapiláry, bazální membránou a výběžky podocytů tvořícími štěrbinovou membránu (slit diaphragm). Hlavním příznakem nefrotického syndromu je těžká, tzv. nefrotická proteinurie, charakterizovaná ztrátami bílkoviny do moči nad 1000 mg/m2/24 hodin. Další příznaky, otoky, hypoalbuminemie pod 25 g/l a hyperlipidemie, jsou odpovědí organismu na masivní únik sérových bílkovin do moči.
Klinicky se nefrotický syndrom rozděluje podle odpovědi na iniciální kortikoterapii na kortikosenzitivní a kortikorezistentní. Většina dětských pacientů s NS zareaguje na iniciální léčbu kortikoidy navozením remise a vymizením proteinurie. Tyto kortikosenzitivní formy mají relativně dobrou prognózu. Přibližně 20 % pacientů ale na iniciální kortikoidní léčbu nezareaguje [1]. Tato kortikorezistentní forma onemocnění má závažnější prognózu, často se pojí s obrazem fokálně segmentální glomerulosklerózy (FSGS) a progreduje do chronické renální insuficience. Kortikorezistentní pacienti mohou být léčeni vysokodávkovanými kortikoidy, protože existuje možnost, že u některých forem bude takto přece jen navozena remise. Při kortikorezistenci se léčebně podávají další imunosupresiva, zejména cyklosporin [2].
Příčiny nefrotického syndromu
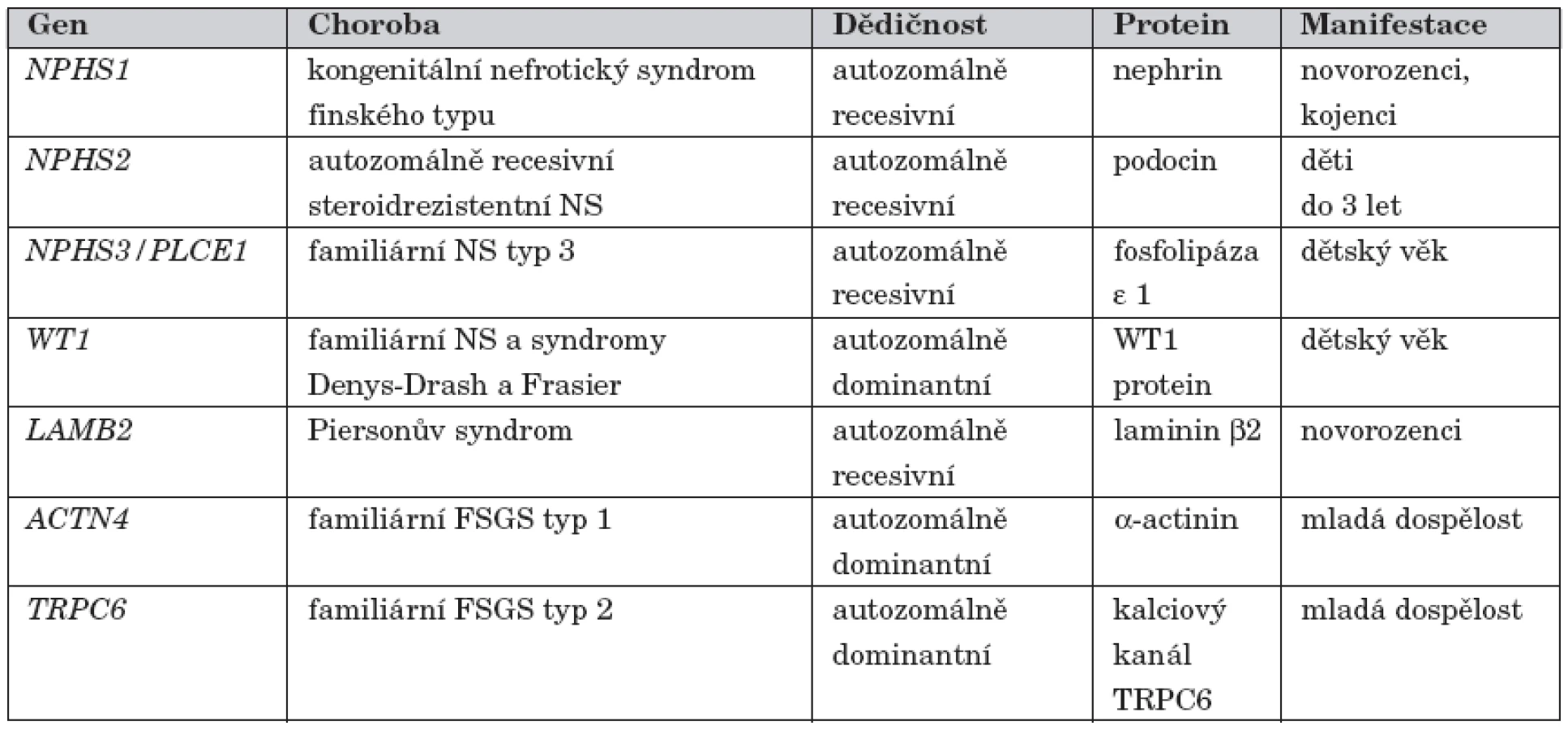
Glomerulární filtrační bariéra je ve většině případů NS narušena imunopatologickým poškozením, jehož příčina dosud není objasněna – idiopatický NS [3]. S rozvojem metod molekulární genetiky se postupně identifikovaly formy NS způsobené mutacemi v genech pro strukturní a regulační proteiny štěrbinové membrány a podocytů [4]. Na těchto geneticky podmíněných formách NS se imunopatologická reakce podílí pouze okrajově. V současné době je známo 6 genů, jejichž mutace způsobují monogenní formy NS v dětském věku. Nejdéle známý je kongenitální NS finského typu (gen NPHS1, OMIM #256300). Dalšími geneticky podmíněnými formami jsou autozomálně recesivní kortikorezistentní NS (NPHS2 gen, OMIM #600995), nefrotický syndrom typ 3 (gen NPHS3/PLCE1 OMIM #610725), Denys-Drashův syndrom a Frasierův syndrom (WT1 gen, OMIM #194080, OMIM #136680), Piersonův syndrom (LAMB2 gen, OMIM #609049) a NS způsobený mutací v genu pro CD2AP (OMIM #607832). U dospělých pacientů jsou nejčastějšími příčinami dědičných forem NS familiární fokálně segmentální glomeruloskleróza typ 1 (ACTN4 gen, OMIM #603278) a familiární fokálně segmentální glomeruloskleróza typ 2 (TRPC6 gen, OMIM #603965).
Všechny tyto genetické formy NS (kromě vzácných případů autozomálně recesivního NS typu 3) jsou kortikorezistentní a téměř vždy rezistentní i k dalším imunosupresivům, jako např. cyklofosfamidu nebo cyklosporinu [5]. U naprosté většiny geneticky podmíněných NS je tedy všechna tato terapie neúčinná a neúčelná a zatěžuje pacienta velkým množstvím nežádoucích účinků [5], mimo jiné zvýšenou náchylností k infekcím, snížením kostní denzity, zpomalením růstu, kataraktou, glaukomem, arteriální hypertenzí nebo nefrotoxicitou cyklosporinu [6]. Průkazem genetické etiologie onemocnění se dá této neúčelné terapii vyhnout a ušetřit tak pacienty těchto závažných nežádoucích účinků léků.
Histologické nálezy u dětí s NS
Biopsie ledviny je indikována u všech pacientů s kortikorezistentním NS a u většiny pacientů s často relabujícím nebo kortikodependentním NS. Histologický obraz neodliší idiopatický od geneticky podmíněného NS. Obě formy NS se mohou histologicky projevovat jako minimální změny glomerulů (minimal change disease – MCD), FSGS nebo difuzní mezangiální skleróza (DMS) [7].
Biopsie ledviny neodliší genetické formy NS od idiopatických a nemůže tedy v diferenciální diagnostice idiopatických a geneticky podmíněných forem nahradit molekulárně genetické vyšetření. Z toho důvodu je již v současné době doporučováno provést molekulárně genetické vyšetření všem dětem s kortikorezistentním NS, aby se vyloučily genetické formy, zabránilo se další zbytečné léčbě a určila se prognóza onemocnění i riziko NS pro další členy rodiny.
