
Molekulová diagnostika dedičných nekonjugovaných hyperbilirubinémií na Slovensku
Molecular Diagnostics of Hereditary Unconjugated Hyperbilirubinemias in Slovakia
Known forms of hereditary unconjugated hyperbilirubinemias are symptomatically and prognostically diverse defects of bilirubin metabolism caused by various alterations of the UGT1 gene, which codes for the enzyme uridine diphosphoglucuronosyl transferase. The aim of the present work was to determine the spectrum of molecular alterations that contribute to their development in Slovak population.
Patients and methods:
Presence of insertion A(TA)7TAA and substitution T-3279G in the regulating areas of the UGT1A1 gene was determined in DNA samples from 110 unrelated subjects with clinically diagnosed Gilbert’s syndrome and from 240 clinically asymptomatic Slovak control subjects of both Gypsy and Non-Gypsy origin (120 and 120 subjects). In addition to this, mutation analysis of the UGT1A1 gene was accomplished in two unrelated Slovak kindreds with clinically diagnosed Crigler-Najjar syndrome type I.
Results:
101 of 110 (91.8%) patients with Gilbert’s syndrome were homozygotes for both the insertional A(TA)7TAA as well as the substitutional T-3279G polymorphism detected in the UGT1A1 gene (allelic frequency 0.95). The allelic frequency of the A(TA)7TAA polymorphism was identical (q = 0.36 and p = 0.64) in control subjects of both Gypsy and Non-Gypsy origin that corresponds in this population to an expected frequency of homozygotes for A(TA)7TAA insertion of 12.9 %. In one of the two unrelated Slovak kindreds with Crigler-Najjar syndrome type I we found a homozygous deletion 1220delA in exon 4 of the UGT1A1 gene (kindred A). Affected subjects from kindred B carried two different deletions (717-718delAG in exon 1 and 1220delA in exon 4) in gene UGT1A1.
Conclusion:
Available molecular analysis of the UGT1A1 gene has an important role for exact clinical diagnosis of Gilbert’s syndrome and contributed to prenatal diagnosis of Criegler-Najjar syndrome in affected kindreds.
Key words:
hereditary unconjugated hyperbilirubinemias, Gilbert’s syndrome, Criegler-Najjar syndrome, genetics
:
I. Zmetáková; I. Čierna; D. Székyová; G. Minárik; A. Ficek; H. Poláková; V. Ferák; E. Feráková; Ľ. Kádaši; L. Kovács
:
2. detská klinika Lekárskej fakulty Univerzity Komenského a Detskej fakultnej nemocnice, Bratislava
prednosta prof. MUDr. L. Kovács, DrSc., MPH
; Katedra molekulovej biológie Prírodovedeckej fakulty UK v Bratislave a Ústav molekulárnej fyziológie a genetiky SAV, Bratislava
vedúci doc. RNDr. Ľ. Kádaši, DrSc.
:
Čes-slov Pediat 2009; 64 (5): 223-229.
:
Original Papers
Známe formy dedičnej nekonjugovanej hyperbilirubinémie sú geneticky príbuzné, ale symptomatologicky a prognosticky značne odlišné poruchy metabolizmu bilirubínu. Vznikajú následkom alterácií génu UGT1A1 kódujúceho enzým bilirubín UDP-glukuronyltransferázy (B-UGT). Cieľom danej práce bolo určiť spektrum genetických zmien, ktoré sa podieľajú na ich vzniku.
Pacienti a metódy:
Autori analyzovali vzorky DNA od 110 nepríbuzných pacientov s Gilbertovým syndrómom a od kontrolnej skupiny 240 klinicky asymptomatických dobrovoľníkov nerómskeho a rómskeho pôvodu (120 a 120 osôb) na prítomnosť promótorovej inzercie A(TA)7TAA a substitúcie T-3279G v regulačnej oblasti génu UGT1A1. Okrem toho v dvoch nepríbuzných slovenských rodinách s Criglerovým-Najjarovým syndrómom typ I sa uskutočnila aj mutačná analýza génu UGT1A1.
Výsledky:
Inzercia A(TA)7TAA a substitúcia T-3279G bola prítomná v homozygotnom stave u 101 zo 110 (91,8 %) pacientov s klinicky diagnostikovaným Gilbertovým syndrómom (alelová frekvencia 0,95). V kontrolných skupinách osôb rómskeho resp. nerómskeho pôvodu sa zistila rovnaká alelová frekvencia inzercie A(TA)7TAA (q = 0,36 a p = 0,64), čomu zodpovedá aj rovnaká, 12,9% očakávaná frekvencia homozygotov pre inzerciu (A(TA)7TAA. V dvoch nepríbuzných slovenských rodinách s Criglerovým-Najjarovým syndrómom typ I sa identifikovala delécia 1220delA v 4. exóne génu UGT1A1 v homozygotnom stave (rodina A) resp. sa našli dve rôzne delécie v géne UGT1A1, a to delécia 717-718delAG v 1. exóne a delécia 1220delA v 4. exóne génu UGT1A1 (rodina B).
Záver:
Dostupná molekulová analýza génu UGT1A1 umožňuje exaktnú klinickú diagnostiku Gilbertovho syndrómu a je prínosom pre prenatálnu diagnostiku Crieglerovho-Najjarovho syndrómu v postihnutých rodinách.
