
Monogenní hypertenze
Monogenic Hypertension
Arterial hypertension (AH) is a common disorder that affects a large patient population including children. Incidence of AH in childhood is above 1%. Essential hypertension is currently the most common cause of AH in children and is usually associated with overweight or obesity.
Among disorders of secondary hypertension is monogenic hypertension caused by mutation of a single gene. This mutation leads to hyperactivity of renal sodium and chloride transporters or mineralocorticoid receptors dysfunction or dysregulation of adrenal steroids synthesis. Increased rates of sodium and water reabsorption lead to plasma volume expansion and AH. Typically, laboratory findings in monogenic forms of hypertension include low plasma renin activity. Hormonal studies coupled with genetic testing can help in the early diagnosis of these disorders as for pediatrician.
Key words:
monogenic hypertension, aldosterone, plasma renin activity, childhood
Authors:
Z. Doležel; J. Štarha; F. Jimramovský; D. Dostálková
Authors‘ workplace:
II. dětská klinika LF MU a FN Brno
přednosta prof. MUDr. Z. Doležel, CSc.
Published in:
Čes-slov Pediat 2009; 64 (2): 89-94.
Category:
Review
Overview
Arteriální hypertenze (HT) je i u dětí závažným zdravotním problémem. Její incidence je obvykle uváděna kolem 1 %. Přibývá však sdělení, která dokládají vzestup primární hypertenze v populaci dětí, a to nejspíše v souvislosti s nárůstem počtů dětí s nadváhou nebo obézních.
Mezi příčinami sekundární hypertenze je skupina chorob označovaných jako monogenní hypertenze (MHT) nebo monogenní hypertenzní syndromy. Jde o choroby podmíněné mutací v jednom genu, která vede k ovlivněním renálního transportu sodíku a chloridů nebo pozměnění funkce mineralokortikoidních receptorů, nebo je porušena syntéza některých hormonů nadledvin. V konečném důsledku pak dochází ke zvýšení reabsorpce sodíku v tubulárním úseku nefronu, objemovému přetížení a HT. Jednotícím laboratorním nálezem chorob skupiny MHT je nízká plazmatická reninová aktivita. I přesto, že MHT jsou v dětském věku vzácné, je důležité, aby o nich byli informováni dětští lékaři, neboť molekulárně genetickými metodami lze tyto choroby přesvědčivě diagnostikovat.
Klíčová slova:
monogenní hypertenze, aldosteron, plazmatická reninová aktivita, dětský věk
Úvod
Arteriální hypertenze (HT) je v dětském věku jasně definována a odborné pediatrické veřejnosti jsou dostatečně známy jak základní principy měření krevního tlaku (TK), tak i příslušné percentilové grafy umožňující správnou interpretaci naměřených hodnot [1]. Literární zdroje opakovaně uvádějí, že HT patří k nejvýznamnějším rizikovým faktorům kardiovaskulárních chorob dospělé populace, zejména pak v industrializovaných zemích [2]. Současně však také uvádějí, že včasná diagnostika a léčba HT je nezbytná již v dětském věku, neboť jde o jedno z opatření, jehož prostřednictvím lze příznivě ovlivnit morbiditu a mortalitu v dospělosti. Podle některých údajů je HT bezprostředně na celém světě ohrožena asi jedna miliarda lidí [3].
U dětí je prevalence HT udávána kolem 1 %. Tento údaj však začíná být podle závěrů některých studií korigován a uváděna je prevalence 4–5 %. Je to především důsledek vzestupu počtu dětí s nadváhou a obézních [4, 5]. V populaci dětí tak pravděpodobně primární HT začíná významným způsobem dominovat. Objektivně je však třeba uvést, že rozsáhlejší studie, které by zjistily skutečnou prevalenci primární a sekundární HT u dětí, jsou ojedinělé.