Epidemiologie a prognóza
Je prokázáno, že zastoupení genetických forem je různé v různých etnických populacích [8]. V populaci českých dětí zatím nebyl výskyt genetických forem NS publikován, lze předpokládat, že výskyt odpovídá evropské populaci. U dětí do jednoho roku věku tvoří v evropské populaci geneticky podmíněné formy 43–75 % případů nefrotického syndromu, v závislosti na etnické příslušnosti [8], v celém dětském věku je to pak 20–30 % [5]. Největší zastoupení mají mutace v genu NPHS2 (10–25 % případů) a genu WT1 (3–7 % případů) [7].
Prognóza těchto pacientů je výrazně horší než u pacientů s idiopatickým NS a relativně časně progredují do chronické renální insuficience a dospívají do terminálního chronického selhání ledvin většinou ještě v dětském věku. Pozitivním zjištěním je fakt, že geneticky podmíněné formy, na rozdíl od idiopatických forem, jen zcela výjimečně rekurují v transplantované ledvině [5].
Přehled genů zodpovědných za geneticky podmíněné NS
Přehled všech dosud popsaných genetických forem nefrotického syndromu u dětí je uveden v tabulce 1.
NPHS1 (nefrinový gen)
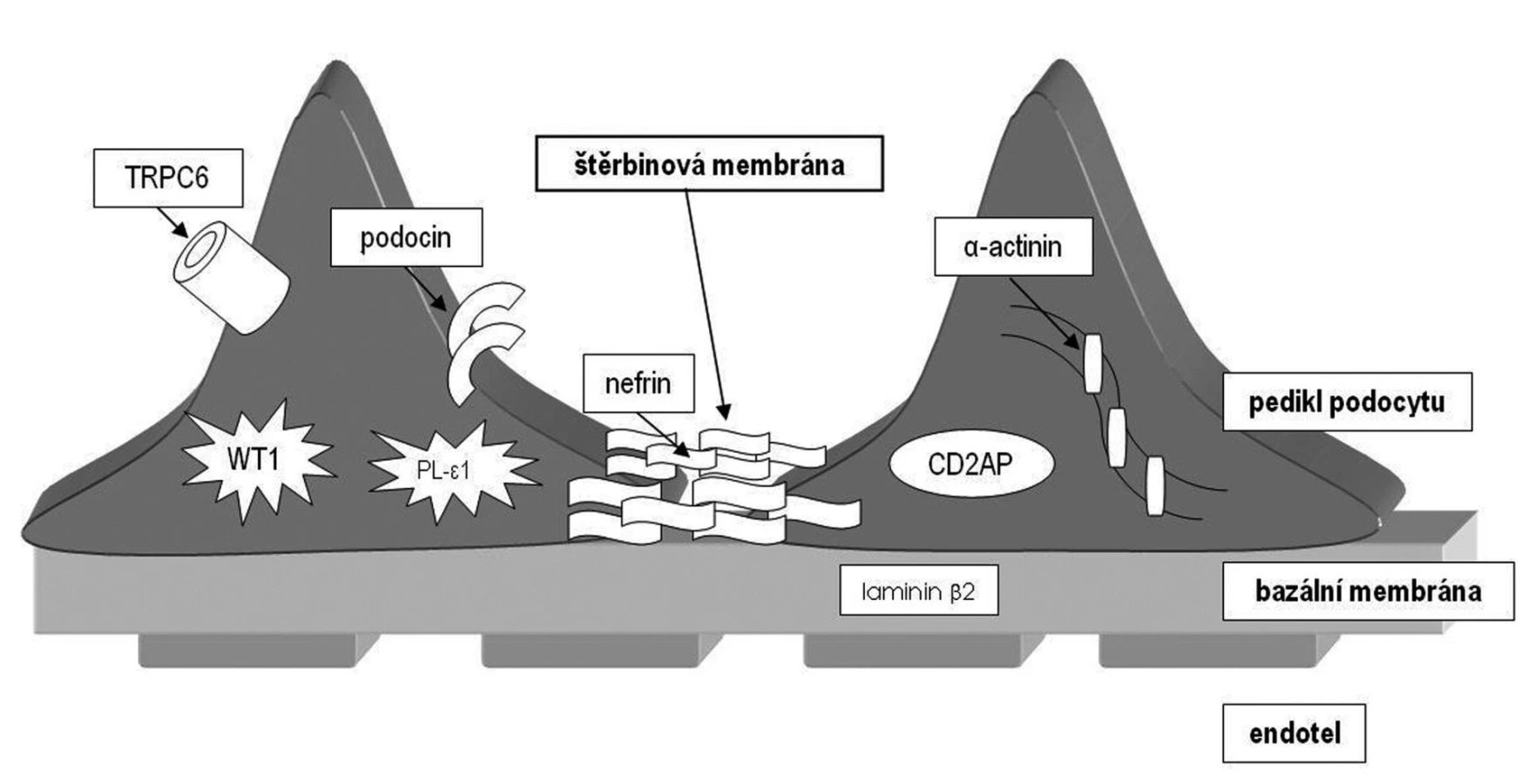
Tento gen (NePHrotic Syndrome 1 = NPHS1) ležící na dlouhém raménku chromozomu 19 kóduje protein zvaný nefrin (obr. 1). Nefrin komunikuje s dalšími proteiny štěrbinové membrány a zajišťuje funkčnost filtrační bariéry. Jedná se o první popsaný geneticky podmíněný nefrotický syndrom. Ve finské populaci byly popsány dvě nejčastější mutace předčasně ukončující syntézu nefrinu (fin-minor a fin-major) zodpovědné za rozvoj NS, každá s různě závažně vyjádřenými příznaky [9]. Choroba se dědí autozomálně recesivně a je nejvíce rozšířena ve Finsku (incidence 1 : 10 000 živě narozených), proto se také označuje jako kongenitální nefrotický syndrom finského typu. Klinicky se manifestuje v novorozeneckém nebo časném kojeneckém věku (kongenitální NS) a někdy i prenatálně. Průběh je těžký s extrémně vysokými ztrátami proteinů do moči.