Kľúčové slová:
hereditárne nekonjugované hyperbilirubinémie, Gilbertov syndróm, Crieglerov-Najjarov syndróm, genetika
Úvod
Dedičné nekonjugované hyperbilirubinémie (Gilbertov syndróm resp. Criglerov-Najjarov syndróm typ I a typ II) reprezentujú rôzne stupne redukcie aktivity enzýmu bilirubín UDP-glukuronyltransferázy (B-UGT), ktorý je u človeka zodpovedný za glukuronizáciu bilirubínu. Ich genetická príbuznosť vyplýva zo skutočnosti, že sú následkom alterácii génu UGT1A1 kódujúceho B-UGT.
Najčastejší typ dedičných nekonjugovaných hyperbilirubinémií, Gilbertov syndróm („juvenilná žltačka“) v podstate nie je choroba, ale skôr benígna odchýlka v regulácii normálnej expresie génu UGT1A1. Vyskytuje sa u 9–13 % kaukazoidnej populácie a charakterizuje sa chronickou miernou hyperbilirubinémiou (60–120 µmol/l) bez prítomnosti bilirubínu v moči, bez známok hemolýzy, resp. postihnutia pečene a žlčových ciest [12, 30]. Najčastejšie sa diagnostikuje v druhej a tretej dekáde života (zriedkavo môže byť odhalený už u novorodencov a batoliat) a intermitentne pretrváva celý život [15]. Frekvencia Gilbertovho genotypu je u oboch pohlaví rovnaká, ale klinicky sa štyrikrát častejšie manifestuje u mužov, zrejme vplyvom mužských pohlavných hormónov (testosterónu) na zníženie aktivity enzýmu B-UGT. Sérová koncentrácia bilirubínu typicky kolíše v závislosti od hladovania, psychickej (napr. študenti v období skúšok) a fyzickej záťaže, interkurentnej infekcie, operácií, úrazov, excesu alkoholu a u žien v predmenštruačnom období. V minulosti používané záťažové testy (napr. hladový test) zaťažujú pacienta a sú veľmi nešpecifické, lebo k vzostupu môže dôjsť u pacientov s chorobami pečene ale aj u zdravých jednotlivcov.
Criglerov-Najjarov syndróm označuje podstatne zriedkavejšiu, no klinicky o to závažnejšiu formu dedičnej nekonjugovanej hyperbilirubinémie [10]. Arias a spol. v roku 1967 [2] navrhli rozlíšiť dva typy syndrómu v závislosti od závažnosti klinického priebehu ako aj od miery zníženia detoxikačnej aktivity enzýmu B-UGT. Criglerov-Najjarov syndróm typ I (CN I) je zriedkavé ochorenie s frekvenciou 1 : 106 po celom svete. Charakterizuje sa chýbaním glukoronidu bilirubínu v žlči a vývojom jadrového ikteru z extrémne vysokých koncentrácii nekonjugovaného bilirubínu v sére už od novorodeneckého veku (310–755 µmol/l), ktoré nereagujú na terapiu fenobarbitálom. Criglerov-Najjarov syndróm typ II (CN II) má miernejší priebeh s koncentráciami sérového bilirubínu v rozmedzí od 120 do 310 µmol/l, ktoré sa dajú terapeuticky modifikovať liečbou fenobarbitálom [8, 30].
Problematika dedičných nekonjugovaných hyperbilirubinémií je na Slovensku málo preštudovaná a doteraz nebola podrobená výskumu založenom na analýze DNA. Našim cieľom bolo preto prispieť k poznaniu problematiky vzťahu medzi fenotypom a genotypom u troch foriem dedičnej nekonjugovanej hyperbilirubinémie, určiť spektrum genetických alterácií, ktoré sa podieľajú na ich vzniku.
Pacienti a metódy
Vyšetrenia sa uskutočnili u pacientov s rôznymi formami hereditárnej nekonjugovanej hyperbilirubinémie a tiež v kontrolnej skupine dobrovoľníkov.
Gilbertov syndróm
Na 110 nepríbuzných pacientov vo veku 12–18 rokov (priemerný vek 15,6 ± 1,2 roky, 18 dievčat, 92 chlapcov) bolo poukázaných pre klinický údaj miernej žltačky. Klinická diagnóza Gilbertovho syndrómu sa opierala o dobu manifestácie miernej nekonjugovanej hyperbilirubinémie (60 až 120 µmol/l) bez iných klinických príznakov, s normálnymi hodnotami pečeňových aminotransferáz a chýbaní laboratórnych príznakov hemolýzy. Po vylúčení ostatných príčin nekonjugovanej hyperbilirubinémie sa od každého jednotlivca odobrali 2 ml krvi na analýzu DNA na prítomnosť promótorovej inzercie (A(TA)7TAA) a enhancerovej (T-3279G) mutácie.
Kontrolnú skupinu tvorilo 240 klinicky asymptomatických dobrovoľníkov nerómskeho aj rómskeho pôvodu (120 a 120 osôb). Od každého z nich sa odobrala vzorka krvi na zisťovanie zastúpenia promótorovej inzercie (A(TA)7TAA) a mutácie T-3279G v celkovej slovenskej populácii.
Criglerov-Najjarov syndróm
Vyšetrili sme vzorky DNA od viacerých jednotlivcov z dvoch rodín s výskytom Criglerovho-Najjarovho syndrómu. Criglerov-Najjarov syndróm typ I sa diagnostikoval u pacientov s extrémne vysokými koncentráciami bilirubínu v sére (310 až 755 µmol/l) od novorodeneckého veku, ktoré nereagovali na fototerapiu. Criglerov-Najjarov syndróm typ II sa určil u pacientov s koncentráciami sérového bilirubínu v rozmedzí od 120 do 310 µmol/l, ktoré pod vplyvom fototerapie poklesli.