Rozvoj molekulárně genetických metod umožnil detailnější poznání některých příčin sekundární HT, které jsou souborně označovány jako monogenní formy HT (MHT). Objasnění některých mechanismů vedoucích k HT u této skupiny chorob jsou velmi intenzivně analyzována, neboť nabízejí vysvětlení vzniku vzestupu TK i u HT esenciální. Přestože vlastní nozologické jednotky doposud známých MHT jsou v dětském věku méně časté, mohli by se dětští lékaři s těmito pacienty ve své praxi setkat. MHT jsou dědičné choroby podmíněné mutací v jednom genu – mutace vede ke změně příslušného genového produktu, který má např. charakter biologického nosiče nebo buněčného receptoru nebo se může jednat o změnu specifického enzymu [6, 7, 8]. Ve vztahu k údajům předchozí věty jsou onemocnění řazená do skupiny MHT charakterizována tím, že je u nich porušen buď transport elektrolytů v tubulárním úseku nefronu, nebo je poškozena tvorba či aktivita některého z hormonů s mineralokortikoidním účinkem. Konstantním laboratorním fenoménem MHT je snížená plazmatická koncentrace reninu vyjadřovaná jako plazmatická reninová aktivita (PRA); proto bývá v těchto případech uváděno i označení nízkoreninová HT.
Klinické jednotky MHT
V následujícím textu jsme se snažili MHT uvést pokud možno stručnou formou, neboť zejména pro běžnou klinickou praxi je detailní znalost patofyziologie méně významná a rozhodující je diferenciálně diagnosticky na tyto choroby pomýšlet. Současně jsme se snažili uvést i jiná označení/ /synonyma, která jsou u těchto nemocí používána. Vybranou charakteristiku MHT uvádí tabulka 1.
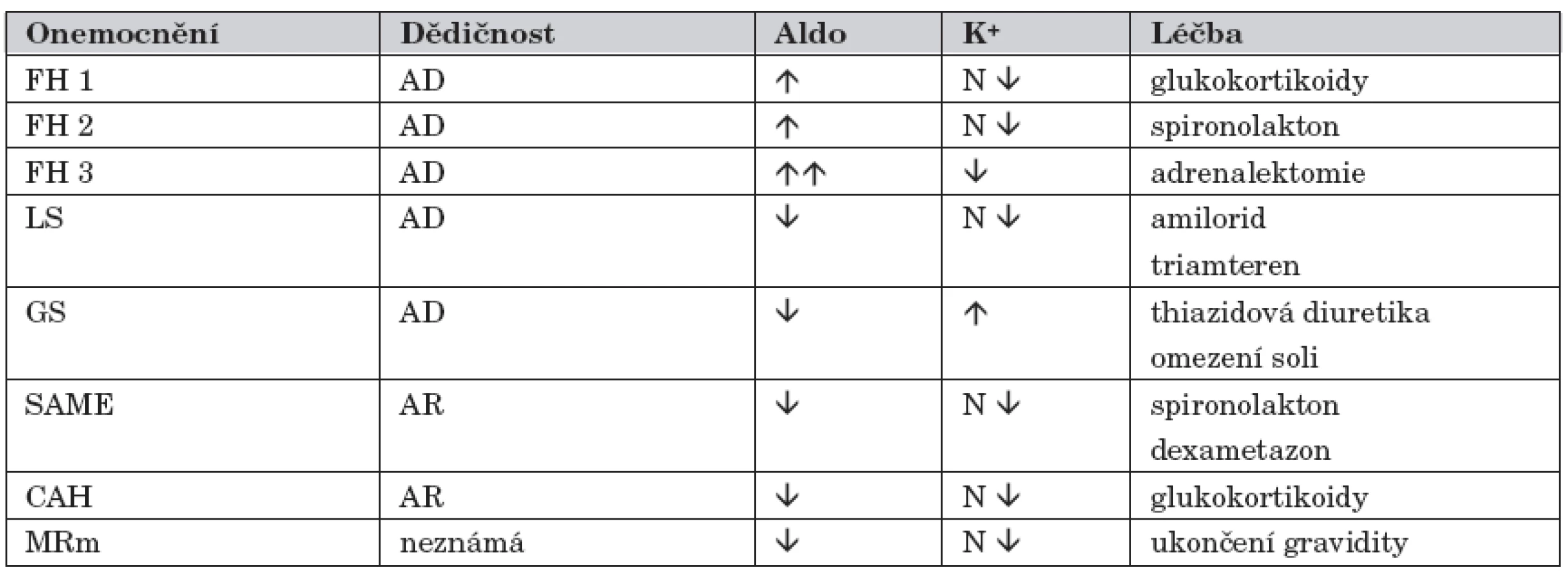
Familiární hyperaldosteronismus typ 1 (FH 1)
Onemocnění bývá v literatuře označováno také jako glucocorticoid-remediable aldosteronism. Za fyziologických podmínek je aldosteron (Aldo) produkován v zona glomerulosa nadledvinové kůry a je to nejvýznamnější mineralokortikoid. Biosyntéza Aldo je výsledkem kaskády enzymatických reakcí, v nichž se v konečné fázi uplatňuje především 11β-hydroxyláza (11β-OH), 18-hydroxyláza a 18-oxidáza. Tyto enzymy jsou kódovány genem CYP11B2. V zona fasciculata nadledvinové kůry přitom probíhají podobné reakce, z nichž na některých se také podílí 11β-OH, ale kódována genem CYP11B1. Oba uvedené geny jsou v těsné blízkosti na 8. chromozomu (8q24.3). A právě toto „genetické sousedství“ vytváří určitý předpoklad snadných mutací mezi CYP11B1 a CYP11B2. V organismu se receptory pro Aldo nacházejí především v distálním tubulu ledvin a ve sběrných kanálcích, byly však identifikovány také v srdci, mozku či hladké svalovině cév.