Děti jsou ohroženy zejména následnou hypogamaglobulinémií s rizikem septických komplikací, které jsou nejčastější příčinou úmrtí dětí s kongenitálním NS finského typu. Proto je standardně doporučovanou léčbou časná jednostranná či oboustranná nefrektomie ke snížení či úplnému zamezení ztrát bílkovin do moči a následná léčba peritoneální dialýzou až do doby, kdy je možná transplantace ledviny. I ta ovšem zvláště u nejtěžších mutací (fin-major) není optimálním řešením, jelikož více než třetina pacientů vykazuje rekurenci ve štěpu, pravděpodobně z důvodu přítomnosti protilátek proti nefrinu. Menší změny v genu (missense mutace) bývají spojovány s rozvojem NS v dětském věku s méně těžkým průběhem.
NPHS2 (podocinový gen)
Gen ležící na 1. chromozomu kóduje bílkovinu podocin. Podocin je bílkovina z rodiny stomatinů, exprimovaná pouze v ledvině. Podílí se na fetálním vývoji ledviny a má zcela zásadní úlohu v membránové signalizaci podocytů. Váže se s nefrinem a je zodpovědná za jeho správné umístění a napojení na membránu podocytu (obr. 1). Mutace v tomto genu způsobují autozomálně recesivní kortikorezistentní typ NS. Spektrum mutací je velmi pestré a stejně tak i histologický obraz, který může vykazovat jak FSGS, tak MCD. Zatím nebyla objevena korelace mezi různými mutacemi a histologickým obrazem (minimální změny či FSGS). Mutace v NPHS2 genu jsou zodpovědné u evropské populace za cca 30 % případů výskytu kortikorezistentního NS. Jedná se tedy o nejčastější genetickou formu NS v evropské populaci mimo Finsko.
Klinicky se vždy projevuje kortikorezistentním NS. První projevy se objevují většinou v předškolním věku, nejsou vzácné ani novorozenecké nebo kojenecké manifestace. Věk prvních projevů koreluje s typem mutace. Mutace, které předčasně ukončují syntézu proteinu, a mutace R138Q v homozygotním stavu mají časnější manifestaci [10]. Zatím nebyla nalezena korelace mezi typem mutace a rychlostí progrese choroby. Pacienti pozvolna spějí do chronické renální insuficience. Veškerá kortikosteroidní léčba je u těchto pacientů neúčinná. Jedinou možnou léčbou je transplantace ledvin, kdy tito pacienti jsou téměř bez rizika rekurence NS ve štěpu.