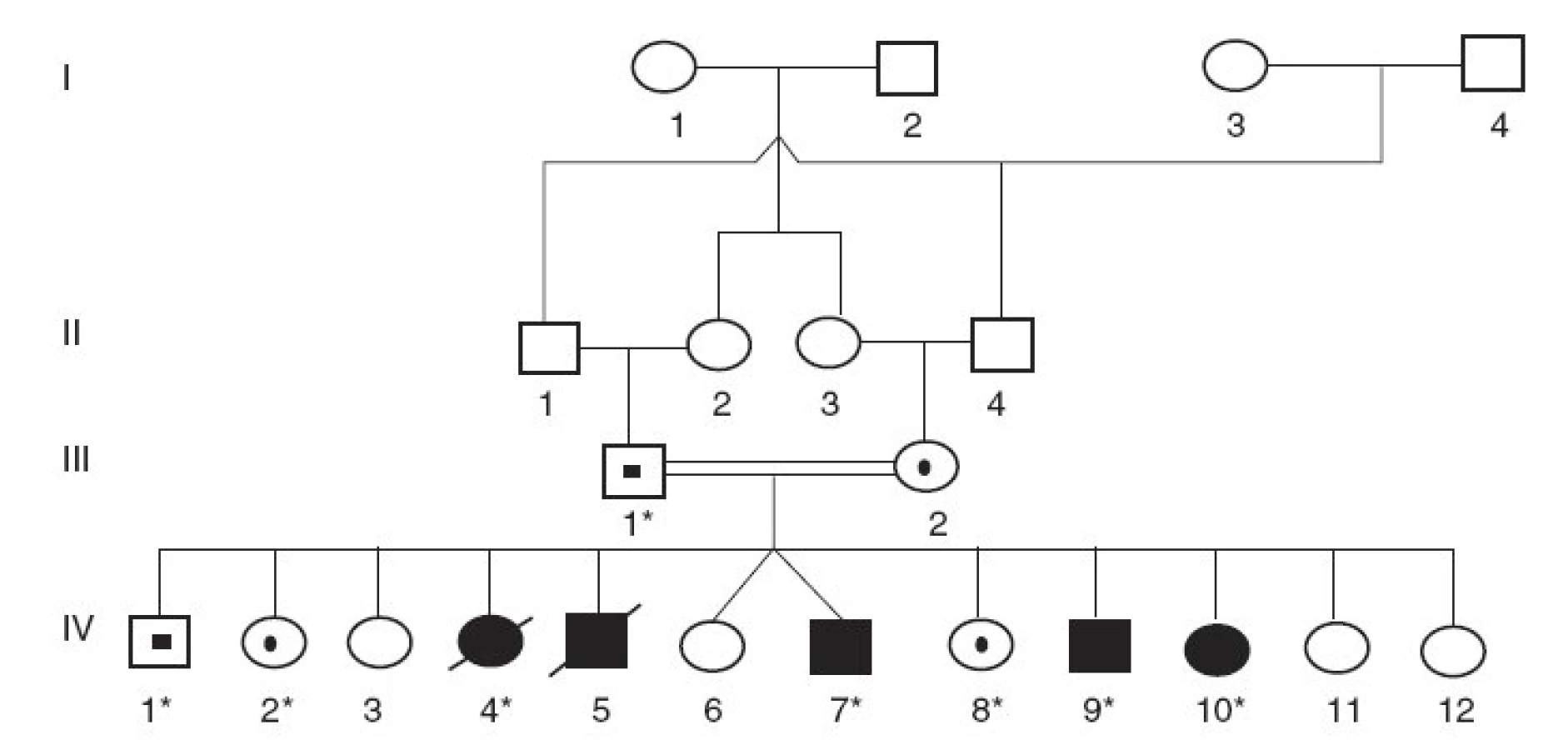
V prvej konsangvinnej rodine rómskeho pôvodu (rodina A) dokázala analýza rodokmeňa genetickú príbuznosť rodičov (dvojnásobný bratranec a sesternica, obr. 1). Z ich 12 potomkov vo veku 5 až 17 rokov je päť postihnutých, z ktorých dve deti už exitovali. U všetkých 5 postihnutých detí sa prejavila závažná žltačka už na 2. deň po narodení a extrémna nekonjugovaná hyperbilirubinémia pretrvávala aj napriek fenobarbitálovej terapii. Aj pri 12-hodinovej fototerapii denne sa dosiahla iba mierna redukcia koncentrácie nekonjugovaného bilirubínu v sére, ktorá sa pri tejto liečbe ustálila na úrovni 370–480 µmol/l. Na vyšetrenie sa získali vzorky DNA od 4 postihnutých a troch zdravých detí a tiež od ich otca (sú označení znakom * na obrázku 1).