Podkladem FH 1 je rekombinace mezi geny CYP11B1 a CYP11B2. Tím na 8. chromozomu vzniká hybridní gen, který obsahuje sekvence CYP11B1 a CYP11B2. To je provázeno nejen produkcí Aldo, ale především tím, že současně v zona fasciculata dochází ke zvýšené tvorbě hybridních 18-hydroxy - a 18-oxo-steroidů, které mají mineralokortikoidní účinek [9, 10]. I za této situace ale zůstává tvorba Aldo pod kontrolou ACTH, ale syntéza hybridních steroidů pod tímto zpětnovazebním kontrolním mechanismem není. Na rozdíl od fyziologické situace nemá na syntézu uvedených působků žádný vliv ani sérová koncentrace draslíku, ani hladina angiotenzinu II.
K manifestaci FH 1 sice může docházet bez závislosti na věku, většina nemocných je však obvykle poprvé diagnostikována mezi 12.–14. rokem života. V rodinné anamnéze bývá často údaj o výskytu závažné HT u mladších osob, podobně jako údaje související s komplikacemi, které obvykle HT doprovází (např. neobjasněné srdeční selhání, předčasné cerebrovaskulární příhody, nejasné preeklampsie). Klinická symptomatologie může být nenápadná a rozvoj příznaků plíživý, neboť někteří nemocní v rodinách s výskytem FH 1 nemají výraznou HT a dokonce mohou dlouhou dobu mít hodnoty TK zcela normální. Hyperaldosteronismus provází objemové přetížení s HT (bolesti hlavy, tinitus, epistaxe, poruchy zrakové ostrosti). Hypokalémie (svalová slabost, parestézie, obstipace, srdeční dysrytmie) není konstantním laboratorním nálezem a bývá prokazatelná jen asi u poloviny nemocných, podobně jako metabolická alkalóza. Laboratorně je pro diagnózu významné stanovení vysokých odpadů Aldo v moči, vysoký poměr (>30) mezi sérovým Aldo a PRA a také zvýšená koncentrace 18-OH derivátů kortizolu v moči. Nezřídka bývají používány k upřesnění diagnostiky i další testy (infuze NaCl, test s captoprilem, separovaný odběr krve ze suprarenálních žil). Vyšetření nadledvin některou zobrazovací technikou není pro diagnostiku FH 1 rozhodující, někdy však může být prokazatelná mírná hyperplazie nadledvinové kůry. Naopak molekulárně genetické vyšetření je pro diagnózu nezbytné a určující.
Léčebně se u FH 1 podávají především glukokortikoidy, k nimž lze přidat spironolakton anebo amilorid. Tato farmakoterapie velmi příznivě ovlivní HT, hypokalémii a metabolickou alkalózu. Některá doporučení uvádějí, že u pacientů s prokázaným FH 1 se má od dosažení dospělosti v 5letých cyklech provádět mozková MR angiografie s cílem pátrat po aneuryzmatech. Tento postup však není běžně akceptován.