NPHS3 (PLCE1)
Nejnověji objevený gen z rodiny autozomálně recesivně dědičných nefrotických syndromů je NPHS3/PLCE1. Tento gen kóduje enzym fosfolipázu C-epsilon (PLCE), která se podílí na nitrobuněčné signalizaci generováním druhých poslů (obr. 1). Mutace, které způsobí předčasné ukončení syntézy proteinu, se projevují histologickým obrazem difuzní mezangiální sklerózy (DMS). Bylo zjištěno, že téměř třetina případů DMS u dětí s NS je způsobena mutací v PLCE1 genu a je tedy třikrát častější genetickou příčinou DMS u dětí než mutace v genu WT1 [11]. Missense mutace se projevují spíše obrazem FSGS.
Klinicky nejzajímavější na tomto geneticky podmíněném NS je fakt, že existuje popsaný případ pozitivní odpovědi na kortikoidy (kortikosenzitivní NS) i přes velmi závažnou mutaci, která u příbuzných způsobila NS s DMS. Kortikoidy zde nemají pravděpodobně imunosupresivní efekt, ale spekuluje se o přímém ovlivnění metabolismu podocytů [12].
WT1 (Wilms tumor 1)
WT1 gen kóduje protein s motivem zinkového prstu vážící se na DNA, který funguje jako transkripční faktor. Je zcela zásadní pro normální vývoj urogenitálního ústrojí a mezotelu. Jeho mutace způsobují embryonální nefroblastom, známý jako Wilmsův tumor. Mutace lokalizované výlučně v exonech 8 a 9 způsobují též rozvoj kongenitálního nefrotického syndromu [7] a jsou zodpovědné asi za 9 % NS s obrazem DMS [11]. Mutace jsou velmi různorodé a vznikají většinou de novo. Familiární formy vykazují na rozdíl od předešlých genů autozomálně dominantní dědičnost.
Kombinace výskytu Wilmsova tumoru, nefrotického syndromu s rozvojem chronického renálního selhání a mužského pseudohermafroditismu se označuje jako Denys-Drashův syndrom nebo Frasierův syndrom. Dnes převládá názor, že u pacientů s mutací v tomto genu by měla být indikována oboustranná nefrektomie a zahájena dialyzační léčba s následnou transplantací. Na načasování doby nefrektomie jsou různé názory. Většinou je nefrektomie indikována až při zahájení chronické dialyzační léčby [13].
LAMB2 (Piersonův syndrom)
Jedná se o gen, jehož mutace způsobují Piersonův syndrom. Tento syndrom je charakterizován nefrotickým syndromem, těžkou mentální retardací a typickým postižením oka s mikrokorií [14]. Gen kóduje protein laminin β-2.
Nefrotický syndrom je velmi těžký, srovnatelný s finským typem nefrotického syndromu. Postižení většinou umírají v novorozeneckém nebo kojeneckém věku.
CD2AP (s CD2 asociovaný protein)
Jedná se o delší dobu známý protein, identifikovaný poprvé v T-lymfocytech. Později bylo zjištěno, že myši s vyřazeným genem pro CD2AP v heterozygotním stavu vyvinou renální léze histologicky srovnatelné s lidskou FSGS. V ledvinách CD2 asociovaný protein kódovaný stejnojmenným genem přímo interaguje s nefrinem a f-aktinem (obr. 1). Teprve v nedávné době byl objeven první pacient s homozygotní mutací v tomto genu, u kterého se vyvinul těžký nefrotický syndrom v dětském věku s obrazem FSGS [15].
ACTN4 (α-actinin 4)
Tento gen kóduje protein α-actinin 4, který je součástí aktinového cytoskeletu a váže se s f-aktinem (obr. 1). Aktinová cytoskeletální síť je nutná pro správnou prostorovou konfiguraci buněk a jejich pohyb. Správné prostorové uspořádání podocytů a jejich výběžků pravděpodobně umožňuje správnou funkci štěrbinové membrány.