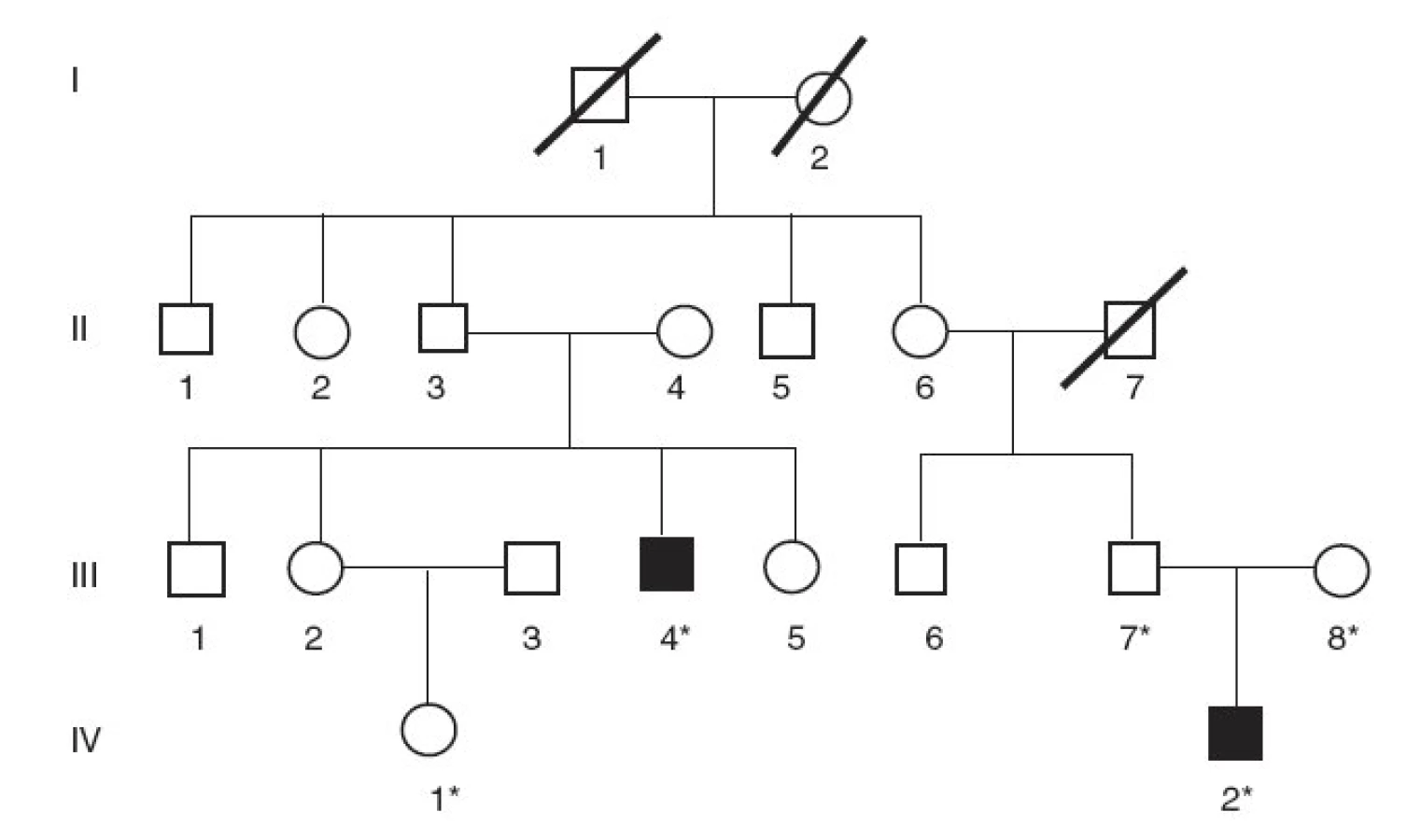
V rodine B nerómskeho pôvodu bez konsangvinity sa analyzovala vzorka DNA od probanda s CN typ I (obr. 2, III/4). Proband mal od narodenia trvalo vysoké hodnoty nekonjugovaného bilirubínu napriek fenobarbitálovej liečbe i fototerapii (560–610 µmol/l). Ako 19-ročný podstúpil transplantáciu pečene, po ktorej sa hodnoty nekonjugovaného bilirubínu za krátku dobu normalizovali. Z tejto rodiny sa vyšetrila aj DNA od netere probanda s klinickou diagnózou CN II (obr. 2, IV/1), stanovenou na základe vysokých hodnôt sérového bilirubínu do 295 µmol/l.
Genetická analýza
DNA sa izolovala z venóznej krvi štandardnou fenol-chloroformovovou extrakciou. PCR reakcie na amplifikáciu všetkých 5 exónov génu UGTIA ako aj jeho regulačných oblastí (promótor resp. „enhancer“) prebehli podľa publikovaného postupu [33]. Amplifikáty boli po precipitácii a špeciálnej príprave sekvenované v automatických genetických analyzátoroch – ABI PRISMR 310 Genetic Analyser a ABI PRISMR 3100-Avant Genetic Analyser (Applied Biosystems). Na detekciu substitúcie T-3279G sa zaviedla reakcia ARMS PCR, výsledok sa vyhodnocoval v 1,5% agarózovom géli. Na detekciu inzercie A(TA)7TAA v oblasti promótora génu sa zaviedla reakcia PCR, ktorá umožňovala amplifikáciu dostatočne krátkeho PCR fragmentu 119 resp. 121 bp, a tým rozlíšenie 2 bp rozdielu medzi jednotlivými genotypmi elektroforézou v 15% nedenaturačnom polyakrylamidovom géle [32]. Alelové a genotypové frekvencie sa vypočítali podľa Hardyho-Weinbergovho zákona.
Projekt bol odsúhlasený Ústavnou etickou komisiou Detskej fakultnej nemocnice s poliklinikou v Bratislave. Od každého pacienta resp. od ich zákonných zástupcov sa vyžiadal písomný informovaný súhlas s účasťou na danom projekte a vyšetrením vzorky DNA.