Familiární hyperaldosteronismus typ 2 (FH 2)
I české literární zdroje běžně uvádějí anglickou mutaci FH 2, tj. nonglucocorticoid-remediable aldosteronism. Onemocnění nebylo dříve jasně vymezeno a nemocní byli zařazeni ve skupině pacientů s primárním hyperaldosteronismem. Větší pozornosti se této skupině nemocných dostalo až s pozorováním, že zvýšenou produkci Aldo se u nich nepodařilo na rozdíl od primárního hyperaldosteronismu snížit podáváním dexametazonu. Děti s FH 2 jsou v naprosté většině případů normotenzní, k rozvoji HT dochází až v dospělosti. Přes pokrok genetických znalostí u FH 1 je však jasná genetická abnormalita u formy FH 2 doposud nejasná. Původní představa, že FH 2 je podmíněna mutací genu CYP11B2, byla opuštěna. Toto podporují některá ojedinělá sdělení, která „chybný gen“ pro FH 2 nalezli na chromozomu 7p22 [11, 12]. Doposud však není k dispozici žádná molekulárně genetická analýza, která by byla rozhodující pro diagnostiku FH 2. Svůj diagnostický význam si tak stále udržuje hodnocení reakce při testu s dexametazonem, průkaz vysokého poměru sérového Aldo k PRA, podobně jako to, že je uváděn častý nález oboustranné adrenální hyperplazie nebo sekrečně aktivní adenom.
Samozřejmostí pro diagnózu FH 2 je vyloučit přítomnost hybridního genu, který je podkladem formy FH 1. Pro klinickou praxi je rozlišení obou forem FH nezbytné, neboť léčba je zcela rozdílná – v terapii FH 2 se používají pouze antagonisté Aldo.
Familiární hyperaldosteronismus typ 3 (FH 3)
Jde o zcela novou variantu [13], u které jen některé parametry odpovídají FH 1, ale není žádný efekt po podávání glukokortikoidů a především není přítomen chimerický gen se sekvencemi CYP11B1 a CYP11B2. U postižených jedinců dominuje závažná HT již v dětském věku s časným rozvojem orgánových změn (zejména hypertrofie levé srdeční komory) a také výrazná hyperplazie nadledvin. Sérová koncentrace Aldo je velmi vysoká, PRA snížená a konstantní je hypokalémie.
I velmi agresivní antihypertenzivní léčba je neúčinná, podobně žádný efekt nemá podávání spironolaktonu či amiloridu. Léčebně je u těchto nemocných nezbytná oboustranná adrenalektomie.
Liddleův syndrom (LS)
Podkladem choroby je mutace β nebo γ podjednotky amilorid senzitivního sodíkového kanálu (ENaC; kódující gen pro ENaC je na chromozomu 16p) ve sběrných kanálcích ledvin. Mutací způsobená hyperaktivita ENaC vede ke zvýšené reabsorpci sodíku. To má za následek objemové přetížení a rozvoj HT. Zvýšená reabsorpce sodíku stimuluje sekreci draslíku a vodíkových iontů do tubulárního lumen s rozvojem hypokalémie a metabolické alkalózy.
Onemocnění se může manifestovat již u kojenců, častěji však až ve věku školním či adolescenci. Pro nemocné s LS je typická HT, hypokalémie, metabolická alkalóza a měnlivá hypokalcémie [14]. Déletrvající hypokalémie může vést k poruše koncentrační schopnosti ledvin a u nemocných se tak manifestuje polyurie a polydipsie. Sérová koncentrace Aldo je snížena, podobně jako PRA. Přesvědčivý diagnostický závěr LS se opírá o vyšetření metodami molekulární biologie.
Literárně je uváděna značná variabilita v tíži HT a hypokalémie mezi jednotlivými nemocnými, což podporuje domněnku, že LS je nedostatečně diagnostikován a mnozí nemocní se skrývají ve skupině pacientů s esenciální HT [15]. Typické pro nemocné s LS je, že pokud je u nich z diagnostických rozpaků podáván spironolakton nebo glukokortikoidy, je tento postup bez odezvy. Základem léčby LS je podávání amiloridu či triamterenu; u některých pacientů je nezbytná ještě substituce draslíkem a omezení příjmu sodíku. Prognóza LS je příznivá.