Mutace v tomto genu způsobují nefrotický syndrom s histologickým obrazem FSGS, který se manifestuje v mladé dospělosti. Familiární formy mají autozomálně dominantní dědičnost a označují se jako familiární FSGS typ 1 [16].
TRPC6
Další gen zodpovědný za rozvoj nefrotického syndromu s obrazem FSGS kóduje kalciový kanál (obr. 1). Mutace způsobují vyšší aktivaci tohoto kanálu a zvýšený vstup kalcia do podocytů. Přesné vysvětlení, jak může mutace v tomto kanálu způsobit glomerulosklerózu, není zatím známo.
Onemocnění je označováno jako familiární FSGS typ 2. Manifestuje se podobně jako familiární FSGS typ 1 až v mladé dospělosti [17, 18].
Molekulárně genetická diagnostika
Indikace k molekulárně genetickému vyšetření dětí s NS
K molekulárně genetickému vyšetření jsou indikovány všechny děti s kortikorezistentním NS, a to bez ohledu na věk při manifestaci či histologický obraz. Molekulárně genetické vyšetření by mělo být provedeno před nebo současně s renální biopsií, neboť výsledek tohoto vyšetření má zásadní význam pro další léčbu dítěte se SRNS.
Vlastní zkušenost s molekulárně genetickou diagnostikou NS na naší klinice
Od října 2007 jsme na naší klinice molekulárně geneticky vyšetřili 15 dětí s kortikorezistentním nefrotickým syndromem z celé České republiky. Všichni pacienti byli molekulárně geneticky testováni v naší laboratoři přímým sekvenováním všech 8 exonů NPHS2 genu a exony 8 a 9 genu WT1 pomocí standardních metod [1, 8].
U 6 dětí ze 4 rodin jsme nalezli mutace v genu NPHS2 a u 3 pacientů byla nalezena mutace ve WT1 genu. U 6 pacientů jsme mutaci v těchto 2 genech neprokázali.
Perspektivy molekulárně genetické diagnostiky NS
S rozvojem metod molekulární diagnostiky se mnoho forem nefrotického syndromu označovaných dříve jako idiopatické daří etiologicky a patofyziologicky objasnit. Jelikož stále existují familiární formy, kde neznáme genetické pozadí, je zcela jasné, že existují další dosud neznámé geny, které způsobují či modifikují projevy nefrotického syndromu. Z nejnovějších výzkumů vyplývá, že nefrotický syndrom, obzvláště formy, které se manifestují v prvním roce života, jsou ve velké míře podmíněné geneticky a že nefrotický syndrom je zastřešujícím projevem mnoha monogenních chorob [8, 12].
V současné době známe většinou formy geneticky podmíněné, které nereagují na kortikoidy. Existují ovšem i rodiny, kde je značně vyšší výskyt nefrotického syndromu oproti normální populaci a NS je kortikosenzitivní. Objev genu PLCE1, kde je u některých pacientů prokázaná léčebná odpověď na kortikoidy, ukazuje, že pravděpodobně i určitá část steroidsenzitivních nefrotických syndromů (SSNS), které vykazují familiární výskyt, může být podmíněna mutacemi v zatím neznámých genech. Pátrá se po nových genech, jak pro SRNS, tak pro SSNS. Zatím bylo lokalizováno několik kandidátních lokusů, kde by se mohly nacházet geny pro steroidsenzitivní NS [19, 20].
Závěr
Steroidrezistentní nefrotický syndrom je choroba, která v dětském věku, obzvláště u dětí ve věku jednoho roku, vykazuje velmi vysoký podíl geneticky podmíněných forem. Je známo celkem 8 genů, které mohou vyvolat projevy SRNS. Molekulární diagnostika u pacientů se SRNS má navíc přímý význam terapeutický a prognostický. Většina SRNS geneticky podmíněných nereaguje ani na podávání imunosupresiv a lze tedy pacienty s těmito chorobami ušetřit vedlejších účinků těchto léků.
Molekulárně genetické vyšetření by tedy mělo být standardním vyšetřením, prováděným u všech dětí se SRNS. Naše klinika je schopna vyšetřit mutace v genech NPHS2 a WT1, které působí až třetinu případů SRNS. U ostatních syndromů máme zajištěnou spolupráci s českými či zahraničními genetickými pracovišti. Praktický význam má samozřejmě i prenatální diagnostika, pokud se již v rodině dříve narodilo postižené dítě, kde byla genetická odchylka prokázána.