Výsledky
Kontrolná skupina
Alelová frekvencia inzercie A(TA)7TAA bola identická v kontrolných skupinách osôb rómskeho resp. nerómskeho pôvodu (q = 0,36 a p = 0,64). Tomu zodpovedá aj rovnaká očakávaná distribúcia jednotlivých genotypov v zdravej časti populácie pre oba kontrolné súbory (A(TA)7TAA = 12,9 %, A(TA)6TAA/A(TA)7TAA = 46,1 %, A(TA)6TAA = 41 %).
Alelová frekvencia substitúcie T-3279G v oblasti zosilňovača („enhancer“) génu UGT1A1 sa iba minimálne odlišovala medzi testovanými kontrolnými súbormi (q = 0,51 a p = 0,49 pre osoby nerómskeho resp. q = 0,53 a p = 0,47 pre osoby rómskeho pôvodu). Očakávaná frekvencia homozygotov pre sledovanú mutáciu je podľa týchto údajov 26 % v rámci rómskeho a 28 % v rámci nerómskeho segmentu slovenskej populácie.
Z uvedených výsledkov vyplýva, že takmer všetci homozygoti pre inzerciu A(TA)7TAA boli zároveň aj homozygoti pre mutáciu T-3279G (pričom opačné tvrdenie neplatí). Inými slovami, takmer na každom chromozóme s alelou A(TA)7TAA bola prítomná aj alela T-3279G. Výnimkou z 240-členného kontrolného súboru boli len 4 osoby (1,6 %): dve osoby s genotypom A(TA)6TAA/A(TA)7TAA a T-3279T; dve osoby s genotypom A(TA)7TAA/A(TA)7TAA a T-3279T/T-3279G. Dá sa z toho usudzovať, že pre stanovenie zastúpenia Gilbertovho genotypu v populácii je smerodajná frekvencia inzercie A(TA)7TAA (q = 0,36), na základe ktorej môžeme v slovenskej populácii očakávať 12,9% zastúpenie homozygotov s Gilbertovým genotypom.
Gilbertov syndróm
Analýza DNA na zistenie zodpovedných polymorfizmov A(TA)7TAA resp. T-3279G v regulačnej oblasti génu UGT1A1 sa uskutočnila u 110 nepríbuzných pacientov s klinickou diagnózou Gilbertovho syndrómu. U 101 osôb (91,8 %) boli v homozygotnom stave prítomné obe (A(TA)7TAA a T-3279G) polymorfizmy, čo zodpovedá ich alelovej frekvencii 0,95. U zvyšných 9 pacientov s mierne zvýšenými hodnotami celkového sérového bilirubínu v rozsahu od 23 do 102 µmol.l-1 sa detegovali 4 rozdielne genotypy v oblasti promótora resp. zosilňovača („enhancer“) génu.
Criglerov-Najjarov syndróm
Vyšetrenia sa uskutočnili v dvoch nepríbuzných rodinách s Criglerovým-Najjarovým syndrómom. V rodine A sa u všetkých pacientov s klinickou diagnózou CN typ I identifikovala delécia 1220delA v 4. exóne génu UGT1A1 v homozygotnom stave. Traja vyšetrení zdraví súrodenci a ich otec boli heterozygoti pre túto deléciu ako aj pre obe vyšetrené mutácie v regulačnej oblasti génu (A(TA)7TAA resp. T-3279G) (obr. 1). Všetci vyšetrení členovia rodiny (postihnutí aj nepostihnutí) boli homozygoti aj pre dve ďalšie substitúcie (C-3440A a T-3279G) v oblasti zosilňovača génu UGT1A1.
V rodine B bol proband s CN typ I (obr. 2, III/4) zložený heterozygot na dve rôzne delécie v géne UGT1A1 – v 1. génu UGT1A1 sa odhalila delécia 717-718delAG, kým na druhej alele sa našla delécia 1220delA v 4. exóne, rovnaká, ako sa dokázala aj v rodine A. Sekvenovaním regulačnej oblasti génu UGT1A1 sa zistilo, že pacient je heterozygot pre inzerciu A(TA)6TAA/A(TA)7TAA ako aj pre subtitúciu C-3440A a homozygot pre substitúciu T-3279G.
Diskusia
Gilbertov syndróm
Podstatou Gilbertovho syndrómu v kaukazoidnej a africkej populácii je polymorfizmus v regulačných oblastiach proximálne od génu UGT1, menovite a) v oblasti „TATA“ v polohe -37 až -23 bp (tzv. promótor génu), respektíve b) v oblasti „zosilňovača“ (tzv. „enhancer“), ktorý sa nachádza v polohe -3194 až -3483 bp [8, 11]. Keďže v populácii ázijského pôvodu sa tento typ polymorfizmu vyskytuje podstatne zriedkavejšie, v danej práci sa samostatne hodnotili genetické výsledky v rómskej populácii na Slovensku.
Najčastejší (fyziologický) variant promótora „TATA“ obsahuje šesť opakovaní motívu TA – A(TA)6TAA – a je spojený s normálnou intenzitou vyjadrenia enzýmu. Inzercia ďalšieho motívu TA do sekvencie so vznikom alely A(TA)7TAA je spojená s asi 30% redukciou expresie génu oproti zdravým kontrolám [7, 29]. V našej populácii sme zistili rovnakú alelovú frekvenciu inzerčnej alely A(TA)7TAA v kontrolných skupinách osôb rómskeho resp. nerómskeho pôvodu, čomu zodpovedá 12,9% výskyt homozygotov v zdravej časti slovenskej populácie nezávisle od etnického pôvodu. Naše výsledky sú v súlade s literárnymi údajmi, ktoré uvádzajú výskyt tejto alely TATA tzv. „Gilbertovho“ typu v homozygotnom stave u 9 až 15 % Európanov, 36 % Afričanov, ale iba u 2–3 % príslušníkov východoázijských populácií [5, 6]. Čo sa týka rómskej populácie, ktorá je indického pôvodu, výsledky našej práce sa zhodujú s údajmi Balrama a spol., ktorí dokázali výskyt promótorovej inzercie v homozygotnom stave u 13,1 % indickej populácie [1, 4].