Gordonův syndrom (GS)
Manifestace v dětském věku je vzácná, většina případů bývá diagnostikována mezi 20.–30. rokem života. Přesný výskyt choroby však není znám. Onemocnění bývá literárně uváděno pod názvy pseudohypoaldosteronismus typ 2 nebo familiární hypertenze s hyperkalémií.
S ohledem na delikátní procesy homeostázy draslíku (K+) ovlivňované nepoškozenou funkcí ledvin uvádíme stručnou formou některé poznámky, které jsou pro pochopení patofyziologie GS, ale i jiných tubulopatií nezbytné. Renální exkrece a reabsorpce K+ jsou ovlivňovány činností transportních systémů, které se nacházejí v tubulárním úseku nefronu a jsou schopny reagovat na řadu měnících se podmínek/potřeb organismu. Za fyziologických okolností proniká K+ glomerulární filtrací do ultrafiltrátu a je téměř z 90 % resorbován v tubulárním systému. V proximálním tubulu se resorpce děje převážně pasivně mezibuněčnými štěrbinami. V tlusté části ascendentního raménka Henleovy kličky (HEK) je K+ transportován kontransportním systémem Na+ K+ 2Cl–. Tento systém, podobně jako jiné, je membránový protein a nese označení NKCC2. Funkci NKCC2 významně ovlivňuje kaliový kanál označovaný ROMK (rat outer medullary K+ channel). ROMK se nachází ve stejné části HEK jako NKCC2, dále však také v kortikální části sběrného kanálku. ROMK za fyziologických podmínek zajišťuje recyklaci K+ z nitra buněk do lumen v oblasti HEK a v distálním tubulu. Současně je aktivita NKCC2 ovlivňována velikostí nitrobuněčné koncentrace chloridových aniontů (Cl–). Tato koncentrace závisí na intenzitě transportu Cl– chloridovým kanálem, který se nachází na bazolaterální membráně tubulárních buněk HEK, v distálním tubulu a v kortikální části sběrných kanálků; příslušný membránový protein chloridového kanálu nese označení CLC-Kb. V distálním tubulu je transport K+ ovlivňován kontransportem K+/Cl–, dále však opět průnikem přes ROMK. V distálním tubulu se dále významně funkčně uplatňuje i kotransport Na+/Cl– (membránový protein NCCT). Všechny z uvedených transportních a kotransportních systémů jsou geneticky determinovány a je známa i řada jejich mutací.
GS je důsledkem mutace dvou kináz s označením WNK 1 a WNK 4. Za fyziologických podmínek WNK 4 inhibuje v ledvinách iontový kanál NCCT (thiazid senzitivní kotransport Na+/Cl–). Tím dochází k tomu, že je potlačena zpětná reabsorpce Na+ a Cl– a ty mohou být vyloučeny močí. Mutovaná WNK 4 tedy nijak neovlivňuje kanál NCCT, což vede ke zvýšení zpětné resorpce Na+ a Cl– s následným objemovým přetížením, vzestupem TK a k poklesu PRA. Mutovaná WNK 4 však v ledvinách současně potlačuje činnost draslíkového kanálu ROMK. Jeho inhibicí tak snadno dochází k rozvoji hyperkalémie. Doposud nejsou u GS zcela přesvědčivě poznány mechanismy, které vznikají mutací kinázy WNK 1. Pravděpodobně tato mutovaná WNK 1 „spolupracuje“ prozatím ne zcela objasněným mechanismem s WNK 4, ovlivňuje však vedle kanálu NCCT i sodíkový kanál ENaC. V konečném důsledku tedy i WNK 1 zvyšuje zpětnou resorpci sodíku, to je provázeno objemovým přetížením a vznikem HT [16, 17]. Určitá rozdílnost mutovaných forem WNK 1 a WNK 4 se projevuje v tom, že u pacientů s mutací WNK 4 je konstantně přítomna hyperkalciurie, u mutace WNK 1 tento laboratorní fenomén chybí. Genetické analýzy u pacientů s GS prokázaly lokus na chromozomu 12, ale i 1 a 17. V současné době jsou velmi intenzivně studovány další možné mechanismy působení obou kináz, neboť jejich polymorfismus se pravděpodobně významně podílí na rozvoji esenciální HT.