Došlo: 28. 9. 2008
Přijato: 12. 12. 2008
MUDr. Michal Malina
Pediatrická klinika UK 2. LF
FN Motol
V Úvalu 84
150 06 Praha 5
e-mail: michal.malina@lfmotol.cuni.cz
Sources
1. Cho HY, et al. WT1 and NPHS2 mutations in Korean children with steroid-resistant nephrotic syndrome. Pediatr. Nephrol. 2008;23(1): 63–70.
2. Ehrich JH, et al. Steroid-resistant idiopathic childhood nephrosis: overdiagnosed and undertreated. Nephrol. Dial. Transplant. 2007;22(8): 2183–2193.
3. van den Berg JG, Weening JJ. Role of the immune system in the pathogenesis of idiopathic nephrotic syndrome. Clin. Sci. (Lond). 2004;107(2): 125–136.
4. Niaudet P. Genetic forms of nephrotic syndrome. Pediatr. Nephrol. 2004;19(12): 1313–1318.
5. Ruf RG, et al. Patients with mutations in NPHS2 (podocin) do not respond to standard steroid treatment of nephrotic syndrome. J. Am. Soc. Nephrol. 2004;15(3): 722–732.
6. Hodson EM, et al. Corticosteroid therapy for nephrotic syndrome in children. Cochrane Database Syst. Rev. 2004;(2): CD001533.
7. Mucha B, et al. Mutations in the Wilms’ tumor 1 gene cause isolated steroid resistant nephrotic syndrome and occur in exons 8 and 9. Pediatr. Res. 2006;59(2): 325–331.
8. Hinkes BG, et al. Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2). Pediatrics 2007;119(4): e907–e919.
9. Kestila M, et al. Positionally cloned gene for a novel glomerular protein – nephrin – is mutated in congenital nephrotic syndrome. Mol. Cell 1998;1(4): 575–582.
10. Hinkes B, et al. Specific podocin mutations correlate with age of onset in steroid-resistant nephrotic syndrome. J. Am. Soc. Nephrol. 2008;19(2): 365–371.
11. Gbadegesin R, et al. Mutations in PLCE1 are a major cause of isolated diffuse mesangial sclerosis (IDMS). Nephrol. Dial. Transplant. 2008;23(4): 1291–1297.
12. Hinkes BG. NPHS3: new clues for understanding idiopathic nephrotic syndrome. Pediatr. Nephrol. 2008; 23/6) 847–850.
13. Hu M, et al. Prophylactic bilateral nephrectomies in two paediatric patients with missense mutations in the WT1 gene. Nephrol. Dial. Transplant. 2004;19(1): 223–226.
14. Zenker M, et al. Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum. Mol. Genet. 2004;13(21): 2625–2632.
15. Lowik MM, et al. Focal segmental glomerulosclerosis in a patient homozygous for a CD2AP mutation. Kidney Int. 2007;72(10): 1198–1203.
16. Kaplan JM, et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet. 2000;24(3): 251–256.
17. Mukerji N, Damodaran TV, Winn MP. TRPC6 and FSGS: the latest TRP channelopathy. Biochim. Biophys. Acta 2007;1772(8): 859–868.
18. Winn MP, et al. Unexpected role of TRPC6 channel in familial nephrotic syndrome: does it have clinical implications? J. Am. Soc. Nephrol. 2006;17(2): 378–387.
19. Ruf RG, et al. Identification of the first gene locus (SSNS1) for steroid-sensitive nephrotic syndrome on chromosome 2p. J. Am. Soc. Nephrol. 2003;14(7): 1897–1900.
20. Fuchshuber A, et al. Clinical and genetic evaluation of familial steroid-responsive nephrotic syndrome in childhood. J. Am. Soc. Nephrol. 2001;12(2): 374–378.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2009 Issue 2
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Očkování proti tuberkulóze
- Geneticky podmíněné formy nefrotického syndromu u dětí
- Refluxní striktury jícnu u dětí – léčba a výsledky
- Monogenní hypertenze