V roku 2001 sa identifikoval ďalší významný polymorfizmus v regulačnej oblasti génu, menovite v oblasti tzv. „zosilňovača“ („enhancer“) génu UGT1A1, označovaného aj ako PBREM („phenobarbital responsive enhancer module“), menovite substitúcia T-3279G [28]. Z našich výsledkov vyplýva 26% resp. 28% očakávaná frekvencia homozygotov s polymorfnou alelou T-3279G v nerómskej resp. v rómskej populácii na Slovensku. Ukazuje sa pritom, že takmer všetci homozygoti pre inzerciu A(TA)7TAA boli zároveň aj homozygoti pre polymorfnú alelu T-3279G (pričom opačné tvrdenie neplatí!). Inými slovami, takmer na každom chromozóme s alelou A(TA)7TAA bola prítomná aj alela T-3279G [20, 33]. Z toho sa dá usudzovať, že pre patogenézu GS je esenciálna práve spojitosť medzi oboma uvedenými polymorfizmami. Kombináciou ich efektov dochádza k redukcii syntézy enzýmu B-UGT až o 60–80 %, teda na úroveň ako bolo pôvodne zistené u pacientov s Gilbertovým syndrómom. Prítomnosť oboch polymorfizmov však určuje iba sklon ku zvýšeným hodnotám sérového bilirubínu. Fenotypový prejav, teda klinická manifestácia hyperbilirubinémie závisí v značnej miere aj od iných faktorov, najmä od rýchlosti produkcie bilirubínu [9, 20, 26]. Z tohto dôvodu za normálnych podmienok zhruba 35–50 % nositeľov Gilbertovho genotypu (najmä žien) zostáva celý život asymptomatických [8].
Criglerov-Najjarov syndróm
Criglerov-Najjarov syndróm (CN) sa charakterizuje mutáciami kódujúcej časti génu UGTIA1, ktoré vedú k úplnému vymiznutiu (typ 1) resp. k veľmi ťažkému deficitu (pod 10 %, typ 2) glukuronizačnej aktivity enzýmu B-UGT [16, 25]. Všetky doteraz pozorované delécie, inzercie a substitúcie so vznikom „stop kodónu“ v homozygotnom stave sa vyskytli výlučne u pacientov s CN typ I. Bodové mutácie s následnou zámenou aminokyselín sú častejšie príčinou vzniku CN typ II [19].
V danej práci referujeme výsledky genetickej analýzy v dvoch slovenských rodinách s Criglerovým-Najjarovým syndrómom typ I. V oboch rodinách sa dokázali mutácie génu UGT1A1, ktoré spôsobujú posun čítacieho rámca so vznikom predčasného „stop kodónu“. Tvorba skráteného proteínu s nulovou enzymatickou aktivitou vysvetľuje závažný priebeh ochorenia s rozvojom jadrového ikteru. Proband z konsangvinnej rodiny rómskeho pôvodu (rodina A) je prvým dokázaným homozygotom pre deléciu 1220delA v 4. exóne génu UGT1A (obr. 1). Pre kompletnosť však treba dodať, že táto mutácia už bola v heterozygotnom stave popísaná u dvojčiat s ťažkou novorodeneckou žltačkou vyúsťujúcou do jadrového ikteru. K rozvoju hyperbilirubinémie a chronickej encefalopatie však v týchto prípadoch prispela i zvýšená produkcia bilirubínu následkom inkompatibility v systéme AB0 s pozitívnym Coombsovým testom [16]. V druhej slovenskej rodine B s CN typ I bol proband zložený heterozygot pre dve rôzne delécie v géne UGT1A1 (obr. 2). Okrem mutácii 1220delA identickej ako v rodine A sa v exóne 1 génu UGT1A dokázala delécia 717-718delAG, ktorá spôsobuje posun čítacieho rámca a vznik predčasného „stop kodónu“. Preto u pacienta dochádza k syntéze skráteného enzýmu B-UGT s nulovou enzymatickou aktivitou. Táto mutácia 717-718delAG už bola opísaná v homozygotnom stave u pacienta s CN typ I [15] a tiež u pacientov s CN typ II v heterozygotnom stave, spolu s dvoma rôznymi missense mutáciami [21].
Záver
Novšie molekulovo-genetické práce sú okrem ich teoretického významu prínosom aj pre klinickú diagnostiku nekonjugovaných hyperbilirubinémií. V rodinách s Criglerovým-Najjarovým syndrómom prispeje prenatálna molekulová diagnostika k redukcii závažných foriem nekonjugovanej novorodeneckej hyperbilirubinémie s potenciálnym vývojom jadrového ikteru a mentálneho poškodenia probanda.
Gilbertov syndróm si v princípe nevyžaduje žiadnu liečbu ani diétu, povoľuje sa aj antikoncepcia a nie je dôvod k obmedzovaniu telesnej aktivity (hyperbilirubinémiu má veľa vrcholových športovcov). Fyzickú zaťaž určuje pacient podľa svojho pocitu a tolerancie. Hlavnou úlohou lekára je, aby pacientovi vysvetlil benígnosť metabolickej zmeny, to že je „viac žltý než chorý“ a že je najlepšie netraumatizovať sa sledovaním výraznosti ikteru.