Klinicky nemocné s GS charakterizuje malý vzrůst, svalová slabost, abnormality zubů a porucha intelektu; HT je obvykle přítomna ve druhé či třetí dekádě života. Laboratorními abnormalitami jsou hyperkalémie, hyperchlomerická metabolická acidóza, nízká sérová hladina Aldo i PRA. Funkce ledvin je intaktní. V léčbě se používají thiazidová diuretika a omezení příjmu soli.
Syndrom zdánlivé nadprodukce/nadbytku mineralokortikoidů
Není žádnou výjimkou českých odborných sdělení, že při popisu tohoto syndromu běžně používají anglickou mutaci, tj. syndrom of apparent mineralocorticoid excess (SAME). Za normálních okolností Aldo a kortizol svého mineralokortikoidního účinku dosahují v ledvinách vazbou s příslušným receptorem, který je v cytoplazmě buněk kortikálního úseku sběrných kanálků ledvin. Po vazbě s receptorem dosahuje konečného efektu Aldo působením přes kanál ENaC a kortizol přes kanál ROMK. SAME je podmíněn mutací genu HSD11B2 (je na chromozomu 16), který kóduje enzym 11β-OH steroid dehydrogenázu typ 2 (11β-OHSD2). Tento enzym v cytoplazmě buněk sběrných kanálků ledvin katalyzuje přeměnu kortizolu na kortizon a v přeneseném smyslu slova tak chrání mineralokortikoidní receptor před jeho vystupňovanou aktivací. Kortizon není schopen vazby na příslušný receptor. V důsledku mutace genu HSD11B2 a nedostatku či chybění 11β-OHSD2 proto dochází k vzestupu koncentrace cytoplazmatického kortizolu a tím vyšší aktivaci mineralokortikoidního receptoru.
Manifestace SAME je obvyklá již v dětském věku, dominuje hypokalémie, metabolická alkalóza, nízký sérový Aldo a nízká PRA. V moči je typický nález zvýšeného poměru volný kortizol : volný kortizon, resp. poměru metabolitů obou těchto hormonů. SAME může být diagnostikován u novozenců anebo kojenců – v popsaných případech dominovaly u dětí nižší porodní hmotnost, polyurie, polydipsie, nefrokalcinóza a neprospívání. Molekulárně genetické vyšetření je nezbytné, vždy je vhodné vyšetřit i jiné rodinné příslušníky, neboť jsou možné heterozygotní mutace HSD11B2 (heterozygoti mají obvykle mírnější projevy choroby a zvýšení TK není často výrazné) [18].
V terapii SAME se používá spironolakton, někdy je nezbytná suplementace draslíku a omezení přívodu sodíku. Bývá také používán dexametazon, ale je třeba určité opatrnosti, neboť může již přítomnou HT udržovat.
Vedle výše uvedené formy SAME existuje i kongenitální varianta označovaná někdy SAME typ 2, u níž je pravděpodobně vlivem mutace HSD11B2 inaktivní enzym 5β-reduktáza. Tento enzym normálně konvertuje kortizol na neúčinný tetrahydrokortizol. Klinické projevy choroby jsou méně vyjádřeny než u SAME, chybí hypokalémie i metabolická alkalóza.
Pro zajímavost uvádíme, že existují také získané varianty SAME, jejichž klinická a laboratorní podoba s vrozenými formami je identická (tj. přítomnost HT, hypokalémie, metabolické alkalózy) a diferenciální diagnostika proto může být obtížná. Získaný SAME vzniká např. při ektopické produkci ACTH, při nadměrném užívání lékořice (v potravinách a pochutinách nebo při pravidelném žvýkání tabáku). Hypersekrece ACTH vede k nadbytku kortizolu, tím je „přetížena“ kapacita 11β-OHSD2 a tak je umocněna aktivace receptoru pro mineralokortikoidy. Lékořice a tabák obsahují kyselinu glycyrrhetinovou, která je inhibitorem 11β-OHSD2.
Vrozená adrenální hyperplazie (CAH)
Pouze dvě formy CAH charakterizuje přítomnost HT, a to deficit 11β-OH (kódující gen CYP11B1) a 17α-hydroxylázy (17α-OH; kódující gen CYP17). Obě enzymopatie provází zvýšená produkce deoxykortikosteronu (DOC), který má výrazný mineralokortikoidní účinek a současně je snížena syntéza kortizolu. Zpětnou vazbou se za této situace zvyšuje sekrece ACTH, což má za následek další vzestup DOC. K projevům zřetelné a život ohrožující hypofunkce nadledvin ale nedochází [19]. V případě nedostatku 11β-OH mají postižení jedinci známky ženského pseudohermafroditismu, u dívek dochází k virilizaci, u chlapců k rozvoji předčasné puberty; laboratorně dominuje hypokalémie, sérový Aldo je v mezích normy, PRA snížena. Základem léčby je podávání glukokortikoidů, plastika genitálu vyžaduje spolupráci dětského endokrinologa, gynekologa a chirurga. Při nedostatku 17α-OH jsou u chlapců projevy mužského pseudohermafroditismu nebo známky nekompletní virilizace s genotypickými projevy ženského pohlaví [20]. U dívek je významné opoždění sexuální maturace. Deficit 17α-OH provází hypokalémie a snížení sérového Aldo i PRA. Léčebně je nezbytné především podávání glukokortikoidů, aplikace dalších hormonů (estrogeny/androgeny) patří do péče dětského endokrinologa. Tento odborník také spolupracuje s dalšími při chirurgické úpravě genitálu.
Mutace mineralokortikoidního receptoru (MRm)
Jak již bylo uvedeno v předchozím textu, nachází se receptor pro mineralokortikoidy (MR) intracelulárně v buňkách kortikální části sběrných kanálků ledvin a za fyziologických podmínek má afinitu především pro Aldo a kortizol. U žen, které jsou nositelkami mutace MRm (kodon S810L), nejsou žádné její fenotypické projevy. Teprve v těhotenství, kdy se významně zvyšují hladiny progesteronu (ale i jiných steroidů), dochází k iniciaci mutace a následné aktivaci MR. Sérový Aldo i PRA jsou sníženy, koncentrace draslíku v séru je normální nebo snížená. Klinickým korelátem je rozvoj velmi závažné HT a stav tak značně napodobuje jiné příčiny pre-/eklampsie. Na rozdíl od nich však při MRm chybí edémy, proteinurie a případně neurologická symptomatologie [21, 22].
Farmakoterapie HT je u těchto žen neúčinná, prakticky vždy je nezbytné ukončení gravidity. Zda je MRm vázána jen na některé rasy, nelze jednoznačně říci, podobně jako to, že lze předpokládat i jiné prozatím neurčené MRm [23].
Závěr
U doposud známých onemocnění řazených do skupiny MHT není přesně známa jejich incidence, stále patří mezi choroby málo frekventní/vzácné. Rozvoj molekulárně biologických metod však přispěl k poznání některých mechanismů, které vedou k rozvoji arteriální hypertenze u těchto onemocnění, současně se však toto poznání stalo impulzem pro výzkum patofyziologie esenciální HT. Hypoteticky tak lze očekávat i nalezení nových léků s antihypertenzním účinkem.
I přes skutečnost, že u MHT dochází k rozvoji HT spíše až v adolescenci nebo v dospělém věku, mohou k včasné diagnóze MHT přispívat také dětští lékaři. Mohou to být právě oni, kdo arteriální hypertenzi u dítěte zjistí anebo na základě pečlivých anamnestických údajů mohou napomoci k odhalení některých dospělých příslušníků, u nichž přesvědčivý diagnostický závěr jejich HT nebyl učiněn.
Došlo: 8. 10. 2008
Přijato: 9. 12. 2008
Prof. MUDr. Zdeněk Doležel, CSc.
II. dětská klinika LF MU a FN Brno
Černopolní 9
625 00 Brno
e-mail: zdoleze@fnbrno.cz
Sources
1. Seeman T, Dušek J, Janda J. Arteriální hypertenze v dětském věku. Čes.-slov. Pediat. 2003;58(9): 566–578.
2. O’Shaugnessy KM, Karet FE. Salt handling and hypertension. Annu Rev. Nutr. 2006;26 : 343–365.
3. Rowan S, Adrogue H, Mathur A, et al. Pediatric hypertension: a review for the primary care provider. Clin. Pediatr. 2005;44 : 289–296.