Aj keď ide o benígny stav, jeho presná molekulovo-genetická diagnostika sa nemôže považovať za samoúčelnú. Pacient má byť plne informovaný o podstate svojho stavu, lebo v dobe akejkoľvek záťaže (choroba, operácia, úraz) môže dôjsť k zvýrazneniu ikteru, ktorý môže byť nesprávne interpretovaný (napr. jednotlivec môže byť omylom odoslaný na infekčné oddelenie s podozrením na infekčnú hepatitídu). Existujú aj situácie spojené so zvýšenou produkciou bilirubínu (napr. novorodenecká hyperbilirubinémia, ikterus dojčeného dieťaťa, talasémia, hereditárna sférocytóza, atď.), kedy Gilbertov genotyp môže byť rizikovým faktorom pre vznik výrazného ikteru. Molekulárne genetické vyšetrenie je preto opodstatnené pri objasnení príčiny u prolongovaného novorodeneckého ikteru, diferenciálnej diagnostike neobjasnenej hyperbilirubinémie. Informácia o genotype by mala byť k dispozícii aj u pacientov s predpokladanou dlhodobou terapiou farmakami metabolizovanými prostredníctvom UGT1 (napr. olanzapin, tranilast, irinotekán, paracetamol, atď.). Pri Gilbertovom syndróme by mali byť dávky týchto farmák upravené s ohľadom na ich pomalšiu glukuronidáciu [3, 8, 18, 22, 23, 26].
Výskum bol podporovaný grantovou úlohou MZ SR 2005/4-DFNSPBA-02.
Došlo: 1. 11. 2008
Přijato: 3. 2. 2009
Prof. MUDr. László Kovács, DrSc., MPH
2. detská klinika LFUK a DFNsP
Limbová 1
833 40 Bratislava
Slovenská republika
e-mail: kovacs@dfnsp.sk
Sources
1. Ando Y, Chida M, Nakayama K, Saka H, Kamataki T. The UGT1A1*28 allele is relatively rare in a Japanese population. Pharmacogenetics 1998;8 : 357–360.
2. Arias IM, Gartner LM, Cohen M, Ezzer JB, Levi AJ. Chronic nonhemolytic unconjugated hyperbilirubinemia with glucuronyl transferase deficiency. Clinical, biochemical, pharmacologic and genetic evidence for heterogeneity. Am. J. Med. 1969;47 : 395–409.
3. Arias IM, Gartner LM, Seifter S, Furman M. Prolonged neonatal unconjugated hyperbilirubinemia associated with breast feeding and a steroid, pregnane-3 (alpha), 20(beta)-diol, in maternal milk that inhibits glucuronide formation in vitro. J. Clin. Invest. 1964;43 : 2037–2047.
4. Balram C, Sabapathy K, Fei G, Khoo KS, Lee EJD. Genetic polymorphisms of UDP-glucuronosyltransferase in Asians: UGT1A1*28 is a common allele in Indians. Pharmacogenetics 2002;12 : 81–83.
5. Beutler E, Gelbart T, Demina A. Racial variability in the UDP-glucuronosyltransferase 1 (UGT1A1) promoter: a balanced polymorphism for regulation of bilirubin metabolism? Proc. Natl. Acad. Sci. U.S.A. 1998;5 : 8170–8174.
6. Borlak J, Thum T, Landt O, Erb K, Hermann R. Molecular diagnosis of a familial nonhemolytic hyperbilirubinemia (Gilbert´s syndrome) in healthy subjects. Hepatology 2000;32 : 792–795.
7. Bosma PJ, Chowdhury JR, Bakker C, Gantla S, de Boer A, Oostra BA, Lindhout D, Tytgat GN, Jansen PL, Oude Elferink RP, Chowdhury R. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert’s syndrome. N. Engl. J. Med. 1995;333 : 1171–1175.
8. Bosma PJ. Inherited disorders of bilirubin metabolism. J. Hepatol. 2003;38 : 107–117.
9. Cebecauerova D, Jirasek T, Budisova L, Mandys V, Volf V, Novotna Z, Subhanova I, Hrebicek M, Elleder M, Jirsa M. Dual hereditary jaundice: simultaneous occurrence of mutations causing Gilbert´s and Dubin-Johnson syndrome. Gastroenterology 2005;129 : 315–320.
10. Crigler JF, Jr, Najjar VA. Congenital familial nonhemolytic jaundice with kernicterus. Pediatrics 1952;10 : 169–179.
11. Farago B, Melegh B. Gilbert szindróma. Orvosi Hetilap. 2008;149 : 1277–1282.
12. Gilbert A, Lereboullet P. La Cholémie simple familiale. Sem. Méd. 1901;21 : 241–245.
13. Hawes EM. N-glucuronidation, a common pathway in human metabolism of drugs with a tertiary amine group. Drug Metab. Dispos. 1998;26 : 830–837.
14. Jirsa M, Petrasek J, Vitek L. Linkage between A(TA)7TAA and -3279T>G mutations in UGT1A1 is not essential for pathogenesis of Gilbert syndrome. Liver Int. 2006;10 : 1302–1303.
15. Kabícek P, Barnincová L. Juvenile hyperbilirubinaemia and its early manifestation in adolescence. Čas. Lék. čes. 2007;146 : 528–532.