4. Lisá L, Kytnarová J, Stožický F, et al. Doporučený postup prevence a léčby dětské obezity. Čes.-slov. Pediat. 2008;63(9): 501–507.
5. Mráz M, Spurný P, Pytliak M, et al. Endotelová dysfunkcia. Čes.-slov. Pediat. 2004;59(6): 311–315.
6. Scott SW. Advances in genetic hypertension. Curr. Opin. Pediatr. 2007;19 : 192–198.
7. New MI, Geller DS, Fallo F, et al. Monogenic low renin hypertension. Trends Endocrinol. Metab. 2005;16 : 92–97.
8. Tobin MD, Tomaszewski M, Braund PS, et al. Common variants in genes underlying monogenic hypertension and hypotension and blood pressure in the general population. Hypertension 2008;51 : 1658–1664.
9. Mosso L, et al. Primary aldosteronism and hypertensive disease. Hypertension 2003;42 : 161–165.
10. Mulatero P, Morello F, Veglio F. Genetics of primary aldosteronism. Trends Endocrinol. Metab. 2004;22 : 663–670.
11. Lafferty AR, Torpy DJ, Stowasser M, et al. A novel genetic locus for low renin hypertension: familial hyperaldosteronism type II maps to chromosome 7 (7p22). J. Med. Genet. 2000;37 : 831–835.
12. New MI, et al. Monogenic low renin hypertension. Trends Endocrinol. Metab. 2005;16 : 92–97.
13. Mulatero P. A new form of hereditary primary aldosteronism: familial hyperaldosteronism type III. J. Clin. Endocrinol. Metab. 2008;93(8): 2972–2974.
14. Štarha J, Doležel Z, Dostálková D. Liddleův syndrom (LS) – neobvyklá příčina arteriální hypertenze. Čes.-slov. Pediat. 2000;55(4): 244–247.
15. Ciechanowitz A, Doležel Z, Placha G, et al. Liddle syndrome caused by P616R mutation of the epithelial sodium channel beta subunit. Pediatr. Nephrol. 2005;20(6): 837–838.
16. Kahle KT, et al. WNK4 regulates the balance between renal NaCl reabsorption and K+ secretion. Nat. Genet. 2003;88 : 372–376.
17. Xie J, Craig L, Coby MH, et al. Role of with-no-lysine [K] kinases in the pathogenesis of Gordon’s syndrome. Pediatr. Nephrol. 2006;21(9): 1231–1236.
18. Mullins LJ, Bailey MA, Mullins JJ. Hypertension, kidney, and transgenics: a fresh perspective. Physiol. Rev. 2006;86 : 709–746.
19. Luft FC. Mendelian forms of human hypertension and mechanisms of disease. Clin. Med. Res. 2003;1(4): 291–300.
20. Šumník Z, Koloušková S, Šnajderová M. Neobvyklá příčina primární amenorey. In Lebl J, Šnajderová M, Novotná D. Kazuistiky z dětské endokrinologie. Praha: Galén, 2001 : 75–76.
21. Geller DS, Farhi A, Pinkerton N, et al. Activating mineralocorticoid receptor mutation in hypertension exacerbated by pregnancy. Science 2000;289(7): 119–123.
22. Rafestin-Oblin M-E, Souque A, Bocchi B, et al. The severe form of hypertension caused by the activating S810L mutation in the mineralocorticoid receptor is cortisone related. Endocrinology 2003;144 : 528–533.
23. Schmider-Ross A, Wirsing M, Büscher U, et al. Analysis of the S810L point mutation of the mineralocorticoid receptor in patiens with pregnancy-induced hypertension. Hypertens. Pregnancy 2004;23(1): 113–119.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2009 Issue 2
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Očkování proti tuberkulóze
- Geneticky podmíněné formy nefrotického syndromu u dětí
- Refluxní striktury jícnu u dětí – léčba a výsledky
- Monogenní hypertenze