16. Kadakol A, Ghosh SS, Sappal BS, Sharma G, Chowdhury JR, Chowdhury NR. Genetic lesions of bilirubin uridine-diphosphoglucuronate glucuronosyl transferase (UGT1A1) causing Crigler-Najjar and Gilbert syndromes: correlation of genotype to phenotype. Hum. Mutat. 2000;16 : 297–306.
17. Kadakol A, Sappal BS, Ghosh SS, Lowenheim M, Chowdhury A, Chowdhury S, Santra A, Arias IM, Chowdhury JR, Chowdhury NR. Interaction of coding region mutations and the Gilbert-type promoter abnormality of the UGT1A1 gene causes moderate degrees of unconjugated hyperbilirubinaemia and may lead to neonatal kernicterus. J. Med. Genet. 2001;38 : 244–249.
18. Kaplan M, Hammerman C, Renbaum P, Klein G, Levy-Lahad E. Gilbert´s syndrome and hyperbilirubinaemia in AB0-incompatible neonates. Lancet 2000;356 : 652–653.
19. Koiwai O, Aono S, Adachi Y, Kamisako T, Yasui Y, Nishizawa M, Sato H. Crigler-Najjar syndrome type II is inherited both as a dominant and as a recessive trait. Hum. Mol. Genet. 1996;5 : 645–647.
20. Maruo Y, D´Addario C, Mori A, Iwai M, Takahashi H, Sato H, Takeuchi Y. Two linked polymorphic mutations (A(TA)7TAA and T-3279G) of UGT1A1 as the principal cause of Gilbert syndrome. Hum. Genet. 2004;115 : 525–526.
21. Maruo Y, Serdaroglu E, Iwai M, Takahashi H, Mori A, Bak M, Calkavur S, Sato H, Takeuchi Y. A novel missense mutation of the bilirubin UDP-glucuronosyltransferase gene in a Turkish patient with Crigler-Najjar syndrome type I. J. Pediatr. Gastroenterol. Nutr. 2003;37 : 627–630.
22. Monaghan G, McLellan A, McGeehan A, Li Volti S, Mollica F, Salemi I, Din Z, Cassidy A, Hume R, Burchell B. Gilbert’s syndrome is a contributory factor in prolonged unconjugated hyperbilirubinemia of the newborn. J. Pediatr. 1999;134 : 441–446.
23. Muchová L, Kráslová I, Lenícek M, Vítek L. Gilbert’s syndrome – myths and reality. Čas. Lék. čes. 2004;143 : 375–380.
24. Peters WHM, Morsche RHM, Roelofs HMJ. Combined polymorphisms in UDP-glucuronosyltransferases 1A1 and 1A6: implications for patients with Gilbert´s syndrome. J. Hepat. 2003;38 : 3–8.
25. Servedio V, d´Apolito M, Maiorano N, Minuti B, Torricelli F, Ronchi F, Zancan L, Perrotta S, Vajro P, Boschetto L, Iolascon A. Spectrum of UGT1A1 mutations in Crigler-Najjar (CN) syndrome patients: Identification of twelve novel alleles and genotype-phenotype correlation. Hum. Mutat. 2005;25 : 325–333.
26, Strassburg CP, Manns MP. Jaundice, genes and promoters. J. Hepat. 2000;33 : 476–479.
27. Strassburg CP. Pharmacogenetics of Gilbert’s syndrome. Pharmacogenomics 2008;9 : 703–715.
28. Sugatani J, Kojima H, Ueda A, Kakizaki S, Yoshinari K, Gong QH, Owens IS, Negishi M, Sueyoshi T. The phenobarbital response enhancer module in the human bilirubin UDP-glucuronosyltransferase UGT1A1 gene and regulation by the nuclear receptor CAR. Hepatology 2001;33 : 1232–1238.
29. Sugatani J, Yamakawa K, Yoshinari K, Machida T, Takagi H, Mori M, Kakizaki S, Sueyoshi T, Negishi M, Miwa M. Identification of a defect in the UGT1A1 gene promoter and its association with hyperbilirubinemia. Biochem. Biophys. Res. Commun. 2002;292 : 492–497.
30. Šašinka M. Choroby pečene. In: Šašinka M, Šagát T, Kovács L. Pediatria. Bratislava: Herba, 2007 : 417–446.
31. Tukey RH, Strassburg CP. Genetic multiplicity of the human UDP-glucuronosyltransferases and regulation in the gastrointestinal tract. Mol. Pharmacol. 2001;59 : 405–414.
32. von Ahsen N, Oellerich M, Schütz E. DNA base bulge vs unmatched end formation in probe-based diagnostic insertion/deletion genotyping: genotyping the UGT1A1 (TA)(n) polymorphism by real-time fluorescence PCR. Clin. Chem. 2000;46 : 1939–1945.
33. Zmetáková I, Ferák V, Minárik G, Ficek A, Poláková H, Feráková E, Kádasi L. Identification of the deletions in the UGT1A1 gene of the patients with Crigler-Najjar syndrome type I from Slovakia. Gen. Physiol. Biophys. 2007;26 : 306–310.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2009 Issue 5
- What Effect Can Be Expected from Limosilactobacillus reuteri in Mucositis and Peri-Implantitis?
- The Importance of Limosilactobacillus reuteri in Administration to Diabetics with Gingivitis
Most read in this issue
- Acute Neuroborrelioses in Children
- Recent Trends in Diagnosis of Fetal Alcohol Syndrome
- Molecular Diagnostics of Hereditary Unconjugated Hyperbilirubinemias in Slovakia
- Standards of the Multidisciplinary Treatment of a Child with Orofacial Cleft